
Abstract
This article reports the oxidation of cumene by H2O2 in the presence of CO2. It describes the role played by carbon dioxide on the course of the reaction. The reaction was catalyzed by unsupported Co1.5PW12O40 and supported on activated carbon. The prepared materials were characterized by ICP, IR, XRD and UV. Analysis of the reaction products by gas chromatography coupled with mass spectrometry and gas chromatography showed that 2-phenyl-2-propanol and acetophenone were the main products of the reaction. Activated carbon significantly increases conversion by increasing Co1.5PW12O40 accessibility to cumene molecules. The improvement in the conversion and selectivities of 2-phenyl-2-propanol and acetophenone by the use of the oxidizing system H2O2/CO2compared to the use of H2O2 alone or CO2 alone is due to the role of the percarbonate entity  formed by reaction between H2O2 and CO2. Oxidation by large amounts of H2O2 decreases the conversion by decreasing the solubility of cumene in the resulting aqueous medium. An increase in the reaction time resulting in a decrease in the concentration of H2O2 and leaving the effect of the predominant CO2 accentuates the cracking reactions.
formed by reaction between H2O2 and CO2. Oxidation by large amounts of H2O2 decreases the conversion by decreasing the solubility of cumene in the resulting aqueous medium. An increase in the reaction time resulting in a decrease in the concentration of H2O2 and leaving the effect of the predominant CO2 accentuates the cracking reactions.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Oxidation of hydrocarbons is a key challenge in petrochemical industrial processes due to the efficient conversion from cheap petroleum feed stocks to value-added oxygenated compounds (Liao et al 2014, Labinger 2016, Silva et al 2017, Nesterov et al 2018, Xu et al 2018). Oxidation of cumene into 2-phenyl-2- propanol (Ph-PrOH) and acetophenone (Ph = O) is one of such reactions. Ph-PrOH and Ph = O products are important intermediates. Ph-PrOH is used as a fragrance ingredient in many compounds such as cosmetics, shampoos, toilet soaps household cleaners and detergents (Fahlbusch et al 2003). Ph = O is a raw material for the production of pesticides perfumes, pharmaceuticals, resins and alcohols (Alcántara et al 2000). Currently, 2-phenyl-2-propanol is obtained as a by-product in the production of propylene oxide by using Cumene hydroperoxide in an industrial process (Liu et al 2006, Tsuji and Oku 2006). Currently Cumene hydroperoxide was industrially synthesized by using air as oxidant and Cumene hydroperoxide as initiator under high temperature and pressure. (the operating temperatures are 353–393 K; the pressures are 100–600 kPa of air). To prevent the decomposition of the hydroperoxde produced, alkaline solution was used to neutralize the acids formed (Hsu and Cheng 1998, Suresh et al 2000) . Besides Low degrees of conversion equal to 20% and selectivity to hydroperoxide of 90%–95%, are achieved there are still some drawbacks such as poor safety (Wu et al 2008, Dobras and Orlińska 2018). This method, although selective produces large amounts of waste water alkaline or sulfur. The expensive and unecological post-treatment of wastewater and the 2-step synthesis of 2-phenyl-2-propanol make the process unattractive. Thus, it would be more beneficial to develop a cleaner synthetic route by using an environmentally friendly oxidizing agent. The most commonly used oxidants for oxidation reactions are tert-butyl hydroperoxide, hydrogen peroxide and nitric acid. However, these oxidants have serious drawbacks and are not very desirable for use in industrial applications. The use of tert-butyl hydroperoxide and nitric acid produces a large amount of organic pollution and waste. Hydrogen peroxide produces water by decomposition, which contributes to the deactivation of several catalysts (Sundaravel et al 2013, Liu et al 2016). In term of efficiency and eco-friendly sustainability, hydrogen peroxide is the suitable oxidant compared to these oxidizing agents, but its cost limits its application at industrial scale. To minimize its cost, it can be used in a mixture with CO2. In fact, it has been reported that when hydrogen peroxide is used with carbon dioxide its efficiency increased (Hâncu et al 2002, Nolen et al 2002). Among the catalysts used in oxidation reactions, polyoxometalates have proved their effectiveness thanks to their oxidizing and acidic properties that can be adjusted to catalyze specific reactions by the choice of heteroatom, counter-anion and addenda atoms (Katryniok et al 2013). This is why they have been widely used in the field of oxidation catalysis. (Lechner et al 2016, Chen et al 2018). In addition, they are water tolerant (Okuhara 2002). Unfortunately, when they are used in the liquid phase and in the polar medium, they have the disadvantages of homogeneous catalysts, that is to say, difficulties of recycling and purification of the products. Therefore, their utilization as heterogeneous catalytic systems are promising ways. One of the methods used to heterogenize polyoxometalates is to load them on a support. In this work, we report the catalytic activity of a Keggin-type polyoxometalate (CoPW12O40) supported on activated carbon for the oxidation of cumene by H2O2 in the presence of CO2. Cobalt was selected as a cation, because cobalt based catalysts are among the catalysts that have shown high catalytic activities in oxidation reactions. Li et al 2012 reported that that the addition of Со(ОАс)2 as a cocatalyst enhanced significantly the oxidation of cyclohexene. Satokawa et al reported that Co(II) complexe of 2-pyrazine carboxylic acid encapsulated in the Y-zeolite showed higher conversion of cyclohexene leading to 2-cyclohexen-l-one and 1,2-epoxycyclohexane as the major products (Chutia et al 2009). Moreover, the industrial production of cyclohexanone and cyclohexanol mixture used in the manufacture of nylon is achieved by oxidation of cyclohexane using cobalt naphthenate salt catalyst (Musser 2000). Thus, it would be interesting to explore a catalyst having advantages of PW12O40]3− with the cobalt element as counter-cation.
2. Experimental
2.1. Materials
Cumene, C9H12 99.8% purchased from Sigma Aldrich and sodium tungstate, Na2WO4.2H2O, (96%). Tetraethyle ammonium bromide (TEABr), (>99%) from Merck-Schuchardt. Activated carbon (activated decolorizing powder) and CoSO4.7H2O from British Drug Houses Ltd (BDH) Chemicals Ltd, Poole, England).
2.2. Preparation of the catalysts
2.2.1. Bulk Co1.5PW12O40
12-Tungstophosphoric acid H3PW12O40.13H2O was prepared according to a well-known method (Rocchiccioli-Deltcheff et al 1983).
Co1.5PW12O40 (abbreviated CoPW) was prepared by proton substitution of the H3PW12O40 heteropolyacid. This substitution is as follows. To an aqueous solution of H3PW12O40, a required amount of Ba(OH)2.8H2O (to neutralize the 3 protons) is added slowly. Then, the required amount of Ba(OH)2.8H2O was added, resulting in the formation of insoluble barium salt, which was removed by filtration. The filtrate is allowed to stand for a few days at 4 °C to allow precipitation of the CoPW salt which is then recovered from the solution by filtration.
2.2.2. Supported Co1.5PW12O40
The attachment of the CoPW heteropolyanions onto the activated carbon (AC) support necessitates the creation of oxygenated groups on the support, and what was done by oxidation was created by oxidation with concentrated nitric acid.
The preparation of the supported catalyst was achieved as follow:100 mg sample of AC was dispersed in 100 ml of nitric acid (65%) and heated for 5 h at 80 °C with stirring. The desired amount of carbon support was dispersed in 100 ml of nitric acid (65%) and heated for 5 h at 80 °C with stirring. The mixture containing the activated carbon is allowed to cool to room temperature, then filtered and washed with deionized water at pH 7 and dried at 100 °C overnight. A desired amount oxidized AC was then added to the desired amount of CoPW already dissolved in acetone and the mixture was kept under stirring for 30 min. Then after it was heated at about 60 °C to remove the excess acetone and dried overnight in an oven at 80 °C. the supported catalyst was denoted AC-CoPW.
2.3. Characterization of the catalysts
The catalysts were characterized by Fourier transforms infrared (FT-IR) spectra, x-ray diffraction (XRD), inductively coupled plasma spectrometry (ICP) and UV-Vis. IR spectra were recorded by means of an infrared spectrometer SHIMADZU FT- IR NICOLET- 6700 (4000−400 cm−1). XRD characterization were carried out employing an Ultima IV x-ray Rigaku diffractometer using Cu-Kα radiation. ICP measurements using a Perkin Elmer Nexion 300D Spectrometer. UV-Vis characterization was achieved by means of double beam UV-Vis spectrophotometer (Philips 8800). Surface area measurement BET was determined using NOVA 2200e analyzer (Quantachrome, Japan) by the adsorption of N2 at 77 K. Catalysts Surface morphology was analyzed by using a JSM-7600F (JEOL Ltd, Japan) scanning electron microscope (SEM).
2.4. Catalytic oxidation
The experiments were carried out in a stainless steel autoclave equipped with a pressure gauge and manometer controls for pressure settings. The temperature of the autoclave was adjusted by a heating jacket. The temperature of the autoclave is controlled by a heating jacket. The procedure is as follow: a given amount of catalyst and co-catalyst (TBABr) were added to the cumene and hydrogen peroxide (30% in aqueous solution) solution. The mixture was stirred magnetically and heated to the desired temperature under CO2 pressure. After the required time, the resulting mixture was cooled, sampled and analyzed. Qualitative analysis was achieved by a Gas Phase Chromatograph (Thermo Scientific Trace GC Ultra) equipped with a capillary column (TR 5, ID 0.53 mm Film 1 μM). Quantitative analysis was done occasionally by Gas chromatography-mass spectrometry (GC-MS) using a Thermo Trace GC Ultra gas chromatograph AI 3000 equipped with a TR-5 MS-SQC capillary column (30 m × 0.25 mm i.d., phase thickness 0.25 μm) was used with helium as the carrier gas (at a flow rate of 1 ml min−1).
3. Results and discussion
3.1. Catalysts characterization
3.1.1. Infrared spectroscopy
Figure 1 shows the FT-IR spectra of the oxidized AC, CoPW and AC-CoPW catalysts. The spectrum of the oxidized AC (figure 1(b)) shows bands at 1346, 1730, and 1536 cm−1 which were assigned to C—O, C=O, and C=C groups, respectively. The peak at around 1730 cm−1 correspond to the C=O strength vibration in the COOH group (Fanning and Vannice 1993, Zhuang et al 1994, Shanmugharaj et al 2007, Yang et al 2017). This band could be due to either lactone groups (Fanning and Vannice 1993) or to carboxyl groups in aromatic compounds (Zhuang et al 1994, Yang et al 2017). Thus, these results show that the oxidation treatments have created oxygen groups, such as carboxylic and hydroxyl groups, on the carbon surface.

Figure 1. IR spectra of (a) CoPW; (b) AC-CoPW (c) AC.
Download figure:
Standard image High-resolution imageThe spectrum of CoPW (figure 1(a)) shows bands at 1080.2, 982.9, 883.2 and 766.8 cm−1. These bands which are assigned to the stretching modes vas (P–Od), vas (W–Od), vas (W–Ob–W), and vas (W–Oc–W), (Oa, oxygen atom bound to 3W atoms and the central P atom; Ob and Oc, bridging oxygen atoms; Od, terminal oxygen) are characteristic of the keggin structure (Rocchiccioli-Deltcheff et al 1983). As for the FT-IR spectrum of AC-CoPW, it can be seen in figure 1(b), that besides the bands related to the carbon, the four characteristic bands of keggin structure are present. These latter indiquent that CoPW loaded on the AC has preserved its Keggin structure.
3.1.2. X-ray diffraction
Figure 2 the x-ray diffraction patterns of the CoPW and AC-CoPW. The reflections at 9.8°, 21.0°, 26.2°, and 34.9°, observed in the patterns of CoPW (figure 2(a)), indicated that this latter has both a Keggin structure (Fournier et al 1992, Yoshimune et al 2002). Indeed, the x-ray diffractions for the keggin structure show peaks in each of the four ranges of 2θ, namely 7°–10°, 16 –23°, 25°–30° and 31°–38°. The reflections observed at 26.1°, 43.1°, 53.5° and 78.7° 2θ for AC-CoPW (figure 2(b)) correspond to (002), (100), (004) and (110) diffractions respectively of hexagonal graphite (Zhang et al 2002, Sun et al 2005, Delidovich and Palkovits 2016). These reflections indicated that the graphitic structure of AC was preserved after oxidation treatment. Besides the reflections assigned to AC, characteristic peaks of CoPW are also observed. These results indicated that Keggin structure was preserved after loading on AC support. Thus, both XRD and FTIR characterizations showed that AC-CoPW catalyst was successfully synthesized.
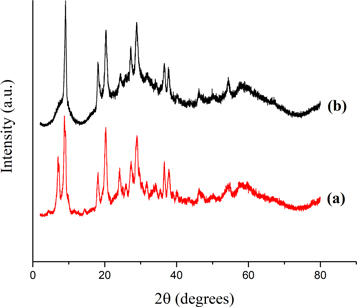
Figure 2. XRD spectra of (a) CoPW and (b) AC-CoPW.
Download figure:
Standard image High-resolution image3.1.3. Elemental analyses
Elemental analysis of CoPW was done by ICP and the results were reported in table 1(a). The results which were determined by considering 1 atom of phosphorous per Keggin unit were found in a good agreement with the expected ones for tungsten and counter ion. Analysis of CoPW loaded on AC support was done by UV-visible in aqueous solution. It has been reported that CoPW in aqueous solution showed an absorbance at 254 nm (Eid et al 2013, Grama et al 2014). The amount of CoPW loaded on AC support was determined by considering the intensity of this band. The results (table 1(b)) showed that the experimental amount of CoPW (0.315 g/0.100g) was very close to the nominal one loaded on AC support (0.350 g/0.100g)
Table 1. Elemental analysis data of the prepared CoPW and AC-CoPW.
| Catalyst | Nominal | Experimental |
|---|---|---|
| (a) P/Co/W (% mol) | 1/1.5/12 | 1/1.52/11.97 |
| (b) CoPW-AC (Wt%) | 0.350/0.100 | 0.315/0.100 |
3.1.4. Surface morphology and Surface Area
The surface morphology of the activated carbon before and after treatment with HNO3 was analyzed by SEM. The results obtained (figure 3) showed that the activated carbon before treatment consisted of particles of variable sizes forming spaces between them (pores) whereas, for the activated carbon treated with nitric acid, the particles are agglomerated and the surface appeared as a porous mass. These results are confirmed by the surface area measurement. In fact, the activated carbon before treatment has a surface area of 1019.66 m2 g−1, whereas the acid treated activated carbon has a surface area of 628.69 m2/g. The treatment of the activated carbon with HNO3 reduces considerably the specific surface area. These results are corroborated by those reported in the literature. In fact, many studies reported that nitric acid treatment reduces the specific surface area (Soudani et al 2013, Bernal et al 2018, Rehman et al 2019). Soudani et al 2013 reported that this reduction depends on the concentration of HNO3. In the opinion of the author, this reduction is also due to the oxygen groups created by the treatment with HNO3 which are probably fixed at the entrance of the micropores leading to a reduction in the pore volume.

Figure 3. The SEM images of (a) activated carbon (AC) (b) oxidized activated carbon with HNO3 (OAC).
Download figure:
Standard image High-resolution image3.2. Catalytic activity
AC-CoPW activity was evaluated for the oxidation of cumene by H2O2/CO2 oxidizing system. The experiments were conducted at 75°C for 7 h under different pressures of CO2. Analysis by GC-MS showed that the reaction led to acetophenone (Ph = O), 2-Phenyl-2-propanol (Ph-PrOH) and phenol (Ph-OH) as major products. Ethylbenzene, toluene, benzene and acetone were obtained as minor products (scheme

Scheme 1. Main products obtained by oxidation of cumene with H2O2 in the presence of CO2 over AC-CoPW catalyst.
Download figure:
Standard image High-resolution image3.3. Effect of the support
The effect of AC support on cumene oxidation was studied by comparing the unsupported CoPW catalytic activity and CoPW supported on AC. Catalytic tests were carried out at 75 °C for 7 h under a CO2 pressure of 0.55 MPa. The results (table 2) show that AC support improved conversion. In fact, the conversion per gram of CoPW obtained over the bulk CoPW (29.1%) increased to (55.6%) when CoPW was supported on AC. This result indicated that the conversion was almost doubled (1.9 times). This considerable increase in conversion can be explained by the fact that the AC support increases the accessibility of the CoPW heteropolyanion to the cumene molecules (organic phase). As for the selectivities, the results show that of Ph = O and that of Ph-CHOH remain almost unchanged. On the other hand, for the reaction catalyzed by the supported catalyst, the selectivity for PhOH decreased in favor of those of the other cracking products (ethylbenzene, toluene and benzene). In fact, when the reaction has been catalyzed by the unsupported CoPW catalyst, the selectivities for toluene, benzene and ethylbenzene obtained are respectively 0%, 0.6% and 1.1%. When the reaction was catalyzed by the supported catalyst OAC-CoPW, the selectivities obtained become 1.3%, 2.4% and 0.9% respectively. This could be due to the acidic oxygen groups (hydroxyl, phenolic and carboxyl) created on the surface of the activated carbon (Gokce and Aktas 2014, Abdulrasheed et al 2018). Indeed, the unsupported CoPW catalyst has only Lewis type acid sites, whereas the supported catalyst ACA-CoPW has, in addition to these Lewis type acid sites, Brønsted type acid sites on the surface of the activated carbon. This clearly indicates that the Brønsted acid sites on the carbon support are responsible for the cracking reactions. This result is in agreement with those reported in the literature where it has been mentioned that the acidity of the support promotes cracking reactions (Wang et al 2004). It has also been reported that Brønsted type acid sites effectively catalyze the cracking of cumene (Yue et al 1998).
Table 2. Effect of AC support on cumene oxidation. Reaction conditions: RT = 75 °C; P(CO2) = 0.55 MPa; (H2O2/ Cumene: 2.5) volume ratio; Rt = 7 h; m(cat) = 0.75 g and m(co-catalyst) = 0.25 g.
| Selectivitiesa (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Conversion (%) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Conversion per g of CoPW |
| CoPW | 21.8 | 33.1 | 57.0 | 6.4 | 1.9 | 0 | 0.6 | 1.1 | 29.1 |
| CoPW/AC | 31.7 | 33.7 | 58.1 | 2.6 | 1.1 | 1.3 | 2.4 | 0.9 | 55.6 |
a1, Ph = O; 2, Ph-PrOH; 3, PhOH; 4, Acet; 5, Toluene; 6, Benzene; 7, Ethylbenzene.
3.4. Effect of reaction conditions
In order to improve the yields of Ph-PrOH and Ph = O, an optimization of the reaction was carried out. For this purpose, the effect of the oxidizing system composition, the reaction temperature, the H2O2/Cumene ratio, the amount of catalyst, the reaction time and the CO2 pressure on the oxidation of Cumene was examined. The catalyst supports AC-CoPW, has been selected to catalyze the reactions.
3.4.1. Effect of H2O2/CO2 oxidizing system
The effect of the oxidizing system composition on the conversion and selectivities was examined by oxidizing cumene by CO2 alone, H2O2 alone and H2O2/CO2 mixture. The results (table 3) show that in the case of oxidation by CO2 alone and H2O2 alone, the conversions obtained are 3.53% and 12.9% respectively. Interestingly when cumene was oxidized by H2O2/CO2 mixture, the conversion increased significantly (31.7%). The improvement in the oxidation of cumene by the H2O2/CO2 oxidizing system is the result of two factors. The first is the effective activation of H2O2 by Co1.5PW12 and the second is the formation of a percarbonate species, a strong oxidant formed by the reaction of H2O2 with CO2. For the activation of H2O2 by Co1.5PW12, it has been reported that in the presence of tungstate-based systems, oxidation by H2O2 leads to high reactivities and low activities for the non-productive decomposition of H2O2 (Mizuno et al 2008). Among the tungstate-based systems, the Peroxotungstate [PO4(WO(O2)2)4]3−, also called Venturelo complex has been postulated as being a catalytically active species in the oxidation of various reactions (Ishii et al 1988, Duncan et al 1995, Qi et al 2011). Venturello et al 1983 reported that in aqueous H2O2, [PW12O40]3− led to the formation of the complex [PO4{W(O)(O2)2}4]3−. This complex which is composed of two species [W2O2(O2)4] attached to the PO43− anion- reacts with the olefin to give an intermediate which subsequently loses a molecule of water to lead to the reaction products and to regeneration of the catalyst.
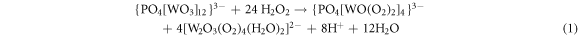
As for the effect of the percarbonate entity ( ) on the oxidation, it has been reported (Richardson et al 2000) that this entity which is formed from hydrogen peroxide and sodium carbonate can oxidize alkene into epoxide. Beckman also postulated the formation of
) on the oxidation, it has been reported (Richardson et al 2000) that this entity which is formed from hydrogen peroxide and sodium carbonate can oxidize alkene into epoxide. Beckman also postulated the formation of  by the reaction of aqueous CO2 and hydrogen peroxide (Beckman 2003). In the opinion of the author, the percarbonate species (HCO4−) can be formed by the reaction of H2O2 with aqueous bicarbonate (
by the reaction of aqueous CO2 and hydrogen peroxide (Beckman 2003). In the opinion of the author, the percarbonate species (HCO4−) can be formed by the reaction of H2O2 with aqueous bicarbonate ( ) and this entity can epoxidize alkenes. It has also been reported by Hâncu et al 2002 that a biphasic aqueous H2O2/CO2 mixture is an efficient epoxidizing system, where
) and this entity can epoxidize alkenes. It has also been reported by Hâncu et al 2002 that a biphasic aqueous H2O2/CO2 mixture is an efficient epoxidizing system, where  is formed through various reactions of water, CO2, and H2O2 and transfers of oxygen to alkenes (equations (2) and (3))
is formed through various reactions of water, CO2, and H2O2 and transfers of oxygen to alkenes (equations (2) and (3))
Truzzi et al 2019 mentioned that the acceleration of oxidation by  /CO2 on oxidations mediated by H2O2 are due to the percarbonate species (
/CO2 on oxidations mediated by H2O2 are due to the percarbonate species ( ), an oxidant with two electrons stronger than H2O2 which is present in the H2O2/
), an oxidant with two electrons stronger than H2O2 which is present in the H2O2/ /CO2 mixture. It was noted that small concentrations of
/CO2 mixture. It was noted that small concentrations of  present in the mixtures are sufficient to significantly increase oxidation.
present in the mixtures are sufficient to significantly increase oxidation.
Table 3. Effect of the composition of the oxidizing system on the conversion and selectivities of the products for the reaction catalyzed by AC-CoPW. Reactions conditions: T = 75 °C; P (CO2) = 0.55 MPa; (H2O2/cumene: 2.5) volume ratio; tr = 7 h; m (cat) = 0.75 g and m (co-catalyst) = 0.3 g.
| Selectivity (%) | |||||
|---|---|---|---|---|---|
| Oxidant | Conversion (%) | Ph = O | Ph-PrOH | Ph-Ol | Cracking |
| CO2 | 3.5 | 27.7 | 40.0 | 2.8 | 29.5 |
| H2O2 | 12.9 | 31.9 | 53.2 | 6.1 | 8.7 |
| CO2/H2O2 | 31.7 | 33.7 | 58.1 | 2.6 | 5.5 |
3.4.2. Effect of Co-catalyst
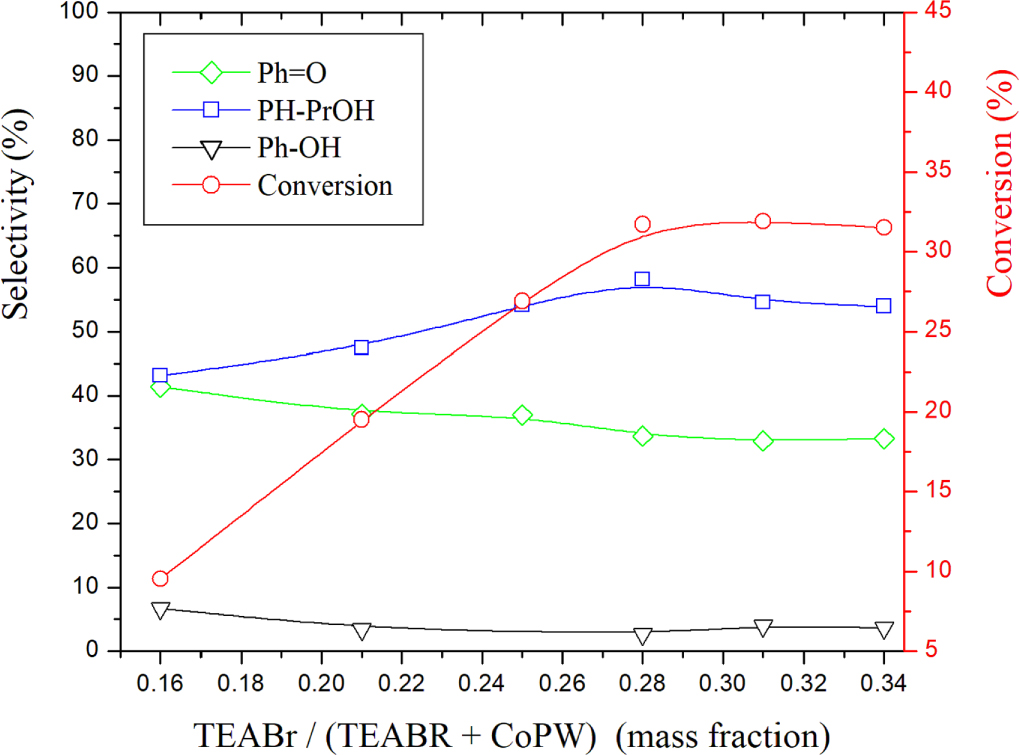
The effect of co-catalyst amount on cumene oxidation was investigated in the mass fraction ranging from 0.16 to 0.34. The results depicted in figure 4 showed that increasing the mass fraction from 0.16 to 0.28 increased the conversion from 9.5% to 31.7%. Beyond 0.28, there is no significant change. The selectivity of Ph-PrOH presented the same trend as the conversion. That is to say it increases when the mass fraction increases from 0.16 to 0.28 and then remains unchanged above 0.28. The increase in the selectivity of Ph-PrOH is to the detriment of that of Ph = O and Ph-OH which clearly indicate that the co-catalyst prevents the deep oxidation but favors the weak oxidation (formation of Ph-PrOH alcohol). Given the above results, it can be deduced that an appropriate amount of the co-catalyst contributes to the activation of CO2. Therefore, the optimal mass fraction 0.29 was selected for all subsequent investigations.
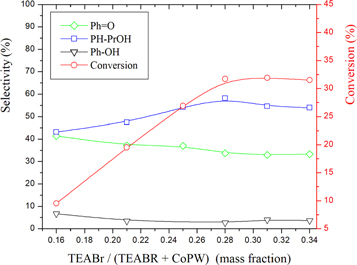
Figure 4. Effect of co-catalyst on the conversion and product selectivities over AC-CoPW catalyst. Reaction conditions: (volume ratio : H2O2/ Cumene = 2.5); RT = 75°C; P(CO2) = 0.55 MPa; Rt = 7h.
Download figure:
Standard image High-resolution image3.4.3. Effect of reaction temperature
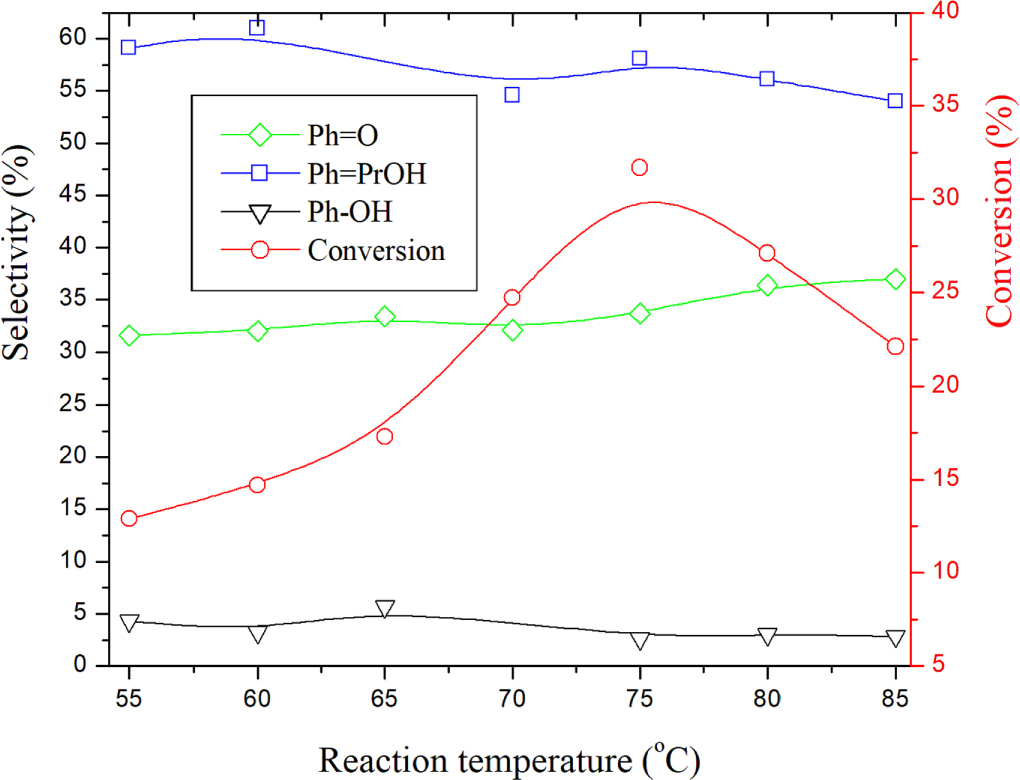
The effect of the reaction temperature (RT) on the conversion and selectivities is depicted in figure 5. The results showed that increasing reaction temperature to 75 °C increased the conversion. Further increase in reaction temperature up to 85 °C caused a decrease in conversion. The conversion decay observed for temperatures above 75 °C could be attributed to the decomposition of H2O2. The selectivity of Ph = O increased slightly, whereas that of Ph-PrOH and Ph-OH decreased slightly throughout the temperature range.

Figure 5. Effect of reaction temperature on the conversion and product selectivities over AC-CoPW catalyst. Reaction conditions: (H2O2/ Cumene: 2.5) volume ratio; P(CO2) = 0.55 MPa; Rt = 7h.
Download figure:
Standard image High-resolution image3.4.4. Effect of H2O2
Heteropolyatungstates are among the most suitable catalysts for H2O2 oxidation reactions. Indeed,the keggin heteropolyoxotungstates have 12 peripheral tungsten atoms (W) and each W atom has a large positive charge. This positive charge makes the heteropolytungstates capable of accepting electron pairs in vacant W orbitals to form a stable complex with H2O2. This makes the oxygen atoms of H2O2 more active and, therefore, the H2O2 activity for the oxidation reactions can be improved (Donoeva et al 2009). This tempted us to examine the effect of H2O2 on cumene oxidation. Experiments with various proportions of H2O2 were carried out. The results (figure 6) show that increasing H2O2/cumene volume ratio increased the conversion to a maximum value of 46.4% for a volume ratio of 2 and then gradually decrease to a value of 20.3% for a volume ratio of 3.5. The decrease in conversion was due to the gradual decrease in cumene solubility in the polar medium which increases with the amount of H2O2. As for the selectivities of the reaction products, it can be noticed that increasing the concentration of H2O2 decreased, the selectivity of Ph-PrOH and PhOH in favor of Ph = O. This is expected since cumene is insoluble in the aqueous phase, it is the alcohols that are oxidized instead.
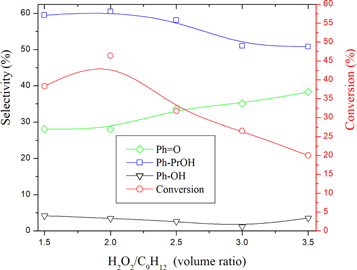
Figure 6. Variation of the conversion and product selectivities with volume ratio over AC-CoPW catalyst. Reaction conditions: RT = 75 °C; P(CO2) = 0.55 MPa; Rt = 7h.
Download figure:
Standard image High-resolution image3.4.5. Effect of catalyst amount
The effect of catalyst amount on cumene reaction was studied in the range 0.50–1.25g and the results are presented in figure 7. As expected, the conversion increases with the increase in the amount of the catalyst. On the other hand, the selectivity of the products depends on the conversion. It is well known that the increase in conversion increases the consecutive reactions. This is in agreement with our results. In fact, increasing the amount of catalyst increased both the conversion and Ph = O selectivity but decreased the selectivity of Ph-PrOH and phenol PhOH. This results indicated that Ph = O might be formed directly from cumene and from Ph-PrOH and/or phenol PhOH by consecutive reactions.
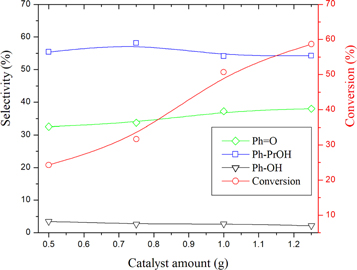
Figure 7. Variation of the conversion and product selectivities with catalyst amount over AC-CoPW catalyst. Reaction conditions: (H2O2/ Cumene) volume ratio =2.5; P(CO2) = 0.55 MPa; RT = 75°C; Rt = 7h.
Download figure:
Standard image High-resolution image3.4.6. Effect of reaction time
The variation in conversion and selectivity as a function of time is shown in figure 8. The results obtained show that the conversion increases linearly with the reaction time. With regard to the selectivity, it is noted that the selectivity for both Ph = O and Ph-PrOH has slightly decreased in favor of that of Ph-OH. This increase in the Ph-OH selectivity as a function of reaction time indicates the progress of the cracking reaction. This is explained by the decrease in the concentration of H2O2, and therefore it is the effect of CO2 that remains predominant (constant CO2 pressure) thus leading to cracking reactions. This interpretation is supported by the increase in the selectivities of the products resulting from the cracking reactions (toluene, ethyl benzene, benzene and toluene) obtained.

Figure 8. Variation of the conversion and product selectivities with reaction time over AC-CoPW catalyst. Reaction conditions: (H2O2/ Cumene) volume ratio=2.5; RT = 75°C; P(CO2) = 0.55 MPa.
Download figure:
Standard image High-resolution image3.4.7. Effect of CO2 pressure
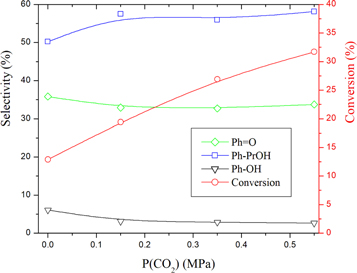
The effect of CO2 on the course of the reaction was studied by varying the CO2 pressure from 0M Pa to 0.55 MPa. The results obtained are illustrated in figure 9. It is noted from these results that the conversion increases rapidly with the pressure of CO2. On the other hand, and what is very interesting is the increase of the selectivity of Ph-PrOH to the detriment of Ph = O and Ph-OH (products resulting from deep oxidation reaction and cracking). This result can be explained by the action of the percarbonate entity (HCO4−) which is responsible for oxidation by oxygen transfer. This result is very interesting because we observe an increase in conversion and selectivity at the same time and this is not the case in the majority of oxidation reactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Variation of conversion and selectivities as a function of CO2 pressure over AC-CoPW catalyst. Reaction conditions: (H2O2/Cumene: 2.5) volume ratio; RT = 75 °C; Rt = 7h.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
The synthesis of 2-Phenyl-2-propanol and acetophenone was achieved by oxidation of cumene by H2O2 in the presence of CO2. The reaction was catalyzed by 12-tungstocobaltate heteropolyanions (Co1.5PW12O40) supported on activated carbon in liquid phase.
Unlike the oxidation of cumene by O2 gas, where cumene hydroperoxide is obtained in significant quantities, the oxidation of cumene by H2O2 leads only to traces of cumene hydroperoxide because of its decomposition in acidic aqueous medium.
By increasing the accessibility of CoPW to cumene molecules (organic phase), the activated carbon support significantly improves conversion while keeping selectivities of Ph = O and Ph-PrOH almost unchanged.
The oxidation of cumene by CO2, H2O2 and H2O2/CO2 shows that when cumene was oxidized by H2O2 in the presence of CO2, the conversion increased considerably. This synegetic effect is due to the formation of the percarbonate entity ( ) which plays a key role in the oxidation reaction.
) which plays a key role in the oxidation reaction.
The conversion of cumene and selectivity of Ph-PrOH can be controlled by selecting appropriate conditions. Higher CO2 pressure and short reaction times favor Ph-PrOH formation, whereas longer reaction times favor Ph = O and Ph-OH.
Acknowledgments
The authors extended their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project no. RGP-VPP-116.
Conflicts of interest
The authors declare no conflict of interest.