

Abstract
We have investigated the structural and elastic properties of Cd1−xMnxTe2 compound using the DFT based FP-LAPW method within a framework of GGA proposed by Perdew–Burke–Ernzerhof. These compounds are found to be stable in the pyrite phase, in agreement with the available experimental reports. The results of the equilibrium lattice constants and bulk moduli are in comparison with experimental and theoretical studies of CdTe2. The analysis of elastic constant at relaxed positions reveals the ductile nature of the compound with ionic contribution in the inter atomic bonding, whereas as the concentration of Mn increases the sample material starts to show a brittle nature with covalent bonding. The substitution of Mn atoms on the Cd site affect their mechanical properties (bulk modulus, young modulus, shear modulus) of the compound which have been reported. The formation energy was also calculated which predicted that the compound is thermodynamically stable and the stability increases with concentration of the Mn.
Export citation and abstract BibTeX RIS
1. Introduction
Transition metal dichalcogenides (TMDCs), with chemical formula MX2 (where M is a transition metal and X is a chalcogen, such as S, Se and Te) have gained much attention due to their novel characteristics, that can be widely used in industrial and engineering fields [1]. TMDCs' crystal structure was first determined by Linus Pauling in 1923 [2], where hexagonally packed atoms form weakly coupled sandwiched X-M-X layers, with one M layer enclosed within two layers of X [3].
The bulk TMDCs possess diverse properties ranging from insulators (such as HfS2), semiconductors (MoS2 and WS2), semimetals (WTe2 and TiSe2) and metals (NbS2 and VSe2) [1] . The relatively high earth abundance of TMDCs and their direct bandgaps in the visible range make them an attractive light-absorbing material that can be used as an alternative thin-film solar cell [4], including flexible photovoltaics that could be used for coating buildings and curved structures. Since, they possess high electron mobilities they can also be used as a catalyst and a good lubricant (for e.g. MoS2 and MoSe2) [5]. The recent reports on TMDCs also suggest their thermoelectric applications with temperature dependent figure of merit defined by (ZT) in range of 0.1 to 0.7 [6, 7]. The ZT value was later on enhanced to 0.8 to 0.95 with doping at moderate temperature [8].
Among the significant member of TMDCs, Cd based candidates (CdX2) are also appreciable and interesting. These materials are mechanically stable in pyrite structure [9] where CdS2 has the strongest resistance to deformation while CdTe2 has the least of them [10]. CdTe2 in its pyrite structure that coexist with CdTe under certain conditions acts as an ohmic contact with large work function. Furthermore, its high absorption coefficient (4 × 104 cm−1–8 × 105 cm−1) with cohesive energy −0.485 eV/c.u. and low band gap semiconducting nature is also regarded as a good photon absorber in the visible range [11]. Similarly the low band gap (0.7 eV) semiconductor MnTe2 [12] also exhibit good thermoelectric properties with high seebeck coefficient of 400 μV K−1 and low hole concentration of 10−19 cm−3 [12]. It also exist in pyrite structure and undergoes a second-order antiferromagnet-paramagnet phase transition at TN = 86.5 K [13].
There exist limited studies on CdTe2 and similarly the previous reports on MnTe2 are also unable to predict the mechanical properties. From the literature it has been observed that p type doping are efficient to enhance the thermoelectric performance and knowing the effect of doping an effort has been made to understand the stability and structural properties of CdTe2 and consequence of subsequent Mn doping. For this purpose an efficient density functional theory (DFT) based method is implemented to explore the structural and elastic properties of Cd1−xMnxTe2 (x = 0.03125, 0.125, 0.25, 0.5, 0.75 and 1) and the results so obtained may provide the reference data for the experimentalists for further investigations.
2. Calculation details
The result reported in the manuscript, are carried out by using full potential linearized augmented plane wave [FP-LAPW] method [14] within the framework of DFT as implemented in the wien2k code [15]. The exchange-correlation effects are treated by using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) [16]. The self consistent potentials are calculated on a 15 × 15 × 15 k-mesh in the Brillouin Zone and the self consistency convergence criteria is set to 10−4 Ry. The energy eigen values were converged with a plane wave cut off parameter of RMT x Kmax = 8 (where RMT is the smallest atomic radii of all atomic spheres and Kmax is the maximum value of the wave vector K = k+G) in the interstitial region and the charge density was Fourier expanded upto Gmax = 13 a.u.−1. The different MT sphere radii (RMT ) used were 2.2 a.u, 2.3 a.u and 2.5 a.u for Cd, Mn and Te, respectively. A 2 × 2 × 2 supercell was constructed for lesser concentration generating a total 32 Cd and 64 Te atoms where a subsequent replacement of Cd atoms by Mn leads to Cd1−xMnxTe2 (x = 0.03125, 0.125) structure and for higher concentration Mn was added in a unit cell with 4 Cd atoms. For x = 0.125 we need to replace 4 Cd atoms which can be done in 35,960 ways which has a high computational cost. After investigating the structure for many possible configuration considering the corner atoms specifically as for these atoms the environment is symmetrically same there was not much difference in the energy, however the energetically most favourable configurations for the Mn atoms was chosen. The same procedure was followed for other lesser concentration.
3. Results and discussion
The (Cd/Mn)Te2 crystallize in pyrite ( space group Pa  ) structure (figure 1) where one Cd/Mn atom takes the atomic positions of 4a (0, 0, 0) and one Te atom is located at 8c (0.389, 0.389, 0.389). The experimental lattice constant of 7.25 Å and 6.95 Å for CdTe2 and MnTe2 were optimized by calculating the total energy of the system by varying the crystal cell volume. The optimized lattice constants are calculated then by fitting the data into Murnaghan's equation of state and a comparitive analysis of the experimental and theoretical results are presented in table 1. The structures were optimized for the magnetic and non-magnetic phases (figure 1), and the optimization curve depicts that CdTe2 crystallizes in non-magnetic phase, whereas the addition of Mn results the magnetic ground state (figure not included). The effect of GGA, that overestimates the results is distinct in CdTe2, however in MnTe2 it is slightly underestimated as compared to experimental report, which can be attributed to the temperature factor existing in experimental and our calculations.
) structure (figure 1) where one Cd/Mn atom takes the atomic positions of 4a (0, 0, 0) and one Te atom is located at 8c (0.389, 0.389, 0.389). The experimental lattice constant of 7.25 Å and 6.95 Å for CdTe2 and MnTe2 were optimized by calculating the total energy of the system by varying the crystal cell volume. The optimized lattice constants are calculated then by fitting the data into Murnaghan's equation of state and a comparitive analysis of the experimental and theoretical results are presented in table 1. The structures were optimized for the magnetic and non-magnetic phases (figure 1), and the optimization curve depicts that CdTe2 crystallizes in non-magnetic phase, whereas the addition of Mn results the magnetic ground state (figure not included). The effect of GGA, that overestimates the results is distinct in CdTe2, however in MnTe2 it is slightly underestimated as compared to experimental report, which can be attributed to the temperature factor existing in experimental and our calculations.
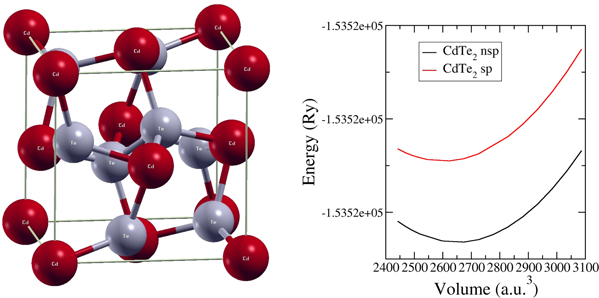
Figure 1. (left) Unit cell structure and (right) volume optimization curve (nsp = non-magnetic, sp = magnetic phase) of CdTe2.
Download figure:
Standard image High-resolution imageTable 1. Calculated, optimized lattice constant (a) and a from Vegards rule (av) in Å, internal parameter (u), elastic constants (C11,C12,C44 ), Bulk modulus (B) , Young's modulus (Y) and isotropic shear modulus (G) in GPa, Poisson's ratio (ν) , Debye temperature (θD) in K.
| Compound | a | u | C11 | C12 | C44 | B | Y | G | ν | θD | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CdTe2 Present | 7.289 | 0.389 | 51.15 | 36.66 | 29.97 | 41.49 | 45.06 | 17.08 | 0.31 | 172.37 | |
| Prev. [10] | 7.24 | 0.389 [11] | 577.1 | 376.8 | 214.7 | 44.35 | 42.14 | 15.72 | 0.29 | 161.95 | |
| 7.264 [17] | |||||||||||
| 7.25 [11] | |||||||||||
| Cd0.96875Mn0.03125Te2 | 7.151 | 0.305 | 27.03 | 23.90 | 11.69 | 24.95 | 15.25 | 5.45 | 0.39 | 100.21 | |
| av = 7.265 | |||||||||||
| Cd0.875Mn0.125Te2 | 7.139 | 0.305 | 88.18 | 35.51 | 24.93 | 53.07 | 65.90 | 25.48 | 0.29 | 209.72 | |
| av = 7.195 | |||||||||||
| Cd0.75Mn0.25Te2 | 7.136 | 0.389 | 77.72 | 22.75 | 39.16 | 41.07 | 79.91 | 33.98 | 0.17 | 241.33 | |
| av = 7.101 | |||||||||||
| Cd0.5Mn0.5Te2 | 6.881 | 0.389 | 114.97 | 33.20 | 52.74 | 60.13 | 112.60 | 47.39 | 0.18 | 286.11 | |
| av = 6.913 | |||||||||||
| Cd0.25Mn0.75Te2 | 6.698 | 0.389 | 131.98 | 34.70 | 68.50 | 66.79 | 137.58 | 59.47 | 0.15 | 322.09 | |
| av = 6.725 | |||||||||||
| MnTe2 Present | 6.537 | 0.368 | 148.61 | 43.77 | 63.14 | 78.38 | 140.31 | 58.38 | 0.20 | 323.99 | |
| Prev. [18] | 6.951 | 0.385 [19] |
The structure Cd1−xMnxTe2 so obtained by adding Mn in CdTe2 supercell was also optimized by minimising their energy corresponding to their volume and the forces between two atoms and as we increase the concentration of Mn atom with slightly small atomic radii (0.67 Å) as compared to Cd (0.95 Å) the lattice constant decreases in regular trend. In lack of experimental results for the complex doped structure , the lattice constant was also verified from the empirical vegard's rule [20] as given by equation (1),
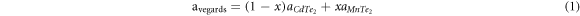
where avegards is the lattice constant calculated by vegards rule,  is the lattice constant of CdTe2 and MnTe2 compound where (1−x) and x denotes the number of Cd and Mn atoms respectively. The qualitative agreement between a and avegards also validates the present data, however a slight deviation (0.2%–0.5%) in the results may be due to mismatch of lattice constants of CdTe2 and MnTe2. In order to find the stable crystal structure that minimises the total energy the internal parameter (u) was optimized, which for CdTe2 agrees well with experimental report. The value of u shows a jump from 0.389 to 0.305 at small x (table 1), and jump back at large x as for lower concentration doping, a supercell was chosen which reduces the value of the internal parameters that defines the position of the Te atom in the cell with minimum forces betwen them. Whereas, for higher concentration a unit cell was considered that does not affect the position of Te atom and hence the internal parameter, however MnTe2 has slightly lower value that follows the trend observed in lattice parameter .
is the lattice constant of CdTe2 and MnTe2 compound where (1−x) and x denotes the number of Cd and Mn atoms respectively. The qualitative agreement between a and avegards also validates the present data, however a slight deviation (0.2%–0.5%) in the results may be due to mismatch of lattice constants of CdTe2 and MnTe2. In order to find the stable crystal structure that minimises the total energy the internal parameter (u) was optimized, which for CdTe2 agrees well with experimental report. The value of u shows a jump from 0.389 to 0.305 at small x (table 1), and jump back at large x as for lower concentration doping, a supercell was chosen which reduces the value of the internal parameters that defines the position of the Te atom in the cell with minimum forces betwen them. Whereas, for higher concentration a unit cell was considered that does not affect the position of Te atom and hence the internal parameter, however MnTe2 has slightly lower value that follows the trend observed in lattice parameter .
The possibility of doping a crystal structure with a particular dopant and their thermodynamical stability is verified by estimating their formation energy (ΔEF ) [21, 22] as given by equation (2),
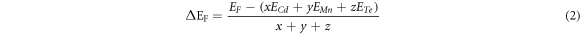
where EF is the total energy of the compound, ECd, EMn and ETe are the equilibrium energy of the individual Cd, Mn and Te atoms, which is found to be –11192.07 Ry, −2316.65 Ry and −13593.64 Ry, and x, y, z denotes the number of Cd, Mn and Te respectively. The formation energy so obtained has been listed down in table 2 and their negative value indicates that they are energetically stable. In addition, the negative values of ΔEF also indicates stronger bonding between the atoms and more alloying stability of the crystal [23]. From the table one can note that as the concentration of Mn increases the value of formation energy also increases suggesting a more stable structure.
Table 2. Total energy (EF) , calculated energy (Ecalc) and formation energy (∆EF) in Ry.
| Compound | EF | ECalc | ∆EF |
|---|---|---|---|
| CdTe2 | −153518.73 | −153517.44 | −0.11 |
| Cd0.96875Mn0.03125Te2 | −1219266.28 | −1219264.10 | −0.02 |
| Cd0.875Mn0.125Te2 | −1192641.29 | −1192637.84 | −0.04 |
| Cd0.75Mn0.25Te2 | −144643.01 | −144642.02 | −0.08 |
| Cd0.5Mn0.5Te2 | −135769.29 | −135766.59 | −0.22 |
| Cd0.25Mn0.75Te2 | −126894.44 | −126891.17 | −0.27 |
| MnTe2 | −118019.54 | −118015.75 | −0.32 |
Elastic constant tells us about the bonding forces and mechanical strength of a system, which will be of great importance in technological applications. Thus, the independent elastic constants Cij which is reduced to C11,C12,C44 due to cubic symmetry of the crystal are estimated at ambient condition after their volume deformation that follow the stability criteria [9], laid by C11 + 2C12 > 0, C44 > 0, C11−C12 > 0. For all the compounds the value of C11 is greater than C12 and C44 which indicates that the compound is hard to compress along the x axis and as C44 relates to shear stress and strain in the same direction, the compound has a weaker resistance against a pure shear deformation.
The Cij(C11, C12, C44) derived parameters such as B, Y, G (table 2) of CdTe2 are in qualitative agreement with the previous reports [10] and similar technique has been applied to compute their values for these materials listed in table 1 that may validate the reliability of the present calculation in the absence of previous theoretical and experimental report .
To shed more light in the mechanical properties of the structure, we calculated the Bulk modulus (B), Young's modulus (Y) and shear modulus (G) using the equations (3)–(5) [24–26],
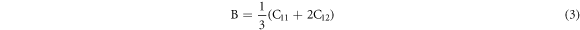
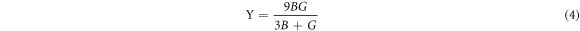
Poisson's ratios depicted in table 1 were calculated from the Voigt-Reuss-Hill (VRH) approximation shown in Eqs.
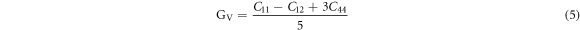
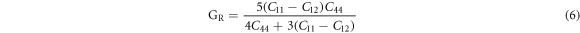
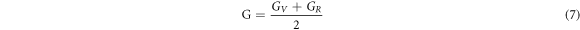
here GV and GR are Voigt's and Reuss's shear moduli respectively.
In comparison with analogous CdX2 (X = S,Se,Te) compound, CdTe2 has the least Bulk modulus specifying that it has the least power of withstanding compressibility. The results also implies that B increases with x (concentration of Mn) and thus the material becomes stiffer.
Higher the value of Y than bulk modulus (B), the material is hard to be broken [27]. The Y value of compounds like CdS2 and CdSe2 has value of 70.075 GPa and 56.445 GPa [10] which is greater than CdTe2. Shear modulus or modulus of rigidity G is also an important parameter that relates to the hardness of a crystal. The calculated values of G for CdTe2 are in qualitative agreement with the theoretical values of analogous compounds and varies as CdS2 > CdSe2 > CdTe2 [10]. Unfortunately for MnTe2 no elastic results are available for comparison.
Poisson's ratio (ν) identifies the compressibility and stretchability of a material before it collapses. The ratio is found to vary as 0.1 ≤ ν ≤ 0.5, which agrees well with the other cadmium based dichalcogenides. It also characterizes the bonding nature of solid material, where for covalent materials the ratio has a small value (typically at 0.1) and for ionic compounds, its value is greater than or equal to 0.25. As the ν value for Cd1−xMnxTe2 (for  ) is greater than 0.25, the ionic nature of bonding can be expected whereas for x >12.5% covalent bonding exists.
) is greater than 0.25, the ionic nature of bonding can be expected whereas for x >12.5% covalent bonding exists.
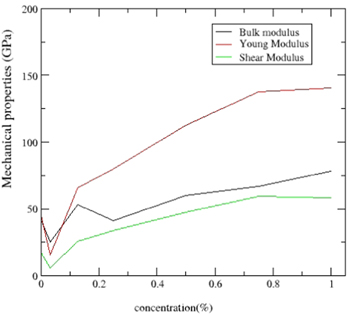
The value of pugh ratio (B/G) measures the ductility or brittleness of a material with cut off value of 1.75. The pure CdTe2 and Mn doped CdTe2 (for x < 0.25) suggest a ductile nature whereas as the concentration of Mn dopant increases the material starts to show a brittle nature. The variation of B,Y and G with respect to concentration are presented in figure 2.
{kind=link}
Figure 2. Variation of bulk modulus (B), shear modulus (G), Young's modulus (Y) as a function of concentration x.
Download figure:
Standard image High-resolution image{kind=link}
Another important parameter, which closely relates to the specific heat and melting point of the solid is the debye temperature (θD) which can be calculated using equation (8),
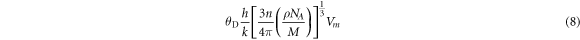
here h is the planck's constant, k is the Boltzmann's constant, n is the number of atoms per molecule, ρ is the density of the unit cell , NA is the Avagadro's number, M is the molar mass and vm is the average sound velocity. High value of Debye temperature indicates that the crystal is stiffer and have a high melting point. The Debye temperature is in reasonable agreement with other theoretical work and other analogous compound like CdS2 and CdSe2 with values 287.25 K and 212.4 K respectively [10]. As the Mn concentration increases the θD also increases with highest value for MnTe2 indicating that MnTe2 is stiffest of all.
4. Conclusions
We have calculated the structural and elastic properties of Cd1−xMnxTe2 using first principles method. The optimized lattice constant and internal parameter are in close agreement with the available results and that have also been validated by Vegards rule. The complex structure Cd1−xMnxTe2 so formed after the addition of Mn in CdTe2 are mechanically and thermodynamically stable and their elastic properties are calculated at stable structure at volume deformation. The Bulk modulus, Young's modulus and Shear modulus of the CdTe2 is in good agreement with available experimental and theoretical work. The elastic constant reveals the ductile nature and ionic bonding of CdTe2 compound with stiffness lower than that of MnTe2 and analogous compound. The debye temperature was also found to be highest for MnTe2 indicating that MnTe2 is stiffest of all. The formation energy calculated reveals that the structure is thermodynamically stable and the stability increases with the increase in the concentration of Mn dopant. Since there have not been any experimental or theoretical works in complex doped structure to compare with our results to the best of our knowledge, we hope these results will be useful for further investigations of their electronic, optical and thermoelectric properties.
Acknowledgments
A Shankhar acknowledges SERB New Delhi (EEQ/2017/000319).