
Abstract
The basic electronic and mechanical properties of 2-Pmmn borophene and their strain and electric field-dependence are studied by the first-principles calculations. The Young's moduli are 236 and 89 GPa in the armchair and zigzag directions, respectively, indicating that the borophene has giant mechanical anisotropy. We also find that the borophene presents anisotropic electronic properties. The borophene is electroconductive in armchair direction but has a bandgap in the zigzag direction. To modulate the band structure, we applied strain and electric fields on borophene, and find that, the resistance of borophene decreases with the increase of applied strain, while the applied electric field has almost no effect on its band structure. The enhanced conductivity of borophene upon applied strain is ascribed to the expansion of the buckled structure through the analysis of the charge density of the strained borophene.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
As a new type of two-dimension (2D) material, borophene is reported to have a serial marvelous nature in electronic, mechanical, optical and thermodynamic properties and can be a powerful competitor to graphene [1–7]. For 2-Pmmn borophene, boron may bond each other neither in a purely covalent nor a purely metallic manner [8]. And due to its unique properties, 2-Pmmn borophene has received widespread attentions in the last five years [9]. The atomic configuration of boron monolayer had been researched and discussed for a long time [10–14]. The earliest study on this issue could be traced back to Ihsan Boustany's work in 1997 [15], in which a series of quasi-planar monolayer boron were proposed and analyzed by theoretical calculations. This method has been developed to evaluate the stability of materials [16]. Besides the binding energy per atom, the phonon dispersion spectrum [17] is another method that we adopted in this work. After the remarkable synthesis of borophene in 2015 [18], the potential of the 2D boron sheet drew the researcher's eyes again. The first question is whether other phases of borophene, except for 2-Pmmn, could be synthesized and the evaluation of the stability. Much more boron allotropes have been proposed [19–23], and the vacancy, such as the honeycomb phase, was found to be an important factor affecting the stability of borophene structures [8, 24, 25]. Borophene structure at a certain vacancy concentration can reach its best stability and flat the quasi-planar buckled triangular sheet [26]. Due to its giant anisotropy in structure and properties, which possesses unique applications, we mainly focus on 2-Pmmn borophene in this work. In further studies, Zhong et al [27] systematically investigate the mechanical and electronic properties of a few-layers 2-Pmmn borophene. For monolayer 2-Pmmn borophene, the Young's modulus in armchair direction can be as high as that in graphene, while it is relatively lower in zigzag direction and a negative Poisson's ratio could be another unusual feature [18]. The high critical strain shows that borophene has good extensibility. Both the mechanical and electronic properties present a giant anisotropy. The investigation of the band structure of borophene indicates that bands that cross the fermi level only exist in armchair direction and may show metallic behavior on the whole. These results are supported by other studies [25–27]. However, for practical applications, the characteristics can usually be optimized and promoted by some methods, such as strain engineering [28–31] or applied electric field [6]. Although such a detail that the electronic properties of borophene have been found out, there is still a lack of this issue.
In this work, the first-principles calculations are used to investigate the structure stability of 2-Pmmn borophene by phonon dispersions. Then, we use both the stiffness constants and stress-strain response to study its mechanical properties. The electronic properties are described by the band structure and density of state (DOS). Finally, we regulated the band structure of 2-Pmmn borophene by the mechanical strain and electric fields and look into the details of the charge density, as well as analyzed the behavior of strain-controlled borophene.
2. Model and method
We performed the calculations using the Vienna ab initio package (VASP), which is based on the Density Functional Theory (DFT) method. The generalized gradient approximation (GGA) using Perdew–Burke–Ernzerhof (PBE) [32–34] was adopted to describe exchange-correlation functional. The GGA-PBE has been found be more suitable for the density inhomogeneity structures and widely applied for the low-dimensional systems [35–37]. The Projector augmented wave method was employed with a cutoff energy of 500 eV. The conjugate gradient method was used for geometry optimizations. A vacuum layer with a thickness of 10 Å was built to avoid image-image interaction in the Z direction. For the evaluation of mechanical properties, the Brillouin zone was sampled 30 × 15 × 1 k-point mesh using the Monkhorst-Pack method. Uniaxial tension condition was applied to evaluate the mechanical properties of the borophene. The lattice size along the loading direction was increased in multiple steps with small engineering strain steps of 0.004. As for the calculation of the band structure of borophene, a k-point path along Γ-Y-M-X-Γ in the Brillouin zone was set and sampled 50 points in each line. The set of strain engineering is the same as that of mechanical properties. The electronic field in the Z direction was applied, which is ranging from 0 to 0.5 V/ step by every 0.1 V/
step by every 0.1 V/ to investigate the relationship of the applied electronic field and the electronic properties of borophene.
to investigate the relationship of the applied electronic field and the electronic properties of borophene.
As shown in figure 1, the 2-Pmmn borophene has an anisotropic structure in the two orthogonal directions. The black arrows, marked as X and Y, are the relative Cartesian coordinate. Along the Y direction, a buckled line can be observed, due to which, the Y direction is usually called the zigzag direction. And the non-corrugations X direction is called the armchair direction. The primitive cell of borophene is marked by the red arrow, and the lattice constants a and b are calculated to be around 1.6136 and 2.8654  respectively, which well consists with the experimental result with a trivial discrepancy of about 1% [18]. As shown in figure 1(b), the borophene presents a quasi-planar structure with corrugations, which is different from graphene. The buckling height is 0.911
respectively, which well consists with the experimental result with a trivial discrepancy of about 1% [18]. As shown in figure 1(b), the borophene presents a quasi-planar structure with corrugations, which is different from graphene. The buckling height is 0.911  The benchmark calculations of borophene are consistent with the references [27]. For the tensile simulation, two types of in-plane strains are considered, i.e., the biaxial and the uniaxial loading, respectively. For the uniaxial tension, a small incremental strain is applied along the stretch direction, while the internal atomic positions along other two directions are fully relaxed. The basic electronic and mechanical properties of 2-Pmmn borophene under applied strain and electric fields will be demonstrated in the next section.
The benchmark calculations of borophene are consistent with the references [27]. For the tensile simulation, two types of in-plane strains are considered, i.e., the biaxial and the uniaxial loading, respectively. For the uniaxial tension, a small incremental strain is applied along the stretch direction, while the internal atomic positions along other two directions are fully relaxed. The basic electronic and mechanical properties of 2-Pmmn borophene under applied strain and electric fields will be demonstrated in the next section.

Figure 1. (a) Top and (b) side views of 2-Pmmn borophene.
Download figure:
Standard image High-resolution image3. Results and discussion
3.1. Structural stability of 2D borophene
Structural stability of borophene is the first issue discussed before further investigations. Herein, we performed the phonon dispersion calculation to confirm the structural stability of 2-Pmmn borophene. As shown in figure 2, the phonon spectrum and projected phonon density of state (PDOS) for in-plane optical branch B(X) and B(Y) as well as out-of-plane acoustic branch B(Z) were calculated by applying the PHONONPY code [38]. In the phonon spectrum of borophene, two small imaginary frequencies, one in near-gamma point and another in  -X, can be found, which indicates that 2-Pmmn borophene could not be dynamically stable against out-of-plane vibrations. This result is different from the previous study of monolayer boron sheet by Peng et al [2], but similar to the investigation of bilayer borophene by Zhong et al [27]. Since 2-Pmmn borophene has been synthesized, a reasonable explanation for this paradox is that the substrates could sustain borophene structure in a certain condition, such as low temperature and a small enough imaginary frequency value. As for projected PDOS, the B(Z) mainly contributes to the three low acoustic phonon branches, indicating that the whole borophene has a trend to translation along the Z direction because there is no constrain in this direction. Then, the B(X) mainly contributes to the high optical phonon branches, suggesting that the interaction between boron atoms in the X direction is stronger than the other two directions. Last, the vibrations in the Y direction are stronger than that in the Z direction but weaker than the X direction which can be ascribed to the buckled structure of borophene. Through the discussion of phonon spectrum and projected PDOS, we can conclude that the 2-Pmmn phase can be not the most energetically favorable structure among allotropes of borophene. However, on one hand, the free-standing 2-Pmmn borophene has not been studied clearly in experiments; on the other hand, for borophene on substrates, applications in microelectronics equally have a promising future. Besides, the phonon dispersions present an anisotropic behavior, which, in kinetics, provides the basis for mechanical and electronic properties.
-X, can be found, which indicates that 2-Pmmn borophene could not be dynamically stable against out-of-plane vibrations. This result is different from the previous study of monolayer boron sheet by Peng et al [2], but similar to the investigation of bilayer borophene by Zhong et al [27]. Since 2-Pmmn borophene has been synthesized, a reasonable explanation for this paradox is that the substrates could sustain borophene structure in a certain condition, such as low temperature and a small enough imaginary frequency value. As for projected PDOS, the B(Z) mainly contributes to the three low acoustic phonon branches, indicating that the whole borophene has a trend to translation along the Z direction because there is no constrain in this direction. Then, the B(X) mainly contributes to the high optical phonon branches, suggesting that the interaction between boron atoms in the X direction is stronger than the other two directions. Last, the vibrations in the Y direction are stronger than that in the Z direction but weaker than the X direction which can be ascribed to the buckled structure of borophene. Through the discussion of phonon spectrum and projected PDOS, we can conclude that the 2-Pmmn phase can be not the most energetically favorable structure among allotropes of borophene. However, on one hand, the free-standing 2-Pmmn borophene has not been studied clearly in experiments; on the other hand, for borophene on substrates, applications in microelectronics equally have a promising future. Besides, the phonon dispersions present an anisotropic behavior, which, in kinetics, provides the basis for mechanical and electronic properties.
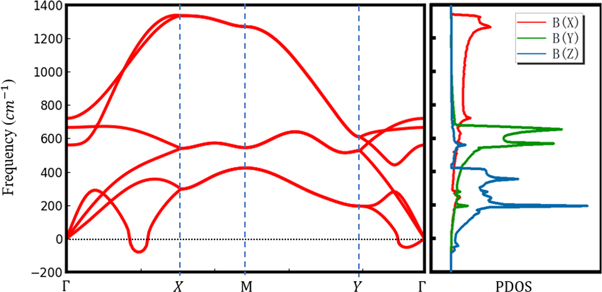
Figure 2. Phonon spectrum and projected PDOS of 2-Pmmn borophene.
Download figure:
Standard image High-resolution image3.2. Mechanical property of borophene
After discussing the structure stability, mechanical properties of borophene are further studied. Mechanical properties can be defined as the performance that materials behave when it is loaded by an external force. Meanwhile, the performance can be characterized by elastic stiffness constants. For orthogonal symmetry under plane stress, the four nonzero elastic stiffness constants can be written into the stress-strain formula as followed [39].
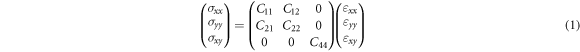
where  and
and  are the stress and strain components, respectively. In our calculations, these elastic stiffness constants
are the stress and strain components, respectively. In our calculations, these elastic stiffness constants 

 and
and  are 247.1, 4.08, 106.5, 525.5 GPa, respectively. The main engineering constants, Young's modulus
E
and Passion's ratio
are 247.1, 4.08, 106.5, 525.5 GPa, respectively. The main engineering constants, Young's modulus
E
and Passion's ratio  can be derived as [40].
can be derived as [40].
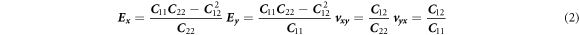
where  = 247 GPa,
= 247 GPa,  = 106.4 GPa,
= 106.4 GPa,  = −0.038,
= −0.038,  = −0.016 in this work.
= −0.016 in this work.
Besides, according to the estimation formula for Young's modulus and Poisson's ratio, we calculated the constants for all angles. For the model of borophene in this work, which possesses orthogonal symmetry, the Young's modulus and Poisson's for all angles can be estimated by the formula followed [41, 42].
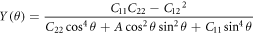
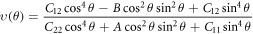
where the A =  −2
−2  B =
B =  In figure 3, the calculated angle-dependent Young's modulus and Poisson's ratio , which vary from 106.4 GPa to 305 GPa and from −0.816 to −0.017, respectively, suggesting the highly anisotropic mechanical properties of borophene. The negative Poisson's ratio is mainly owing to the quasi-planar structure. When the quasi-planar structure is stretched in one direction, and the corrugations are unfolded, leading to the expansion in all the in-plane directions.
In figure 3, the calculated angle-dependent Young's modulus and Poisson's ratio , which vary from 106.4 GPa to 305 GPa and from −0.816 to −0.017, respectively, suggesting the highly anisotropic mechanical properties of borophene. The negative Poisson's ratio is mainly owing to the quasi-planar structure. When the quasi-planar structure is stretched in one direction, and the corrugations are unfolded, leading to the expansion in all the in-plane directions.

Figure 3. The calculated angle-dependent (a) Young's modulus and (b) Poisson's ratio of borophene. The negative Poisson' ratio presents a near-zero value.
Download figure:
Standard image High-resolution imageFurthermore, we investigated the strain-stress response by applied uniaxial and biaxial strain, respectively. Calculated biaxial and uniaxial stress-strain responses of borophene along the armchair and zigzag loading direction are illustrated in figure 4. In figure 4(a), the biaxial stress-strain curves for both armchair and zigzag directions include an elastic stage, yield stage and break stage. The lines gradually trend up to the ultimate tensile strength at which the material yields its maximum load-bearing ability. The critical stain could be easily found to be about 12%. By the linear fitting from 0 to 0.02 in the stress-strain curve, the Young's modulus along the armchair direction is estimated to be about 211 GPa, which is two times larger than that along the zigzag direction (84 GPa), indicating the anisotropic mechanical properties of borophene. Figures 4(c)–(d) shows the uniaxial stress-strain curves. Under uniaxial strain, the Young's moduli are 236 and 89 GPa along the armchair and zigzag directions, respectively, which is slightly larger than those under biaxial strain in both directions.
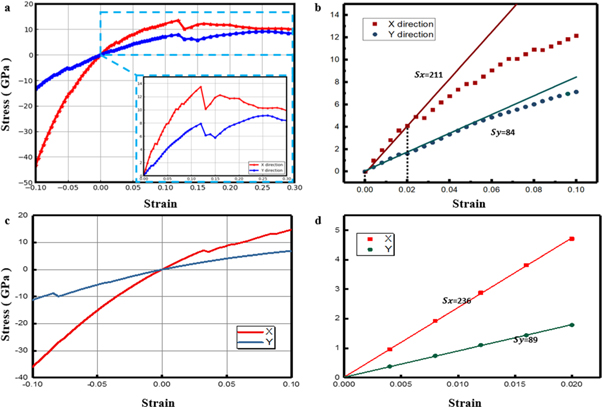
Figure 4. Biaxial (a), (b) and uniaxial (c), (d) stress-strain response of 2-Pmmn borophene.
Download figure:
Standard image High-resolution imageTo investigate the dynamical stability of strained borophene, we investigate the phonon spectrum of borophene with both tension and compression strains, as shown figure 5. For tension and compression strains smaller than 4%, no imaginary frequency is clearly identified, indicating the structural stability of borophene with relatively low strain. However, the tension and compression strains increase and become larger than 8%, obvious phonon imaginary frequency appears in Γ-X and Y-Γ. Therefore, the borophene remains its structural stability with applied strain lower than 4%, which becomes instable when the strain increases and exceeds 8%.

Figure 5. Phonon spectrum of 2-Pmmn borophene under both tension and compression strains of (a) εx = −0.04, (b) εx = 0.04, (c) εx = −0.08, and (d) εx = 0.08, respectively.
Download figure:
Standard image High-resolution image3.3. Electronic property of borophene
The electronic band structure, the density of states (DOS) and project DOS (PDOS) of borophene are shown in figure 6. In figure 6(a), there are two bands cross the Fermi energy level in Γ-Y and M-X, together with DOS (figure 6(b)), in which forbidden band does not exist, indicating its metallic behavior along the armchair direction. On the contrary, band gaps, marked as BG1 and BG2, between the valence and conduction bands exist in Γ-Y and M-X. The anisotropy in electronic property indicates that borophene may behave as a semiconductor in this direction. The electronic properties of borophene are unusual and attractive.
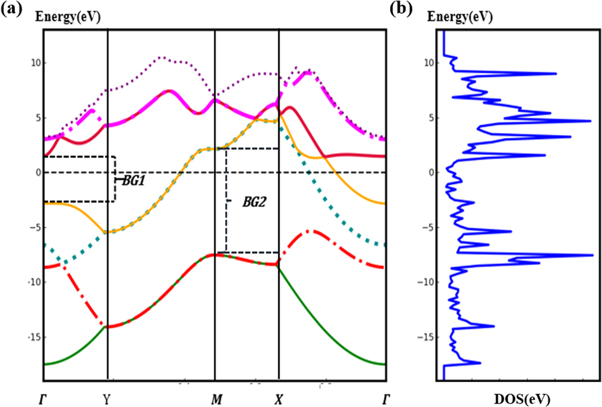
Figure 6. Band structure, DOS and PDOS of borophene. The fermi level corresponds to zero. BG1 and BG2 stand for the value of the bandgap in Γ-Y and M-X.
Download figure:
Standard image High-resolution image3.4. Strain and electric field modulation of borophene
Borophene shows conductivity in the armchair direction, while semi-conductivity along the zigzag direction due to the existence of the band gap. The electronic band structure of borophene with applied uniaxial strain is shown in figure 7. With the increment of tensile strain, both BG1 and BG2 decrease slightly, whereas the overall properties remain unchanged.
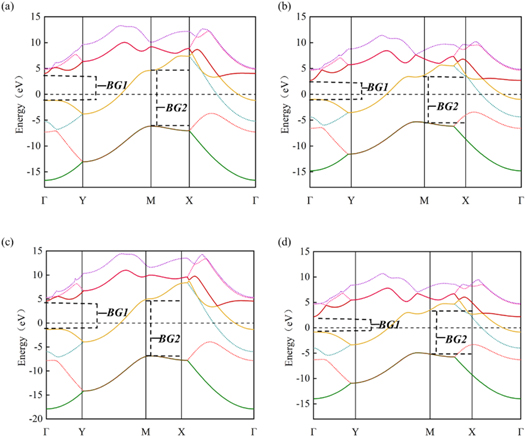
Figure 7. Electronic band structure of 2-Pmmn borophene under axial strain of (a) εx = −0.04, (b) εx = 0.04, (c) εx = −0.08 and (d) εx = 0.08.
Download figure:
Standard image High-resolution imageFor the utilization in the future electronic application of borophene, its band structure regulation via applied strain and the electric fields was further studied to get a better knowledge of the electronic properties of borophene. The tendency of BG1 and BG2 under biaxial strain, armchair uniaxial strain and zigzag uniaxial strain ranging from −0.1 to 0.1, were detailed studied and shown in figure 8. For biaxial strain in figure 8(a), both the BG1 and BG2 decrease when biaxial strains increase. As for uniaxial strain, the bandgap has an orientation dependency. In figure 8(b), with the armchair uniaxial strain increasing, the BG2 decreases, and BG1 nearly remains still. In figure 8(c), zigzag uniaxial strains can lower the BG1 more obviously than BG2. On the other hand, in figure 8(d), when applying an electric field in the Z direction, ranging from 0 V/ to 0.5 V/
to 0.5 V/ the BG1 decreases from 4.03 to 4.06 eV, while BG2 decreases from 9.03 to 9.05 eV. Since the range of change is very small compared to strain engineering, we could conclude that the applied electric field has almost no effect on the borophene band structure.
the BG1 decreases from 4.03 to 4.06 eV, while BG2 decreases from 9.03 to 9.05 eV. Since the range of change is very small compared to strain engineering, we could conclude that the applied electric field has almost no effect on the borophene band structure.
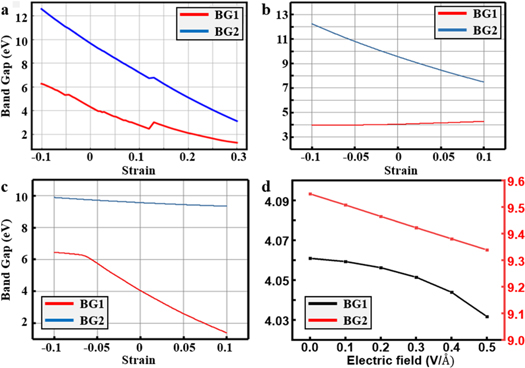
Figure 8. The relationship between bandgap and (a) biaxial strain, (b) armchair uniaxial strain, (c) zigzag uniaxial strain and (d) applied electric field in the Z direction.
Download figure:
Standard image High-resolution imageAs discussed above, the applied strain can be regarded as an effective method to regulate the electronic band structure of borophene. Here, the charge density in two specified cross-sections [010] and [−110], presenting the bonds along the armchair and zigzag directions, respectively, was analyzed to look into stain engineering under biaxial strain. In figures 9(a)–(c), the bonds were gradually elongated and the charge density in the center of the bond decreases, indicating the weaker B-B bonds. In figures 9(d)–(g), the charge density in the B-B bond decreases along the armchair direction. Thus, as the borophene is stretched, the bonds are weakened. This phenomenon can be attributed to corrugations in the zigzag direction. When the corrugations are unfolded, the localized bonds could be changed to a delocalized bond. On the whole, the bandgap can be decreased with the increased strain, and thus the enhancement of borophene conductance by strain engineering.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Charge density in [010] and [−110] crystal plane under −0.1, 0 and 0.5 biaxial strain.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
In this work, we investigate the mechanical and electronic properties of 2-Pmmn borophene using the first-principles calculations. The dynamic stability was studied by the calculations of the phonon spectrum and Projected PDOS, which shows that the 2-Pmmn borophene is not dynamically stable. Then, the mechanical constants were discussed, finding that borophene possesses strong strength in armchair direction and highly anisotropic mechanical properties that the Young's modulus in armchair direction is two times larger than that in the zigzag direction. The band structure and DOS show that borophene exhibits highly anisotropic metallic behavior. And the bandgap has an orientation dependency. In the inspiration of the changes in charge density, we bring together the structural, mechanical and electronic behavior by the language of the valence bond theory. The enhanced conductivity of borophene upon applied strain is ascribed to the expansion of the buckled structure through the analysis of the charge density of the strained borophene.
Acknowledgments
X Wang thanks the support of NSFC (Grant No. 11602252). Y Gao thanks the support of Joint Fund of Ministry of Education of China for Equipment Pre-research (Grant No. 6141A02022136), the Shanghai Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, Shanghai Rising Star Program (A type) (Grant No. 18QA1401300), and the Open Project Program of Wuhan National Laboratory for Optoelectronics (NO. 2020WNLOKF007). T Xu thanks the support of NSFC (Grant No. 11802169).
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.