
Abstract
Designing 2D-materials that exhibit half-metallic properties is crucially important in spintronic devices that are used in low-power high-density logic circuits. The large pores in the C2N morphology can stably accommodate various configurations of transition-metal (TM) atoms that can lead to ferromagnetic (FMC) and anti-ferromagnetic coupling interactions among them, and thus paving the way for achieving half-metallic characteristics. In the present study, we use manganese 'Mn' as a promising catalyst and the spin-polarized density-functional theory to search for suitable configurations of metal atoms that yield half-metallicity. Test samples comprised of single-atom catalyst (SAC) and double-atom catalyst (DAC) of Mn embedded in a C2N sample of size 2 × 2 primitive cells as well as their combinations in neighboring large pores (i.e. SAC–SAC, SAC–DAC, and DAC–DAC). Tests were extended to screen many other TM catalysts and the results showed the existence of half metallicity in just five cases: (a) C2N:Mn (DAC, SAC–SAC, and SAC–DAC); (b) C2N:Fe (DAC); and (c) C2N:Ni (SAC–DAC). Our results further showed the origins of half-metallicity to be attributed to FMC interactions between the catalysts with the six mirror images, formed by the periodic-boundary conditions. The FMC interaction is found to have strength of about 20 meV and critical length scale up to about ∼21–29 Å, dependent on both the type of magnetic impurity and the synergetic effects. The potential relevance of half-metallicity to spintronic device application is discussed. Our theoretical results have been benchmarked to the available data in literature and they were found to be in good agreements.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The breakthrough discovery of graphene had not only fetched the inventors the Nobel prize in physics in 2010 but more importantly, it has established a new field of research in 2D systems [1–3]. Thereafter, many fascinating novel 2D materials have been synthesized and functionalized to tune their properties toward numerous applications, such as C2N and C3N [4–6], black-phosphorus [7], hexagonal BN [8], transition-metal dichalcogenides (TMD) [9], and MXene [10, 11]. The lack of bandgap in graphene has triggered research, with the aim of designing other 2D materials which possess semiconducting properties. For instance, Mahmood et al in 2015 reported their success in synthesizing the nitrogenated holey graphene 'C2N' using simple wet-chemical reaction [4]. In C2N, the presence of the electronegative Nitrogen atoms along with the uniformly distributed pores of 5.5 Å sizes, open up a band gap of 1.97 eV. The reasonably sized pores are suitable sites for stable embedment of transition metal (TM) catalysts which can tune the properties of the material. Another subsequent article by the same group of researchers [6] reported their successful embedment of TM atoms into the big pores of C2N in forms of single-atom catalyst (SAC) and double-atom catalyst (DAC). The functionalization of C2N has made this material strategic with potential of applications in several fields comprising (a) nano-electronics: C2N monolayer (ML) was proposed for metal-oxide-semiconductor field-effect transistor (MOSFET) [12]; (b) photonics: C2N van der Waals-based hetero-structures have been used with enhanced quantum efficiency [13]; (c) batteries: C2N was found suitable for metal-ion batteries in general and lithium–sulfur batteries in particular [5, 14–16]; (d) energy-storage: C2N possesses an enhanced uptake capacity in hydrogen storage [17, 18]; (e) gas-sensing: selective detection of toxic gases [19, 20]; (f) bio-sensing: it was proposed for sensing diabetes mellitus [21]; and (g) spintronics: half-metallicity can be hosted in TM-doped C2N and thus explored in spintronic-device applications [22, 23].
Moore's law had predicted that from 1965 until 2020, the number of transistors on an integrated circuit (IC) would double every two years, thereby shrinking the size of computers, and making them more and more portable over time while simultaneously reducing their price. However, the progress in electronic devices got saturated by the year 2020. Thereafter in order to continue this trend of miniaturization of memory devices, new technologies such as spintronics have to be explored in information's storage, processing and communication [24]. The main property that has to be desired in the host material for spintronic applications is half metallicity [25]. This occurs when one spin is metallic while the other spin channel is semiconducting, making the overall dc conductivity of the material spin polarized. Recently many efforts have been focused in this direction, toward designing or searching suitable materials that exhibit half metallic characteristics. Efforts have also been taken to understand the underlying physics, in order to explain its mechanism or origin. In the field of 2D or quasi 1D systems, the reported findings in literature can be categorized into three main groups: (a) half metallicity that can be obtained upon functionalization with TM atoms [25, 26]; (b) half metallicity obtained in quasi 1D materials by applying transversal electric field [27, 28]; and finally (c) half metallicity that results due to vacancies and point defects in TMDs [29, 30]. In our present investigation we have focused our effort on investigating the effects of TM atom doping C2N (i.e. category #1) on the electronic and magnetic properties while paying special attention to the emergence of half metallicity as attempted to be induced by exploring the ferromagnetic-coupling (FMC) interactions.
In TMDs, half metallicity can be achieved by creating point defects (i.e. category #3) [30, 31]. In our recent research efforts, we have computationally demonstrated that a single Molybdenum vacancy 'VMo' can trigger half metallicity in 2D MoSe2 periodic samples having dimensions of 4 × 4 and 5 × 5 primitive cells (PCs) [31]. The cause of this was attributed to occurrence of FMC interactions among the localized magnetic moments (i.e. in the vicinity of the Molybdenum vacancy), and its mirror images, on account of the periodic boundary conditions that apply on the computational supercell. However, this half metallicity vanishes for larger supercells such as those comprising of 6 × 6 PCs and above, therefore enabling us to determine the critical length for FMC interactions which was found to be about  . This value was found to be a slightly larger than that predicted by Ma et al [29] which was about 12
. This value was found to be a slightly larger than that predicted by Ma et al [29] which was about 12  , since they have taken into consideration only a single sample of 4 × 4 PCs, while overlooking the scaling effects. Also, Hong et al [32] had experimentally reported a maximum vacancy-defect density of
, since they have taken into consideration only a single sample of 4 × 4 PCs, while overlooking the scaling effects. Also, Hong et al [32] had experimentally reported a maximum vacancy-defect density of  cm−2 which is smaller than the densities estimated in our samples 6.4 × 1013 cm−2 and 4.2 × 1013 cm−2. Realistically speaking, experimental efforts based on concrete theory evidence are the only way to achieve novel materials with half metallic characteristics that can be used for robust spintronic applications.
cm−2 which is smaller than the densities estimated in our samples 6.4 × 1013 cm−2 and 4.2 × 1013 cm−2. Realistically speaking, experimental efforts based on concrete theory evidence are the only way to achieve novel materials with half metallic characteristics that can be used for robust spintronic applications.
As a matter of fact, the TM doping (i.e. category #1) is often a preferred route in achieving the half metallicity. Till date, most of the reports in this direction have been based on 3D half-metals and developing their 2D counterparts yet remains a challenge. The biggest hurdle is to establish ferromagnetic (FM) ordering in low dimensional systems [33]. Many computational efforts have suggested designs or different morphologies of 2D materials that are capable of hosting half metallicity [33–35]. Feng et al reported a thorough revision on spintronics in 2D materials (e.g. TM-functionalized graphene, phosphorus, and TMD-ML) [25]. Ahn discussed the superiority of 2D materials thus proposing a superior way to control the carrier spin. He presented graphene and other 2D inorganic semiconductors. He further highlighted the major challenges in integrating 2D materials into spintronic devices as well as provided a future perspective on 2D materials for spin-logic devices [34]. An overview on the unusual physical properties emerging 2D materials as new platform for novel spintronic devices as well as the challenges and future opportunities was recently addressed by Hu and Xiang [35].
Although metal free planar FM half-metallic systems are more favored in spintronic devices, its task remain very challenging from practicality point of view. These systems are important as they possess long spin relaxation times due to the weakness of spin–orbit-coupling interactions. Many researchers suggested just computer-designed structures where both ferromagnetism and half-metallicity can exist simultaneously. For instance, Choudhuri and Pathak [22] in a density-functional theory (DFT) study suggested C-doped C2N as a metal-free FM and half-metallic 2D material. Gong et al [23] showed that the electron-doping via gating in C2N with moderate density (ρ = 4–8 × 1013 cm−3) would not only yield FM but also half-metallicity. Nonetheless, the metal-free systems are very challenging for experimental synthesis. Besides, for practical reasons and relying on the successes achieved in TM-embedment in the large pores of C2N with adequate stability [6], we decided to explore the opportunity to study the effect of embedment of SAC, DAC and their combinations on the emergence of ferromagnetism and half-metallicity. The paper is organized as follows. Next section gives details on the computational model and method. Section 3 elaborates a discussion on the obtained results. The last section summarizes our main findings.
2. Computational model and method
From the perspective of top-view, C2N monolayer has graphitic structure with one benzene ring missing per area of four benzene rings (i.e. porosity = 1:3) and the six carbons most internal in each big pore are converted into nitrogen atoms. The pores are connected just by pyridine rings (i.e. C4N2), as shown in figure 1(a). Figure 1(a) contains samples that comprise of 2 × 2 primitive cells (PCs). The PC is composed of 12 C and 6 N atoms and has a lattice constant of a = 8.33 Å, which is in a good agreement with the experimental value of 8.30 Å reported by Mahmood et al [4]. Most of our work is done using a computational supercell of size 2 × 2 PCs (i.e. it contains 48 C + 24 N atoms) and having dimensions A = B = 16.66 Å and C = 20 Å. The latter C value is maintained sufficiently large to ensure the isolation of the monolayer to interact with the mirror images along the c-axis. The great advantage in studying C2N, over conventional graphene, is that it has pores which are uniformly distributed and having sufficiently large diameters (5.5 Å) [4], which opens up the possibilities of functionalization via embedment of TM atoms in SAC and DAC configurations. These metal atoms are found to be stable inside the pores, and they tune the properties of the host material for specific tasks such as spintronic and selective gas-sensing applications.

Figure 1. Relaxed atomic structures of C2N 2 × 2-PCs supercell with and without Mn-atom catalysis: (a) Pristine, (b) SAC, (c) DAC, (d) SAC–SAC, (e) DAC–SAC, and (f) DAC–DAC configurations. Colors: C (grey), N (blue), and Mn (purple).
Download figure:
Standard image High-resolution imageRegarding the computational method, we used the DFT method as incorporated in the 'Vienna Ab-initio Simulation Package' (VASP) [36]. The ion–electron interaction is treated by projected augmented-wave method [36]. The exchange and correlation interactions were addressed using the generalized gradient approximation with the Perdew–Burke–Ernzerhof functional [37]. For structural optimization, the convergence criteria/tolerances for total energy of 10−8 eV and atomic force of 0.01 eV−1 Å were applied. In sampling the Brillouin zone, a k-mesh of 5 × 5 × 1 Gamma-centered Monkhorst–Pack [38] was utilized in the geometry optimization of the samples 2 × 2, 3 × 3 and 4 × 4 PCs. A denser k-mesh grid of 8 × 8 × 1 (or 5 × 5 × 1) was used in the calculations of density of states (DOS) for the 2 × 2-PC (or 4×4 -PC) samples. The plane-wave cut-off energy was selected to be 550 eV and on-site U-Hubbard parameter U = 4.5 eV and J = 1.0 eV, which are almost the default values in the VASP-package, and in good agreement with the adjusted values done by Mann et al [39]. The charge exchange between dopant and host crystal is calculated based upon the Bader charge analysis within the framework of VASP package [40].
Curie temperature (TC) is defined to be the critical temperature above which the FMC interactions would vanish and thus the system exhibits a phase transition to become paramagnetic. The rigorous calculation of TC requires either Monte Carlo [41, 42] or ab-initio molecular dynamics [43] simulations. Based upon the DFT study done by Gong et al [23], owing to the prominent van Hove singularity in the band structure of C2N, this material exhibits spontaneous ferromagnetism at a relatively low doping density. Over the range of carrier density  , the system becomes half-metallic with carriers fully spin-polarized. The estimated Curie temperature was about TC ≅ 320 K [23]. Many researchers argued that the TM functionalization of for instance graphene can make the Curie temperature much higher than room temperature (RT) and rather at order 500–800 K [44]. As discussed in section 1, our doping density should be within the range studied by Gong et al and this would pave an opportunity of the half-metallicity for room-temperature spintronic device applications.
, the system becomes half-metallic with carriers fully spin-polarized. The estimated Curie temperature was about TC ≅ 320 K [23]. Many researchers argued that the TM functionalization of for instance graphene can make the Curie temperature much higher than room temperature (RT) and rather at order 500–800 K [44]. As discussed in section 1, our doping density should be within the range studied by Gong et al and this would pave an opportunity of the half-metallicity for room-temperature spintronic device applications.
Last but not the least, regarding the potential synthesis of the proposed structures yielding half metallicity, Mahmood et al [4] first reported, in 2015, their successful synthesis of C2N monolayers using wet-chemical reaction between hexaminobenzene trihydrochloride and hexaketocyclohexane octahydrate in N-methyl-2-pyrrolidone in the presence of sulphuric acid. Thereafter, the same group [5, 6] reported, in 2017 and 2019, more progress through presenting an experimental protocol for creating C2N material with embedment of some selected metal atoms (like Ru, Pd, and Co) for applications as anodes in lithium-ion batteries. From our theoretical perspectives, searching for 2D materials hosting half metallicity at RT (i.e. with Curie temperature higher than RT) is by itself a formidable task, especially if the synthesis of the predicted structures is potentially plausible.
3. Results and discussion
3.1. Atomic relaxations
In any DFT study, the calculation of the atomic relaxation should be carried out first. Figure 1 shows the atomic relaxed structures of supercells of C2N with a common size of 2 × 2 PCs. The PC contains 6 N + 12 C atoms (i.e. 18 atoms); so that the pristine supercell would have 72 atoms (i.e. 24 N + 48 C atoms). Table 1 shows the geometrical parameters of relaxed structures in the cases yielding the half-metallicity character. Figure 1 shows six relaxed structures: (a) Pristine C2N; (b) C2N with 1-Mn (SAC) inside the big pore; (c) C2N with 1 DAC-Mn inside the big pore; (d) C2N with double SACs Mn inside two neighboring big pores; (e) C2N with 1 DAC + 1 SAC of Mn atoms embedded inside two neighboring big pores; and (f) C2N with 2 DACs Mn inside two neighboring big pores. The details about the results are summarized in table 1 and should be described as follows: (a) Pristine C2N: The results showing a lattice constant a = 8.34 Å and bond lengths bC–C = 1.43–1.48 Å, bC–N = 1.34 Å, and angle (C–N–C) = 118° are in excellent agreement with the experimental data reported by the inventors Mahmood and et al [4] and the DFT results of Liu et al [45] and Ma et al [46]. In most of the work reported here, we used a supercell of size 2 × 2 PCs as to be optimally suitable to assess the effects of the embedment of 1–4 TM atoms on the acquisition of half-metallicity within the framework and the capability of VASP-package. Basically, we screened all the ten TM atoms with various combinations of SAC and DAC and found only three elements (Mn, Fe, and Ni) to yield the half metallicity. (b) C2N:1Mn (SAC): figure 1(b) shows the case of SAC-Mn embedded in the large pore of C2N. It is very important to emphasize that such embedment is thermodynamically very stable as it indeed corresponds to very strong binding energy (i.e. Ebind = −4.497 eV for Mn in table 2). In the relaxation process, Mn atom was initially placed at the center of the big pore. However, the center seems to be an unstable point and during the relaxation the Mn atom moved closer to make two single covalent bonds with the two nitrogen atoms at the border of the big pore at distance of about 1.88–1.91 Å. This bond length is smaller than the sum of the two atomic radii 2.32 Å [46], revealing the covalent character of the bonding. The final destiny of Mn atom is to stabilize at off center by an off distance of about 0.7 Å with a binding energy deeper than the center by about 55 meV. These results are also in good agreement with those reported by Ma et al [46].
Table 1. Geometrical parameters of relaxed structures of pristine and Mn-doped C2N samples of size 2 × 2 primitive cells with various combinations of SAC and DAC.
| (a) Pristine C2N (2 × 2) | (b) C2N:1Mn (SAC) |
|---|---|
| bC–C = 1.43–1.47 Ŷ, 1.43–1.47 Ň bC–N = 1.34 Ŷ, 1.34 Ň Ang(C–N–C) = 118¶, 118°‡ A = B = 16.685 Ŷ, 16.66 Ň, 16.66 ņ | bMn–N = 1.88–1.91 Ŷ Ang(N–Mn–N) = 85.77°¶ A = B = 16.685 Ŷ |
| (c) C2N:Mn2 (DAC) | (d) C2N:Mn–Mn (SAC–SAC) |
| bMn–N = 1.93–2.03 Ŷ bMn–Mn = 2.02 Ŷ Ang(N–Mn–N) = 82.99°–83.10°¶ A = B = 16.685 Ŷ | bMn–N = 1.85–1.96 Ŷ Ang(N–Mn–N) = 85.52°–85.64°¶ A = B = 16.685 Ŷ |
| (e) C2N:Mn2–Mn (DAC–SAC) | (f) C2N:Mn2–Mn2 (DAC–DAC) |
| DAC: bMn–N = 1.93–2.03 Ŷ bMn–Mn = 2.02 Ŷ Ang(N–Mn–N) = 84.29°–84.97°¶ SAC: bMn–N = 2.12–2.31 Ŷ Ang(N–Mn–N) = 74.12°¶ A = B = 16.685 Ŷ | bMn–N = 1.92–2.02 Ŷ bMn–Mn = 2.03–2.20 Ŷ Ang(N–Mn–N) = 84.35°–89.21°¶ A = B = 16.685 Ŷ |
Table 2. Magnetization and properties in 2 × 2-PC C2N samples with and without Mn, Fe and Ni catalyst cases.
| Structure |
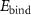 (eV) (eV) |
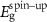 (eV) (eV) |
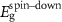 (eV) (eV) | Property | M (μB) |
|---|---|---|---|---|---|
| (a) Pristine C2N | N/A | 1.67 | 1.67 | Semiconductor | 0 |
| (b) C2N:Mn (SAC) | −4.497 | 0 | 0 | Metal | 4.33 |
| (c) C2N:Mn (DAC) | −7.645 | 0.405 | 0 | Half-metal | 4.0 |
| (d) C2N:Mn (SAC–SAC) | −9.126 | 0.20 | 0 | Half-metal | 8.004 |
| (e) C2N:Mn (DAC–SAC) | −11.881 | 0.246 | 0 | Half-metal | 7.0 |
| (f) C2N:Mn (DAC–DAC) | −14.597 | 0 | 0 | Metal | 7.575 |
| (g)C2N:Fe (DAC) | −5.885 | 0 | 0.368 | Half-metal | 2.0 |
| (h)C2N:Ni (DAC–SAC) | −10.711 | 0 | 0.164 | Half-metal | 1.999 |
(c) C2N:Mn2 (DAC): figure 1(c) shows the case of DAC-Mn embedded as a dimer in one large pore of C2N. As it is shown, each Mn atom gets attached to two N atoms, with respective bond length bMn–N = 1.93 Å and 2.03 Å, while the dimer makes an inter Mn–Mn bond bMn–Mn = 2.02 Å. The dimer Mn2 occupy positions inside the big pore at the membrane plane similar to the case of copper dimer reported by Zhao et al [47]. The relaxed configuration of dimer Mn2 has a binding energy Ebind (2Mn) = −7.645 eV dimer−1 (see table 2). Comparing this latter energy value to the binding energy of SAC-Mn, which is Ebind (1Mn) = −4.497 eV, one can extract the recursive energy of the second Mn-atom to be about Erec = −3.148 eV, whose magnitude is still larger than Mn-cohesive energy (i.e. Ecoh = 2.92 eV [48]). So, one can deduce that the DAC-Mn can be very stable inside the pore and contribute in the so-called irreversible capacitance in the language of metal-ion battery perspectives [18]. (d) C2N:Mn–Mn (SAC–SAC): As initial positions, two Mn atoms were put at the centers of two neighboring big pores of C2N. Throughout the relaxation process, they move closer so that each Mn atom makes two bonds with the nitrogen atoms at the wall of the pore as shown in figure 1(d). The bond lengths of Mn–N are about bMn–N = 1.85–1.96 Å and the bond angle is about Ang(N–Mn–N) = 85.52°–85.64° very similar to the case of SAC.
(e) C2N:Mn2–Mn (DAC–SAC): After relaxing a dimer of Mn2 in the central large pore, the third Mn atom was initially placed at the center of the neighboring large pore and the atomic relaxation is again carried out. The results are sown in figure 1(e) and the geometric parameters are displayed in table 1. For SAC-Mn, the bond length Mn–N is about bMn–N = 2.12–2.31 Å (i.e. a bit larger than the case of single SAC) and bond angle Ang(N–Mn–N) = 74.12° (i.e. a bit smaller than the case of single SAC). For DAC-Mn2, the bond length Mn–N is about 1.92–2.03 Å (i.e. same as in the case of single DAC) and bond angle Ang(N–Mn–N) = 84.29°–84.97° (i.e. a bit larger than the case of single DAC). The dimer bond-length is about bMn–Mn = 2.02 Å, definitely smaller than the sum of the two radii (2R = 2.56 Å) [48], revealing the covalent-character aspect. (f) C2N:Mn2–Mn2 (DAC–DAC): Two dimers Mn2 were atomically relaxed in two neighboring large pores of C2N. The results of relaxed structure are shown in figure 1(f) and table 1. The obtained values of bond lengths Mn–N are about bMn–N = 1.92–2.02 Å (i.e. about same as single DAC) and bond angles Ang(N–Mn–N) = 84.35°–89.21° (i.e. a bit larger than the case of single DAC). The dimer bond-length is about bMn–Mn = 2.03–2.20 Å (i.e. similar to single DAC). So, the dimers should be assumed to alter covalent bonding with the C2N lattice.
3.2. Spin-polarized partial density of states (PDOS)
Figure 2 shows the spin-polarized PDOS corresponding to the samples shown in the previous figure 1 with basically the valence-band maximum (VBM) energy and Fermi level are taken as an energy references in cases of pristine and Mn-doped C2N, respectively. (a) Pristine C2N: figure 2(a) shows the contributions of both C and N atoms to the total density of states (TDOS). The results are based on VASP calculations and do yield a bandgap energy of  = 1.67 eV, which is a bit less than the experimentally reported one of 1.96 eV [4]. Such underestimation of bandgap energy is expected in case of using DFT with plane-wave basis set unless further consideration of hybrid functional for the exchange correlation should be involved. Moreover, the nitrogen atoms are shown to predominantly contributing to the structure of the valence band due to their possession of filled dangling bonds. (b) C2N:1Mn (SAC): The embedment of one Mn atom in the big pore of C2N sample of size 2 ×2 PCs would lead to the metallization of the samples. Figure 2(b) shows Fermi level shifting up to cross the dispersive bands originating from Mn atoms. Moreover, the asymmetry existing between spin-up and spin-down TDOS should reveal the existence of magnetization (i.e. magnetic moment). Hence the obtained system is FM metal. (c) C2N:Mn2 (DAC): the embedment of DAC-Mn as a dimer in one big pore, proven to be very stable, is shown in figure 2(c) to yield half-metallicity character. Not only the system gets magnetized, as displayed by the asymmetry between spin-up and spin-down TDOS, but Fermi level crosses some spin-down bands while spin-up states behaving semiconducting and having an energy gap of 0.405 eV. It is worth to emphasize some reduction in magnetization from 4.33 μB to 4.0 μB corresponding to SAC and DAC, respectively (see tables 2 and 3). Such reduction reveals that the two Mn-atoms stabilize into an anti-ferromagnetic (AFM) state. One question should be raised about the origins of 'half-metallicity' is whether will it be due to the existence of such AFMC interaction or due to the FMC interactions of the whole magnetic moment with its (six) mirror images, which are expected to be formed by periodic-boundary conditions. Throughout the present investigation we will come out with answers to this question.
= 1.67 eV, which is a bit less than the experimentally reported one of 1.96 eV [4]. Such underestimation of bandgap energy is expected in case of using DFT with plane-wave basis set unless further consideration of hybrid functional for the exchange correlation should be involved. Moreover, the nitrogen atoms are shown to predominantly contributing to the structure of the valence band due to their possession of filled dangling bonds. (b) C2N:1Mn (SAC): The embedment of one Mn atom in the big pore of C2N sample of size 2 ×2 PCs would lead to the metallization of the samples. Figure 2(b) shows Fermi level shifting up to cross the dispersive bands originating from Mn atoms. Moreover, the asymmetry existing between spin-up and spin-down TDOS should reveal the existence of magnetization (i.e. magnetic moment). Hence the obtained system is FM metal. (c) C2N:Mn2 (DAC): the embedment of DAC-Mn as a dimer in one big pore, proven to be very stable, is shown in figure 2(c) to yield half-metallicity character. Not only the system gets magnetized, as displayed by the asymmetry between spin-up and spin-down TDOS, but Fermi level crosses some spin-down bands while spin-up states behaving semiconducting and having an energy gap of 0.405 eV. It is worth to emphasize some reduction in magnetization from 4.33 μB to 4.0 μB corresponding to SAC and DAC, respectively (see tables 2 and 3). Such reduction reveals that the two Mn-atoms stabilize into an anti-ferromagnetic (AFM) state. One question should be raised about the origins of 'half-metallicity' is whether will it be due to the existence of such AFMC interaction or due to the FMC interactions of the whole magnetic moment with its (six) mirror images, which are expected to be formed by periodic-boundary conditions. Throughout the present investigation we will come out with answers to this question.

Figure 2. Spin-polarized PDOS of C2N 2 × 2-PCs supercell with and without Mn-atom catalysis: (a) Pristine, (b) SAC, (c) DAC, (d) SAC–SAC, (e) DAC–SAC, and (f) DAC–DAC configurations. Colors of curves are: C (blue), N (green), Mn (red), and TDOS (black).
Download figure:
Standard image High-resolution imageTable 3. Magnetization and properties in 3 × 3-PC C2N samples that showed half-metallicity in 2 × 2 sample size.
| Structure |
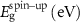
|
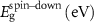
| Property | M (μB) |
|---|---|---|---|---|
| (a) C2N:Mn (DAC) | 0.302 | 0 | Half-metal | 4.0 |
| (b) C2N:Mn (SAC–SAC) | 0 | 0 | Metal | 7.533 |
| (c) C2N:Mn (DAC–SAC) | 0 | 0 | Metal | 7.001 |
| (d) C2N:Fe (DAC) | 0.739 | 0.295 | Magnetic semiconductor | 2.0 |
| (e) C2N:Ni (DAC) | 0.148 | 0.177 | Magnetic semiconductor | 2.0 |
| (f) C2N:Ni (DAC–SAC) | 0 | 0.163 | Half-metal | 1.999 |
(d) C2N:Mn–Mn (SAC–SAC): The embedment of two SAC-Mn atoms in neighboring big pores would also induce the half-metallicity character as it is shown in figure 2(d). Similar to the previous case of C2N:Mn2 (DAC), figure 2(d) shows the asymmetry between spin-up and spin-down states' contributions to TDOS revealing the existence of magnetic moment (M = 8.004 μB), which is about the double of the value of SAC case shown in figure 2(b). Such huge magnetic moment should reveal that the SAC–SAC configuration has stabilized to yield FM state. The second question one should raise here is about the origins of the 'half-metallicity', was it due to the FMC interaction between SAC–SAC or between the whole magnetic moment and the others due to the (six) mirror images?
(e) C2N:Mn2–Mn (DAC–SAC): figure 2(e) shows another case of realization of 'half-metallicity' character due to the embedment of DAC and SAC Mn atoms in two neighboring big pores of C2N. Fermi level is shown to cross the spin-down states as becoming metallic while the spin-up states remaining semiconducting with an energy gap of 0.246 eV. The total magnetization of DAC–SAC is shown in tables 2 and 3 to be about M = 7.0 μB, which is less than the case of SAC–SAC and thus revealing an AFMC to take place between the two atoms of DAC configuration. The acquisition of the half-metallicity characters in the three cases (c–e) will be proven to originate from the FMC interactions with the six mirror images, formed by the periodic boundary conditions. (f) C2N:Mn2–Mn2 (DAC–DAC): figure 2(f) shows that the embedment of DAC–DAC in two neighboring pores of C2N would result in magnetic metal with a magnetization of about M = 7.575 μB, which is larger than the case of DAC–SAC but smaller than the case of SAC–SAC. It might reveal the fact of existence of AFMC interaction within DAC and FMC interaction between DAC–DAC.
3.3. VBM, CBM and Fermi eigenstates
In attempting to dig for further understanding of the half-metallicity characteristics, we planned to perform calculations of the VBM, the conduction-band minimum (CBM), and the Fermi eigenstates corresponding to semiconducting and metallic behaviors of the two spin states, respectively. Besides, we perform the calculations of the orbital density of states (ODOS). Figure 3 shows the eigenstates density plots corresponding to all the samples displayed in the previous figure 2. Of course, only cases (c–e) are previously known to host the 'half-metallicity' character. Details of the distribution of the eigen-states are as follows: (a) Pristine C2N behaves as paramagnetic semiconducting. So there is no distinction between spin-up and spin-down states. The VBM state and the CBM state are shown to be predominated by the N atoms to corroborate the results of PDOS shown in figure 2(a). (b) C2N:1Mn (SAC): The embedment of SAC-Mn in one big pore of C2N would yield a magnetic metal, as shown in the previous figure 2(b). The density plots of figure 3(b) shows both spin states to behave metallic as their Fermi eigenstates are hosted by basically all types of atoms and to be percolating from one side to the other. (c) C2N:Mn2 (DAC): In this case the spin-up states behave semiconducting and one needs to present both their corresponding VBM and CBM eigenstates; whereas the spin-down behaves metallic to yield the 'half-metallicity'. While the VBM/CBM states are hosted basically by all species in the samples (see figure 3(c)), the metallic spin-down states are attributed mainly to the two Mn atoms. So, one would expect some d-orbitals mixing to take place in the spin-down states. (d) C2N:Mn–Mn (SAC–SAC): Similarly to the previous case, the spin-down here also behaves metallic to yield 'half-metallicity'. VBM/CBM eigen-states due spin-up sates are attributed basically by all species of atoms in the sample (see figure 3(d)), while the metallic spin-down states at Fermi level are shown to kind of repel from the vicinities of two SAC sites but clearly shown to be percolating to yield metallicity. Yet, the mixing of orbitals at Fermi level seems to take place in a fashion different from that of DAC-based half-metallicity.

Figure 3. Eigen-functions of VBM, CBM and Fermi-level states of C2N 2 × 2-PCs supercell with and without embedment of Mn-atom catalysts: (a) Pristine, (b) SAC, (c) DAC, (d) SAC–SAC, (e) DAC–SAC, and (f) DAC–DAC configurations. Colors of atoms are: C (grey), N (blue), and Mn (purple). Amplitude of wave-function is in yellow color.
Download figure:
Standard image High-resolution image(e) C2N:Mn2–Mn (DAC–SAC): This is a third case to yield half-metallicity where spin-down states behave metallic while the spin-up remain semiconducting. In this particular case, the spin-up eigen-states of VBM and CBM are shown in figure 3(e) to be mainly attributed to the host-crystal atoms (i.e. C and N atoms). On the other hand, the metallic spin-down states are shown to have contributions more on DAC than SAC in addition to the C and N atoms of the sample. This might reveal the mixing of d-states of the two Mn atoms of DAC structure. (f) C2N:Mn2–Mn2 (DAC–DAC): The embedment of two DACs in two neighboring pores of C2N would yield magnetic metal where both spin-up and spin-down exhibit mixing of d-orbitals of Mn atoms within two DACs. The eigen-states at Fermi levels corresponding to the two spin-states are shown in figure 3(f) to be attributed to both DACs as well as N and C atoms in the sample.
3.4. Assessment of configurations versus half-metallicity
It is important to emphasize that the studied structures (i.e. SAC, DAC, and their combinations) so far presented in the previous figures are not unique. Actually for each case, one can think of many other configurations. For instance, in case of combination of SAC–SAC, there is a total of six different combinations one could consider and should inspect for further checking of the validity of the half-metallicity character. As a matter of fact, we undertook cases of combinations of SAC–SAC, SAC–DAC and DAC–DAC more rigorously and inspected all their possible configurations by performing atomic relaxations and carrying out spin-polarized DFT electronic structures with all the results shown in supplementary documents. (1) Case of SAC–SAC Mn embedded in C2N 2 × 2 PC sample: There are six different possible configurations of embedding two SAC-Mn in two neighboring pores of C2N (i.e. keeping the first SAC stationary, the second SAC can be embedded in six possible locations as indicated by letters A–F shown figure S1). All the corresponding spin-polarized TDOS shown in figure S1 reveal the existence of half-metallicity character and persistently showing spin-down states to behave metallic while spin-up state semiconducting with bandgap varying within the range [0.185–0.215] eV. Consistently, the magnetization of the system is found to be about 8.0 μB, which is double of the case of SAC, confirming the FM interactions between SAC and SAC. Results are also summarized in table S1.
(2) Case of DAC–SAC Mn embedded in C2N 2 × 2-PC sample: Here also, there are three DAC orientations per pore and each orientation could be associated with six different possible configurations of SAC (i.e. by maintaining each DAC stationary, there exist six possible configurations for SAC as indicated by letters A–F in the structures presented in figures S2–S4). So, we have a total of 18 different configurations to investigate. The results of total energy, bandgap energy and magnetization per configuration are summarized in tables S2–S4. The results of all the spin-polarized TDOS presented in figures S2–S4 show the existence of half-metallicity character with spin-down states behaving metallic and spin-up states semiconducting with bandgap energy in the range [0.245–0.280] eV. The results of magnetization are shown in tables S2–S4 to be about 7.0 μB. This estimate consolidates the scenario that an anti-ferromagnetic coupling (AFMC) does take place within DAC but a FMC is governing the DAC–SAC interaction. Yet, the question of which interaction AFMC/FMC causing the half-metallicity remains open until we study the effect of sample scaling below.
(3) Case of DAC–DAC Mn embedded in C2N 2 × 2-PC sample: Keeping first DAC fixed, there are three different orientations to consider for the second DAC and thus yielding three different configurations. The atomic structure illustrating these three configurations and the corresponding spin-polarized TDOS are shown in figure S5 and the results of total energy, bandgap energy, and magnetization for each configuration are summarized in table S5. All the results corroborate the metallic behavior for both spin states. Yet the magnetization is about 7.6 μB, which is about the double of magnetization of DAC and thus revealing FMC interaction to take place between DAC and DAC. Besides, the absence of half-metallicity might be attributed to the lack of FMC interaction, as it will be under focus later below when we study the effect of scaling.
3.5. Screening of TM doping versus half-metallicity
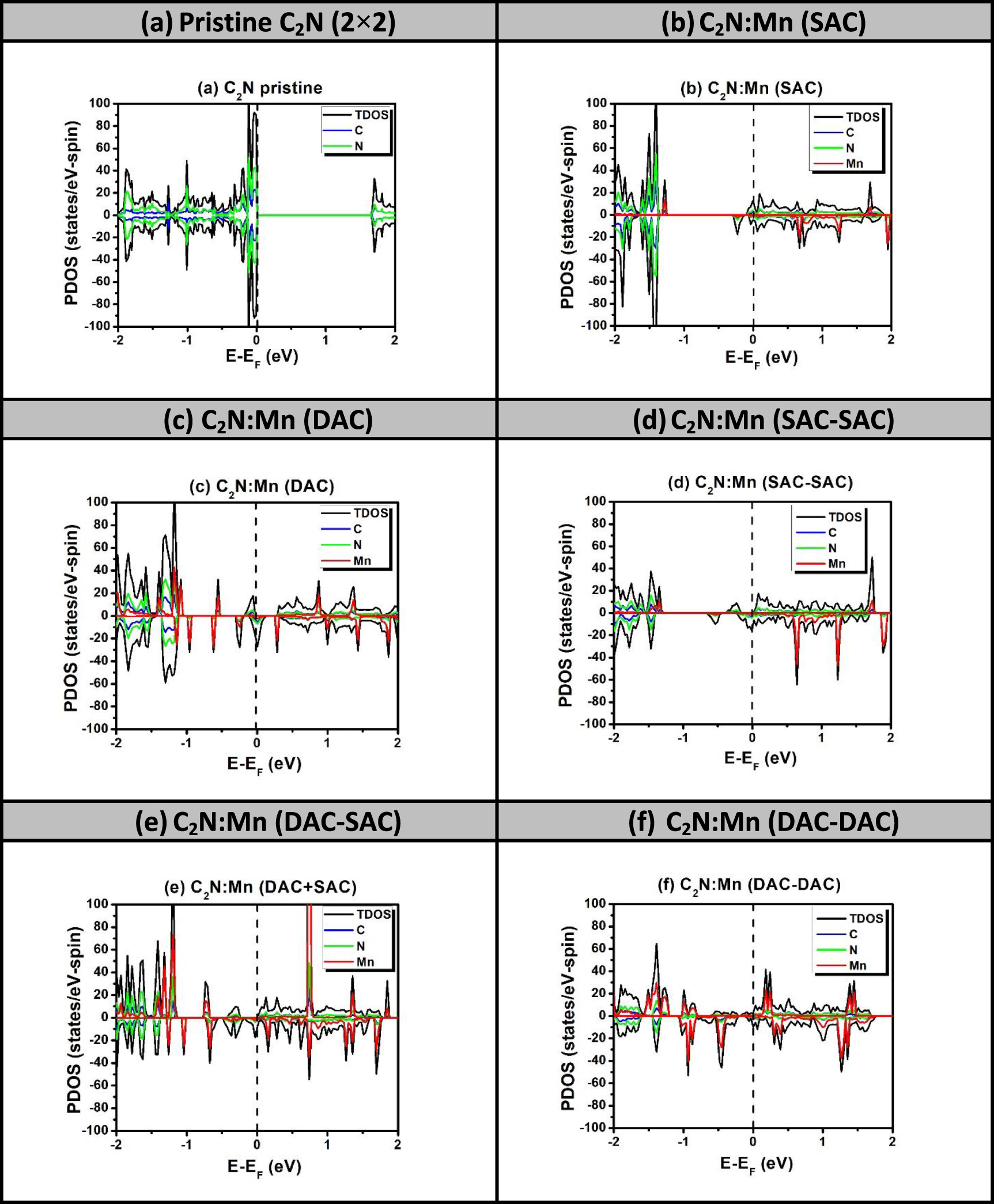
The selection of manganese in our presented study, so far, is based on its high catalytic activity and FM character as it possesses five unpaired d electrons. Actually, we have scanned all the TM atoms ranging from Sc to Zn and searched for possible configurations to obtain half-metallicity. We took the three cases of Mn atoms (DAC, SAC–SAC and SAC–DAC) yielding half-metallicity as model configurations. Nonetheless, we obtained just two further cases exhibiting half metallicity, which correspond to: (a) C2N:Fe2 (DAC) and (b) C2N:Ni2–Ni (DAC–SAC). The results of spin-polarized TDOS for the cases of TM DAC and DAC–SAC for the remaining seven TMs (Sc, Ti, V, Cr, Co, Cu and Zn) are shown in figures S6 and S7, respectively. All the results shown in these latter figures show metallic behavior for both spin states. Throughout the present investigation, the occurrence of half metallicity requires the providence of two simultaneous conditions, which are kind of a compromise between synergetic and FMC-interaction effects. The lack of these conditions would affect the occurrence of half-metallicity. So, having said that, the fact that the half-metallicity found to take place only in special cases, specifically when using the FM elements such as Mn, Fe, and Ni, but not to occur in Co is among the wonders. Furthermore, it is important to emphasize the relevance of magnetic semiconductors for spintronic device applications as was recently reported by Anbuselvan et al [49]. Nonetheless, in the current investigation, we would rather focus on cases of half-metallicity. Figure 4 shows the spin-polarized PDOS of three samples: (a) C2N:Fe2 (DAC): figure 4(a) displays half-metallicity where spin-up states behave as metallic, while the spin-down state remain semiconducting with bandgap energy of about 0.368 eV. The Fe atom is supposed to possess four unpaired d-electrons and 2s-electrons when it is a free atom. In the configuration of DAC-Fe embedded in C2N, three electrons in each Fe-atom are engaged in the bonding. Yet, table 2 shows that the magnetization of the configuration corresponding to figure 4(a) is about M = 2 μB. This in turn suggests that the DAC-Fe stabilizes in an AFM configuration (i.e. similar to Mn2 DAC).
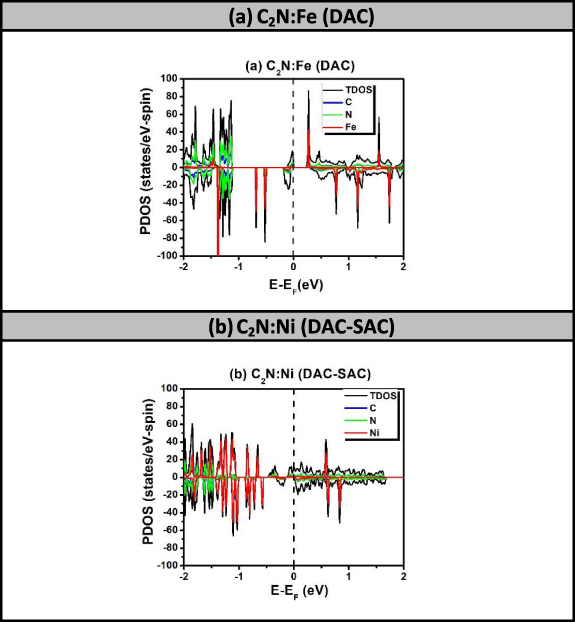
Figure 4. Spin-polarized PDOS of C2N with catalysts Fe and Ni as DAC and DAC–SAC, respectively. These two cases exhibit the half-metallicity.
Download figure:
Standard image High-resolution image(b) C2N:Ni2–Ni (DAC–SAC): figure 4(b) shows the half-metallicity character when Ni2–Ni (DAC–SAC) are embedded in two neighboring large pores of C2N. Fermi level crosses the spin-up states to reveal their metallization while spin-down states remain semiconducting with an energy bandgap of about 0.164 eV. The magnetization value is higher than the case of DAC as FMC between DAC–SAC seems to take place. In a similar way, the trend of half-metallicity seems to remain occurring in similar fashion to Mn-embedment in C2N. We will show below that that trend should rather be attributed to the FMC of the total magnetization with its six mirror images.
Figure 5 displays the VBM/CBM and Fermi eigen-states corresponding to the two samples whose PDOSs were elaborated in the previous figure 4. Namely, the two structures are as follows: (a) C2N:Fe2 (DAC): It exhibits half-metallicity due to spin-up states becoming metallic. So, figure 5(a) presents the eigen-state at Fermi level associated with spin-up states, and the VBM/CBM eigen-states due to the spin-down states exhibiting the semiconducting behavior. The Fe-dimer seems to induce metallization in spin-up states through bands to be carried by C and N atoms around the DAC-Fe. On the other hand, the spin-down states behave semiconducting where the VBM is a very localized state at the DAC-Fe and the CBM is a delocalized state on all sites but not DAC-Fe. Such behaviors of VBM/CBM states should be good model representation of materials with indirect-bandgap transitions (see for instance the band structure in figure 7(e)). (b) C2N:Ni2–Ni (DAC–SAC): This structure behaves as half-metallic with the spin-up states being conductive while spin-down states remain semiconducting. Figure 5(b) shows all the eigenstates of spin-up at Fermi level, and spin-down VBM/CBM to be delocalized. The metallization in spin-up states might be caused by FMC interactions between the total magnetic moment at the DAC–SAC complex and its six mirror images, which are formed by the periodic-boundary conditions.
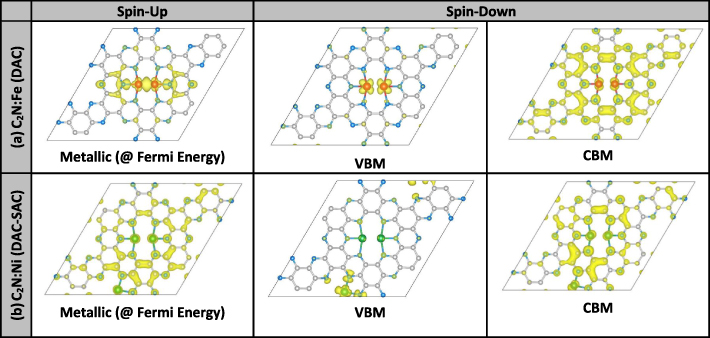
Figure 5. Eigen-functions of VBM, CBM and Fermi-level states of C2N 2 × 2-PCs supercell with Fe and Ni atom catalysts: (a) C2N:Fe (DAC), and (b) C2N:Ni (DAC–SAC). Colors of atoms are: C (grey), N (blue), Fe (red), and Ni (green). Amplitude of wave-function is in yellow color.
Download figure:
Standard image High-resolution image3.6. Bands and ODOS
To pursue further analysis about the origins of the 'half-metallicity', figures 6 and 7 present the spin-polarized band structures and the ODOS for the pristine C2N sample of 2 × 2 PCs in size and the five samples hosting half-metallicity. In figure 6, the spin-up and spin-down are presented by the green-dashed and red-solid curves, respectively. (a) Pristine C2N sample of 2 × 2 PCs shows the evidence of para-magnetism where both spins are indistinguishable. Pristine C2N is shown to have a direct bandgap of Eg = 1.67 eV at Gamma-point (i.e. as being folded from K-point of Brillouin zone, which is the Wigner–Seitz cell of the reciprocal lattice). All the remaining samples behave FM and whose bands are spin-dependent as follows: (b) C2N:Mn2 (DAC): spin-down bands (red-solid curves) are metallic and spin-up bands (green-dashed curves) are semiconducting with an energy bandgap of 0.405 eV. (c) C2N:Mn–Mn (SAC–SAC): spin-down (red-solid curves) are dispersive at Fermi level and thus being metallic; whereas the spin-up bands (green-dashed curves) are semiconducting with an energy bandgap of about 0.20 eV. (d) C2N:Mn2–Mn (DAC–SAC): spin-down bands (red-solid curves) are metallic as crossing Fermi level in dispersive fashion. The spin-up bands (green-dashed curves) are semiconducting having an energy bandgap of 0.25 eV. (e) C2N:Fe2 (DAC): spin-up states (green-dashed curves) behave metallic while spin-down states (red-solid curves) are semiconducting with an energy bandgap of 0.37 eV. (f) C2N:Ni2–Ni (DAC–SAC): spin-up states (green-dashed curves) behave metallic while the spin-down states (red-solid curves) are semiconducting with an energy bandgap of 0.16 eV.

Figure 6. Spin-polarized band structures of C2N with TM-atom catalysts yielding half-metallicity: (a) Pristine C2N, (b) C2N:Mn (DAC), (c) C2N:Mn2 (SAC–SAC), (d) C2N:Mn (DAC–SAC), (e) C2N:Fe (DAC), and (f) C2N:Ni (DAC–SAC). Colors: green (spin-up), red (spin-down) and red-violet shows overlapping of spin up and down.
Download figure:
Standard image High-resolution imageFigure 7 displays the results of spin-polarized ODOS for all the six samples including the pristine C2N 2 × 2-PC sample and all the five samples hosting half-metallicity. Figure 7(a) presents the case or pristine sample and shows the contributions from the s and p orbitals on each C and N atom. The valence band seems to be predominantly composed of C(p) and N(p) orbitals and more specifically attributed to the filled dangling orbitals residing on the N atoms at the big pores. In case of Mn-doped C2N with the catalysts in configurations of DAC, SAC–SAC, and DAC–SAC, shown in figures 7(b)–(d), respectively, the metallization occurs in the spin-down states. The contributions to the spin-down metallic bands are shown to be attributed mainly to the metal catalyst 'Mn' and the neighboring C and N atoms at the perimeter within the big pore containing the catalysts. In the cases of Fe and Ni catalysts, the DAC and DAC–SAC configurations are found to host the half-metallicity, as shown in figures 7(e) and (f). In both these latter cases, the metallization takes place in the spin-up states and attributed to the metal atom and its surrounding C and N atoms located at the perimeter of the large pore.

Figure 7. Spin-polarized ODOS of cases of C2N with embedment of TM-atom catalysts yielding half-metallicity: (a) Pristine C2N, (b) C2N:Mn (DAC), (c) C2N:Mn (SAC–SAC), (d) C2N:Mn (DAC–SAC), (e) C2N:Fe (DAC), and (f) C2N:Ni (DAC–SAC). Curve colors according to what therein legends.
Download figure:
Standard image High-resolution image3.7. FMC versus sample-scaling
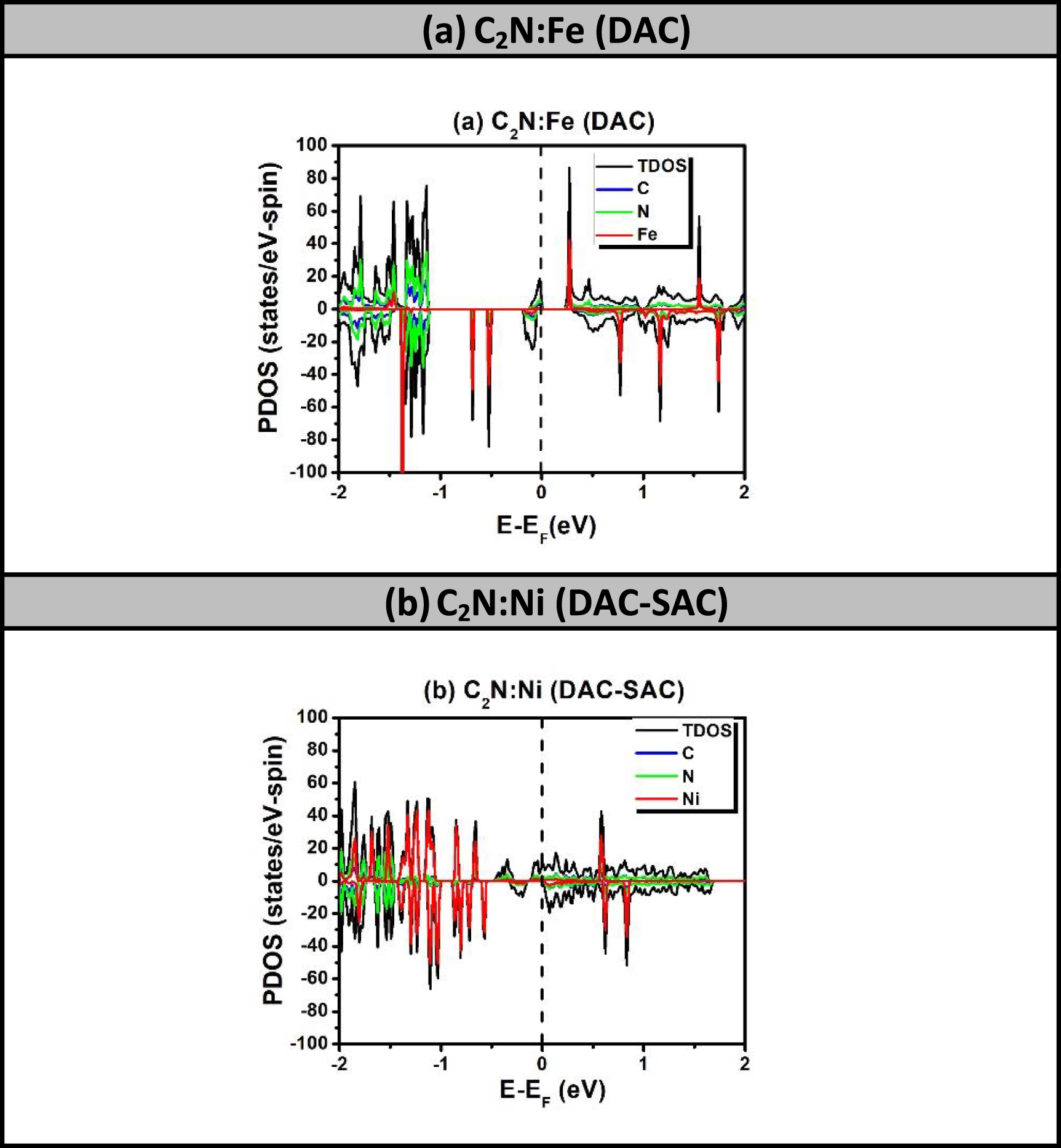
Focusing on the origins of half-metallicity in TM-doped C2N, definitely it is due to synergetic effects of incorporating more than one dopant in adjacent locations. It is clear that SAC-TM alone cannot make it happen. Now, the next decisive question is whether the half-metallicity is a product of an internal FMC/AFMC interactions within the group of TM dopants or external FMC interactions of the group with its six mirror images, formed on the xy-plane by the periodic boundary conditions. In order to assess this latter hypothesis, we have tested the existence of 'half-metallicity' in case of larger samples while keeping the group of TM atoms robust. Furthermore, one should focus on both the existence of 'half-metallicity' and more specifically on the width of the band at Fermi level of the metallic spin. So, we have basically five cases to inspect, for which we calculated the spin-polarized PDOS and bands. The resulting data are extracted and summarized in table 4. (a) C2N:Mn2 (DAC): For the sample of size 3 × 3 PCs, figures 8(a) and 9(a) show the existence of metallic (dispersive) band crossing Fermi level for spin-down states while the other spin is semiconducting with bandgap energy of 0.302 eV. The half-metallicity in such system disappears in the sample of size 4 × 4 PCs, which becomes semiconducting. (b) C2N:Mn-Mn (SAC–SAC): For the sample of size 3 × 3 PCs, figures 8(b) and 9(b) show the disappearance of 'half-metallicity' as both spin states becoming metallic. The bands of both spin-up and spin-down sates shown in figure 9(b) are dispersive. (c) C2N:Mn2-Mn (DAC–SAC): For the sample of size 3 × 3 PCs, figures 8(c) and 9(c) confirm the disappearance of 'half-metallicity' as both spin states becoming metallic with dispersive bands crossing Fermi level. (d) C2N:Fe2 (DAC): For the sample of size 3 × 3 PCs, figures 8(d) and 9(d) show the disappearance of 'half-metallicity' as both spin-up and spin-down states becoming semiconducting. (e) C2N:Ni2–Ni (DAC–SAC): For the sample of size 3 × 3 PCs, figures 8(e) and 9(e) show the half-metallicity to still exist and is due to the metallization of spin-up states while the spin-down states behave as semiconducting with bandgap energy of about 0.163 eV. Nonetheless, for larger sample size 4 × 4 PCs, the 'half-metallicity' disappears as both spins becoming semiconducting. In brief, the disappearance of 'half-metallicity' for larger samples is confirming that it originates from the FMC interactions of the magnetization of the group of dopants with the six mirror images, formed on xy-plane by the implementation of the periodic-boundary conditions. The FMC interaction, inducing the half-metallicity, have potential energies of magnitude up to 20 meV and spatially ranging up to critical lengths of order Lc ∼ 21–29 Å, as shown in table 4.

Figure 8. Spin-polarized PDOS of 3 × 3 scaled up samples of C2N with TM-atom catalysts that exhibited half-metallicity in 2 × 2 sample size: (a) C2N:Mn (DAC), (b) C2N:Mn (SAC–SAC), (c) C2N:Mn (DAC–SAC), (d) C2N:Fe (DAC), and (e) C2N:Ni (DAC–SAC).
Download figure:
Standard image High-resolution image
Figure 9. Spin-polarized band structures of 3 × 3 scaled up samples of C2N with TM-atom catalysts that exhibited half-metallicity in 2 × 2-sample size: (a) C2N:Mn (DAC), (b) C2N:Mn2 (SAC–SAC), (c) C2N:Mn (DAC–SAC), (d) C2N:Fe (DAC), and (e) C2N:Ni (DAC–SAC). Colors: spin-up (green) and spin-down (red).
Download figure:
Standard image High-resolution imageTable 4. The variation of magnetization, spin-polarized bandgap, and FMC potential 'VFMC' versus the distance between the net magnetic-moment center and its mirror images using the scaling of computational sample size (i.e. ranging from 2 × 2 to 3 × 3 and to 4 × 4 PCs).
| Sample 2 × 2 | Sample 3 × 3 | Sample 4 × 4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Host | dM–M (Å) | M (µB) | Property BW (eV), VFMC (meV) |
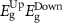
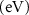
| dM–M (Å) | M (µB) | Property BW (eV), VFMC (meV) |
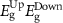
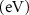
| dM–M (Å) | M (µB) | Property BW (eV), VFMC (meV) |
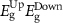
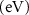
|
| Pristine C2N | N/A | 0 | S/C | 1.67 | N/A | 0 | S/C | 1.67 | N/A | 0 | S/C | 1.67 |
| 1.67 | 1.67 | 1.67 | ||||||||||
| C2N:Mn (DAC) | 16.66 | 4.0 | Half metal | 0.405 | 25.0 | 4.0 | Half metal | 0.302 | 33.33 | 4.33 | S/C | 0.64 |
| 0.150 (12.5) | 0 | 0.050 (4) | 0 | 0.55 | ||||||||
| C2N:Mn (SAC–SAC) | 16.66 | 7.0 | Half metal | 0.20 | 25.0 | 7.533 | Metal | 0.0 | ..... | ..... | ..... | ..... |
| 0.240 (20) | 0 | 0.0 | ||||||||||
| C2N:Mn (DAC–SAC) | 16.66 | 7.0 | Half metal | 0.25 | 25.0 | 7.001 | Metal | 0.0 | ..... | ..... | ..... | ..... |
| 0.230 (19) | 0 | 0.0 | ||||||||||
| C2N:Fe (DAC) | 16.66 | 2.00 | Half metal | 0.0 | 25.0 | 2.00 | Magnetic S/C | 0.739 | ..... | ..... | ..... | ..... |
| 0.122 (10) | 0.368 | 0.29 | ||||||||||
| C2N:Ni (DAC–SAC) | 16.66 | 1.999 | Half metal | 0.0 | 25.0 | 1.999 | Half metal | 0.0 | 33.33 | 1.9224 | S/C | 0.25 |
| 0.185 (15.4) | 0.164 | 0.105 (9) | 0.163 | 0.504 | ||||||||
3.8. Ferromagnetism and anti-ferromagnetism versus half-metallicity
To assess the nature of magnetism taking place in case of occurrence of half-metallicity, it is more convenient to use a larger computational supercell such as 4 × 4 PC of C2N (i.e. in case of pristine, it contains 288 atoms = 192 C + 96 N atoms and in case DAC–SAC the supercell should contain 288 + 4 × 3 = 300 atoms). Basically this supercell should comprise four weights of repetition of the 2 × 2-PC sample (i.e. two simultaneous repetitions along each x and y direction). We have used this large supercell for two tasks. In task #1, we tested to double check the existence of half-metallicity in the case of large 4 × 4-PC samples corresponding to five cases: (a) C2N:Mn (SAC), (b) C2N:Mn (DAC), (c) C2N:Mn (SAC–SAC), (d) C2N:Mn (DAC–SAC), and (e) C2N:Mn (DAC–DAC). The results of the spin-polarized TDOS are shown in figure S6. The results are consistent with those obtained using the smaller supercell of size 2 ×2 PCs, displayed in figures 2(b)–(f), approving the metallicity in the two cases of (a) SAC and (e) DAC–DAC; and confirming the half-metallicity in the other three cases (b) DAC, (c) SAC–SAC, and (d) DAC–SAC revealing the half-metallicity to be attributed to the FMC interactions.
In task #2, the large-sized sample of 4 × 4 PCs is explored to carry out a comparison between the effects of FMC and AFMC interactions on the half-metallicity. For demonstration, we took the case of DAC, which yields half-metallicity in the 2 × 2-PC sample, and we assessed the effects of both FMC and AFMC interactions on the half-metallicity in the large 4 × 4-PC sample. The results of relaxed atomic structures and spin-polarized PDOS and TDOS are shown in figure 10. It is remarkable that the total energy in the sample exhibiting FMC interactions is lower than that of the sample exhibiting AFMC interactions (i.e. ΔE =  ). Consequently, the ground state should correspond to the existence of the FMC interactions. The second interesting trend shown in figure 10 is that the FMC interactions do yield half metallicity whereas the AFMC interactions yield metallic state. The magnetization in the latter case seems to vanish as the AFMC interactions cannot even exist at that range of distance (which is about 16.6 Å). In our simulations, we have considered each DAC as a unity having one magnetic moment as presented in figure 10, and we turned on the magnetic interactions with results displayed therein. Throughout our discussions of various combinations of SACs and DACs and various types of magnetic interactions, it should be concluded that the existence of the half-metallicity requires a compromise between synergetic and FMC effects.
). Consequently, the ground state should correspond to the existence of the FMC interactions. The second interesting trend shown in figure 10 is that the FMC interactions do yield half metallicity whereas the AFMC interactions yield metallic state. The magnetization in the latter case seems to vanish as the AFMC interactions cannot even exist at that range of distance (which is about 16.6 Å). In our simulations, we have considered each DAC as a unity having one magnetic moment as presented in figure 10, and we turned on the magnetic interactions with results displayed therein. Throughout our discussions of various combinations of SACs and DACs and various types of magnetic interactions, it should be concluded that the existence of the half-metallicity requires a compromise between synergetic and FMC effects.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Comparison between the effects of FMC and AFMC interactions on the half-metallicity using a large sample of C2N:Mn 4 × 4 PCs containing four Mn-DACs. (a) Relaxed atomic structures, (b) spin-polarized PDOS and TDOS. Fermi level is taken as an energy reference.
Download figure:
Standard image High-resolution image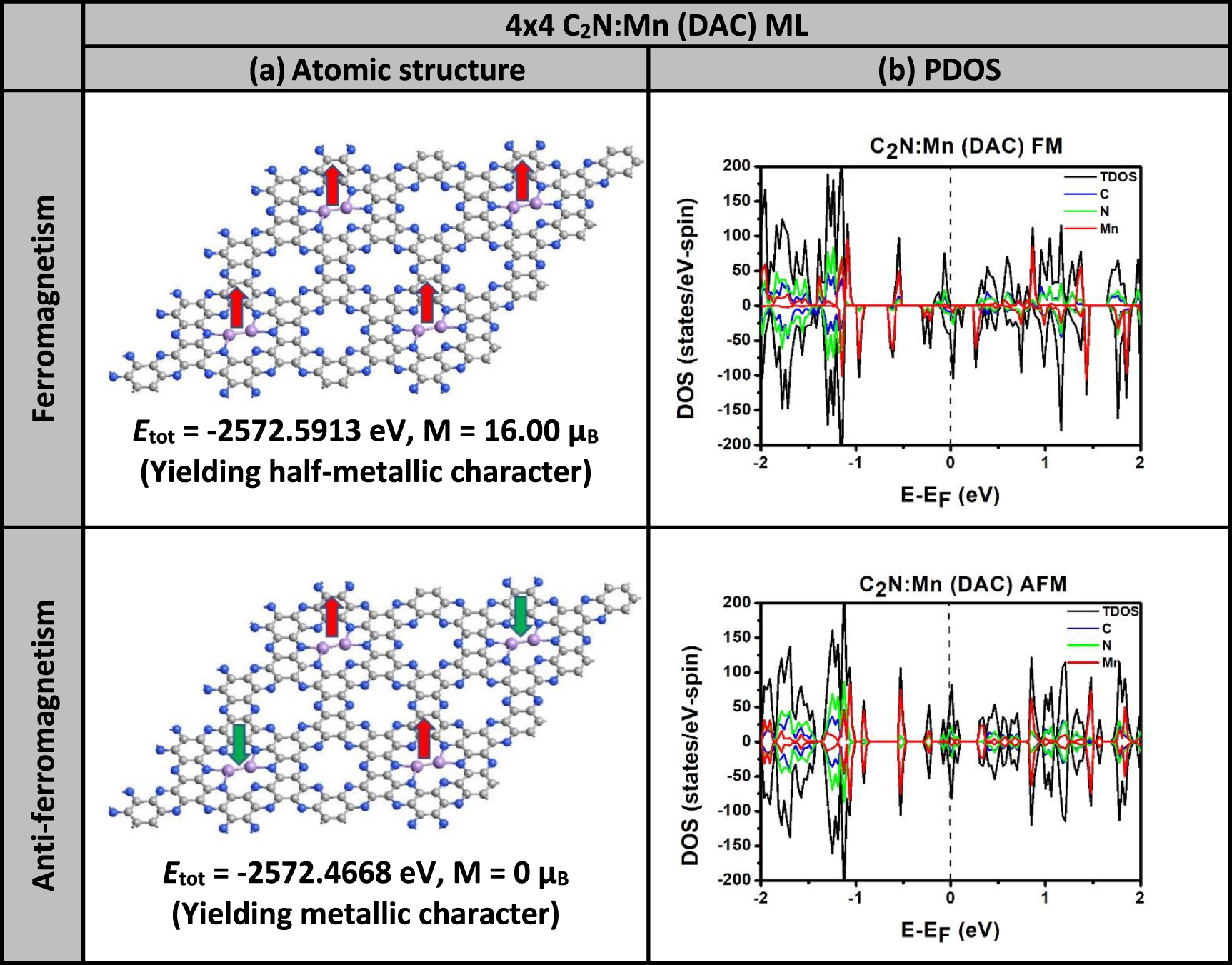{kind=link}
4. Conclusions
We have used spin-polarized DFT to search for the existence of 'half-metallicity' in C2N functionalized by embedding various configurations of TM-atom such as SACs, DACs, and their combinations. We scanned all possible catalysts comprising of the 3D block of TM elements ranging from Sc to Zn. In our calculations we employed VASP-package which is world-wide popular for its reliability for predicting the ground-state properties such as the atomic, electronic, and magnetic properties. We were limited by the system size, but luckily, the task was completely achieved as the 'half-metallicity' was proven to originate from FMC interactions between the group of TM-dopants and its six mirror images, formed by the periodic-boundary conditions. So, the needed samples are of sizes 2 × 2, 3 × 3, and at most 4 × 4 PCs (i.e. comprising four-TM atoms at maximum, and 72, 162, 288 atoms, respectively). The results can be summarized as follows:
- (a)Pristine C2N is paramagnetic semiconductor with bandgap energy of about 1.67 eV a bit underestimated compared to the experimental data of inventors 1.96 eV.
- (b)Three TM atoms (i.e. Mn, Fe and Ni) are found to induce the 'half-metallicity' in C2N sample of size 2 × 2 PCs in five different configurations, namely: (a) C2N:Mn2 (DAC), (b) C2N:Mn–Mn (SAC–SAC), (c) C2N:Mn2–Mn (DAC–SAC), (d) C2N:Fe2 (DAC), and (e) C2N:Ni2–Ni (DAC–SAC).
- (c)The calculations of binding energies confirmed that the embedment of both SAC and DAC were thermodynamically very stable. The recursive energy in DAC is even higher than the cohesive energy in strength.
- (d)The calculations of the magnetization reveal that the DAC in one large pore of C2N exchange AFMC interaction, whereas the inter-pore magnetic interaction between SAC–SAC, or DAC–SAC, or DAC–DAC to be FMC interaction. Yet, such FMC/AFMC intra-group interaction should be the driving force for the induction of the 'half-metallicity'.
- (e)The scaling of sample size to comprise the 3 × 3-PC and 4 × 4-PC samples has shown the disappearance of the half-metallicity and confirmed that its existence would be attributed not to the intra-magnetic coupling interactions but rather to the FMC interactions of the group of TM-dopants with its six mirror images, formed via the implementation of the periodic-boundary conditions.
Thus, our work showed the design of 2D functionalized materials (e.g. TM-catalysts embedded in large pores of C2N as SAC–DAC combinations) hosting 'half-metallicity'. The results further showed that the origins of existence of 'half-metallicity' to be attributed to the existence of FMC interactions between the group of TM-dopants and its six mirror images. Half metallicity is a fundamental pillar in the spintronics and our results should be very relevant to spintronic device applications.
Acknowledgments
The authors are indebted to thank Professor A Qteish for fruitful discussions, Dr M A Sattar for some computational support, Dr Thomas Fowler for the critical reading of the original manuscript, and the National Water and Energy Center (NWEC) at the UAE University for the financial support (Grants Numbers. 31R145 and 12R125).
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Conflict of interest
The authors declare no competing interests. The project was sponsored by the National Water and Energy Center at the UAE University (Grant Nos. 31R145 and 12R125).
Author contributions
This work started during a visit by two of us (S K and N T) to NUS to collaborate with Y P F. One of us (S K) performed DFT calculations using the VASP package and prepared the figures. Y P F was the host in NUS and participated in both the supervision of the project and the writing of the manuscript. N T acted as the supervisor to the PhD student (S K), revised the figures, and wrote the first-draft of manuscript. Thus their contributions can be divided as 60%, 20% and 20%, respectively.
Ethics statement
The authors confirm that they have read and understood the research ethics and integrity policy of the journal.
Supplementary data (3.8 MB PDF)