
Abstract
Thermally activated delayed fluorescence (TADF) has recently emerged as one of the most attractive methods for harvesting triplet states in metal-free organic materials for application in organic light emitting diodes (OLEDs). A large number of TADF molecules have been reported in the literature with the purpose of enhancing the efficiency of OLEDs by converting non-emissive triplet states into emissive singlet states. TADF emitters are able to harvest both singlets and triplet states through fluorescence (prompt and delayed), the latter due to the thermally activated reverse intersystem crossing mechanism that allows up-conversion of low energy triplet states to the emissive singlet level. This allows otherwise pure fluorescent OLEDs to overcome their intrinsic limit of 25% internal quantum efficiency (IQE), which is imposed by the 1:3 singlet–triplet ratio arising from the recombination of charges (electrons and holes). TADF based OLEDS with IQEs close to 100% are now routinely fabricated in the green spectral region. There is also significant progress for blue emitters. However, red emitters still show relatively low efficiencies. Despite the significant progress that has been made in recent years, still significant challenges persist to achieve full understanding of the TADF mechanism and improve the stability of these materials. These questions need to be solved in order to fully implement TADF in OLEDs and expand their application to other areas. To date, TADF has been exploited mainly in the field of OLEDs, but applications in other areas, such as sensing and fluorescence microscopies, are envisaged. In this review, the photophysics of TADF molecules is discussed, summarising current methods to characterise these materials and the current understanding of the TADF mechanism in various molecular systems.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Thermally activated delayed fluorescence (TADF), also known as E-type delayed fluorescence (DF), was first rationalised by Perrin in 1929 [1], and later in 1941 by Lewis in fluorescein solutions [2]. In 1961 Parker and Hatchard also reported E-type DF in eosin and benzyl [3, 4], and in 1968 TADF was the form chosen by Wilkinson and Horrocks to identify the delayed luminescence [5]. This relatively well known mechanism was recently reinvigorated by Adachi, who proposed it as a way to harvest non-emissive triplet excited states in organic light emitting diodes (OLEDs) [6].
The singlet production yield appearing from direct charge recombination in OLEDs is limited to 25%, the remaining 75% of charge recombination events give rise to triplet states that are generally non emissive at room temperature. This represents a major loss mechanism in the efficiency of OLEDs, which has triggered the investigation of different mechanisms for triplet harvesting in organic materials. The most common approach to overcome this limitation is the use of complexes containing heavy-metals, which because of the enhanced spin–orbit coupling (SOC) due to the presence of heavy-atoms, introduce sufficient mixing between the singlet and triplet states to make radiative decay of the triplet state possible [7]. Consequently, heavy-metal complexes became very popular, because they allowed internal quantum efficiencies (IQEs) of OLEDs to reach 100%. However, while transition-metal (predominately iridium or platinum) based materials certainly have many advantages, they also show the following major disadvantages when used in OLEDs: (i) they are unstable, particularly in the blue emission region [8]; (ii) they are expensive and (iii) may be toxic. Consequently, they are not suitable for applications where high-production output is required, such as lighting and display industries, because they are not economically-viable, also due to potential limitations on the global supplies of rare-earth elements [9]. Copper complexes have been introduced as a way of replacing rare-metals in complexes while maintaining the ability to harvest singlet and triplet states in OLEDs. Interestingly some of these complexes show a significant TADF contribution [10, 11]. Their photophysics has recently been reviewed by the Yersin group [12], therefore, herein we focus on purely organic molecules.
Triplet harvesting trough triplet fusion, appearing from triplet–triplet annihilation (TTA) has also been proposed as a way to harvest triplet states in OLEDs [13]. In this case, the interaction between two colliding triplet states leads to one singlet excited state, in the most favourable outcome, which can decay radiatively and give rise to DF. Unfortunately, the outcome of the TTA approach is strongly dependent on the energy ordering of the singlet and triplet excited states, and in the best possible alignment of the energy levels, the maximum IQE possible is around 62.5% [14]. TTA has been already proved to contribute to the electroluminescence in OLEDs [15], however, this approach has still not fully proved its merits.
Reverse intersystem crossing in hot excitons (hot-exciton rISC) has also been proposed as a mechanism for triplet harvesting in OLEDs [16, 17]. Molecules designed for efficient hot-exciton rISC have higher lying singlet and triplet excited states, Sn and Tn respectively, with strong charge transfer (CT) character and close energy levels. In this way, the rISC rate constant, occurring between the Tn and Sn states, is able to compete with the rate of internal conversion (IC) within the triplet manifold. The upper level Tn states are thus able to be converted to Sn states in an efficient manner, and then internally convert to the lower energy singlet state (S1) of local character, from where the emission occurs. In summary, the hot-exciton mechanism is based on reverse intersystem crossing that occurs between the upper singlet and triplet levels of CT character, to ensure a larger singlet production yield, followed by emission from the low energy singlet state, with local character and strong fluorescence yield. In this way the 75% triplets will be converted into emissive singlet states, and hot-exciton rISC has potential to achieve 100% IQE. The difficulty with the hot-exciton mechanism is that in order to ensure a larger production of singlet states, the IC between Tn and T1 needs to be supressed. However, IC is very efficient in most organic molecules, with typical IC rates ∼1010–109 s−1. This makes it extremely unlikely that rISC will be able to compete in an efficient manner and consequently, the design of molecules with strong hot-exciton rISC is very challenging [18].
In contrast to the hot-exciton mechanism, TADF molecules are designed to have the lowest singlet (S1) and triplet (T1) excited states with close energy levels, so rISC occurs between the lower singlet and triplet excited states. In this way, the problems affecting rISC in the hot-exciton mechanism are avoided, because the energy gap between T1 and the ground state, S0, is relatively large in most molecules. In practice, this large energy gap between T1 and S0, strongly supresses non-radiative decay from T1, and allows rISC to compete in a very efficient manner. Triplet harvesting efficiencies close to 100% can be obtained in many molecules that emit in the blue [18–21] and green regions [18, 22, 23], however in the red region the triplet harvesting efficiencies are usually lower, due to more pronounced non-radiative decay.
Broadly speaking, the main challenge when designing TADF molecules is engineering a molecular structure that ensures a small energy splitting between the lowest singlet and triplet states, while simultaneously minimising IC and maintaining strong fluorescence yields. Molecules with a small singlet–triplet energy splitting are relatively easy to achieve using covalently linked electron donor (D) and acceptor (A) units. This yields singlet and triplet excited states with strong CT character, which results in very small overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) frontier orbitals, and leads to a decrease in the electronic exchange energy, and therefore, a small energy gap between the singlet and triplet states [24]. However, while this objective seems relatively easy to achieve, simultaneously minimising IC and obtaining strong fluorescence yield in molecules with strong CT character is not a straightforward task, and many factors, such as molecular geometry, the dielectric medium, the presence of low energy triplet excited states, localised in the D or A units, may influence the photophysics of these molecules and the efficiency of the TADF [22, 25].
This article presents an overview rather than a comprehensive review, and is focused on the current understanding of the complex photophysics of TADF materials. In particular we focus upon the specific methods that are currently in use to characterise the TADF mechanism, and determine the photophysical parameters that are useful to correlate the observation of strong TADF with molecular structure. There are already comprehensive reviews covering the design and synthesis of TADF molecules [18], and therefore, here we just touch this aspect briefly.
2. Basic rules for the design of TADF emitters
Efficient TADF molecules have to simultaneously satisfy the conditions of a small energy gap between the singlet and triplet excited states, 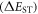 and minimise non-radiative decay to ensure that the triplet excited state lives long enough to maximise the chance of triplet harvesting through the thermally activated reverse intersystem crossing mechanism (rISC), thus maximising the fluorescence yield, see figure 1. A small
and minimise non-radiative decay to ensure that the triplet excited state lives long enough to maximise the chance of triplet harvesting through the thermally activated reverse intersystem crossing mechanism (rISC), thus maximising the fluorescence yield, see figure 1. A small 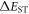 is thus critical to maximise the rISC rate constant
is thus critical to maximise the rISC rate constant 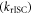 given by equation (1), and it is, therefore, important to understand how this can be minimised in molecular structures.
given by equation (1), and it is, therefore, important to understand how this can be minimised in molecular structures.
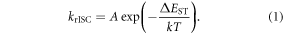
Three different aspects are usually considered when calculating the energy of the lowest singlet 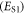 and triplet
and triplet 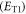 excited states: (i) the orbital energy
excited states: (i) the orbital energy 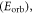 i.e. the energy associated with the one-electron orbital for a fixed nuclear framework in the excited state, (ii) the electron repulsion energy
i.e. the energy associated with the one-electron orbital for a fixed nuclear framework in the excited state, (ii) the electron repulsion energy 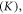 this is the first-order Coulombic correction, and (iii) the exchange energy
this is the first-order Coulombic correction, and (iii) the exchange energy 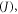 i.e. the first order quantum-mechanical correction involving electron–electron repulsion due to the Pauli principle, which affects the two unpaired electrons in the excited state, i.e. one electron in the HOMO and the other in the LUMO [26]. Therefore, for singlet and triplet electronic states with the same electronic configuration, the three components each contribute equally. However, due to the different spin arrangement of the singlet and triplet excited states, the exchange term increases the energy in the S1 state and decreases it in the T1 state by the same amount, according with equations (2) and (3) [18]. The singlet–triplet energy gap
i.e. the first order quantum-mechanical correction involving electron–electron repulsion due to the Pauli principle, which affects the two unpaired electrons in the excited state, i.e. one electron in the HOMO and the other in the LUMO [26]. Therefore, for singlet and triplet electronic states with the same electronic configuration, the three components each contribute equally. However, due to the different spin arrangement of the singlet and triplet excited states, the exchange term increases the energy in the S1 state and decreases it in the T1 state by the same amount, according with equations (2) and (3) [18]. The singlet–triplet energy gap 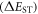 is therefore given by equation (4).
is therefore given by equation (4).
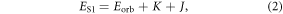
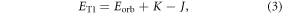

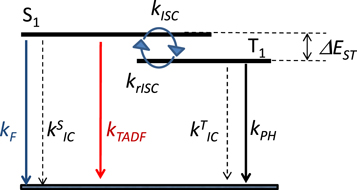
Figure 1. Simplified representation of the electronic energy levels, and rate constants involved in the TADF kinetic mechanism.
Download figure:
Standard image High-resolution imageFrom equation (4) is clear that minimising the singlet–triplet energy gap requires minimisation of the exchange energy 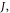 which is calculated using equation (5). Here,
which is calculated using equation (5). Here, 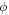 and
and 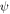 represent the HOMO and LUMO wavefunctions, respectively, and
represent the HOMO and LUMO wavefunctions, respectively, and 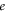 is the electron charge. This of course assumes the excited states under consideration are pure HOMO–LUMO transitions. If this is not the case
is the electron charge. This of course assumes the excited states under consideration are pure HOMO–LUMO transitions. If this is not the case 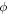 and
and 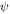 are replaced with the many-body electronic wavefunction for the singlet and triplet states, respectively. This shows that
are replaced with the many-body electronic wavefunction for the singlet and triplet states, respectively. This shows that 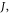 can be minimised by decreasing the overlap between the HOMO and LUMO orbitals, which is achieved in first approximation by spatially separating these frontier orbitals. This is accomplished by molecules containing electron donor (D) and electron acceptor (A) moieties, which favour D–A electron transfer in the excited state. TADF molecules are, therefore, formed in general by D and A units, linked via an aromatic bridge, and form excited states of strong CT character. The singlet–triplet energy splitting can be further reduced by twisting the D and A units around the D–A axis, and obtain D–A relative orientation near-orthogonality, or alternatively, increasing the D–A distance, using a molecular bridge [27].
can be minimised by decreasing the overlap between the HOMO and LUMO orbitals, which is achieved in first approximation by spatially separating these frontier orbitals. This is accomplished by molecules containing electron donor (D) and electron acceptor (A) moieties, which favour D–A electron transfer in the excited state. TADF molecules are, therefore, formed in general by D and A units, linked via an aromatic bridge, and form excited states of strong CT character. The singlet–triplet energy splitting can be further reduced by twisting the D and A units around the D–A axis, and obtain D–A relative orientation near-orthogonality, or alternatively, increasing the D–A distance, using a molecular bridge [27].
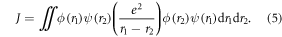
2.1. Evidence for the presence of CT states
Experimental evidence that a particular material forms excited states of CT character comes from the solvatochromism observed in their emission. The pronounced spectral shift with increasing solvent polarity is mainly due to the excited state dipole of the TADF molecules, which arises from the redistribution of electronic density associated with a CT state [28, 29].
Figure 2 shows the solvatochromic effect on the fluorescence spectra of a D–A–D molecule, with fluorene as the electron donor, and dibenzothiophene-S, S-dioxide as the electron acceptor units, (FASAF), which creates excited states of CT character. In low polarity solvents, for example hexane, the FASAF emission spectrum is well resolved, however with increasing polarity, e.g. in ethanol and acetonitrile, the emission spectra redshifts and becomes increasingly structureless and approaching a Gaussian shape, which is typically observed in the emission from excited states with strong charge transfer character (CT) [30].

Figure 2. Emission spectra of FASAF in solvents of increasing polarity [30].
Download figure:
Standard image High-resolution imageThe magnitude of the singlet–triplet energy splitting is in principle directly related with the CT character of the excited state. For example, materials with strong CT excited states, show broad, CT like, emission even in non-polar environments, and 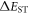 values are often less than 100 meV [22]. However, discrepancies have been reported in the correlation between the HOMO–LUMO overlap extracted from calculations, and
values are often less than 100 meV [22]. However, discrepancies have been reported in the correlation between the HOMO–LUMO overlap extracted from calculations, and 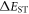 values, which may indicate that other factors may influence
values, which may indicate that other factors may influence 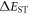 significantly, which require further investigation [31].
significantly, which require further investigation [31].
2.2. Design of TADF molecules
The donor and acceptor moieties used to design TADF emitters have a critical role in the photophysical properties of TADF molecules, and therefore should be carefully selected. The connectivity between the D and A units also has a profound impact on the singlet–triplet energy gap and intersystem crossing rates. The influence of molecular isomerization on TADF has been reported, for example, by Dias et al [22], showing marked difference on the TADF efficiency between D–A–D isomers that differ only in their linkage position of the carbazole or diphenylamine electron donor (D) units to the dibenzothiophene-S, S-dioxide electron acceptor unit (A). Substitution at the C-2, 8 positions of the dibenzothiophene-S, S-dioxide unit gives TADF, whereas with substitution at the C-3, 7 positions the TADF is switched off, see figure 3.

Figure 3. Effect of D–A molecular connectivity on the TADF efficiency, exemplified with two isomers containing carbazole (1 and 2) or diphenylamine (3 and 4) as electron donor (D) units and dibenzothiophene-S, S-dioxide electron acceptor unit (A). Compounds 1 and 3 have strong TADF while 2 and 4 show no TADF emission [22].
Download figure:
Standard image High-resolution imageDepending on the host, the energy difference between the singlet and triplet states in compounds 1–4 in figure 3 is strongly influenced by the different connectivity between the D and A units, with compounds 1 and 3 giving the smallest gaps and consequently the largest TADF contributions.
In general, the singlet–triplet energy splitting is reduced by introducing strong donor and acceptor moieties, as previously discussed. However, further reductions in 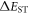 are still achievable. Twisted geometries around the D–A linker have been used to achieve D–A molecules with relative orientation near orthogonality and even smaller
are still achievable. Twisted geometries around the D–A linker have been used to achieve D–A molecules with relative orientation near orthogonality and even smaller 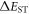 [23, 25, 32], see figure 4 for the molecular structure and molecular geometry of DPTZ-DBTO2, a TADF molecule formed by two phenothizazine donors and the dibenzothiophene-S, S-dioxide acceptor [23]. The single–triplet energy gap in DPTZ-DBTO2 is around 50 meV, depending on the host. Device efficiencies with this green emitter were obtained around 18%.
[23, 25, 32], see figure 4 for the molecular structure and molecular geometry of DPTZ-DBTO2, a TADF molecule formed by two phenothizazine donors and the dibenzothiophene-S, S-dioxide acceptor [23]. The single–triplet energy gap in DPTZ-DBTO2 is around 50 meV, depending on the host. Device efficiencies with this green emitter were obtained around 18%.

Figure 4. Chemical structure, HOMO and LUMO and and x-ray molecular structure of DPTZ-DBTO2 [23].
Download figure:
Standard image High-resolution imageSimilar effects can be obtained by increasing the D–A distance [33, 34]. These molecular geometries lead to strongly localised HOMO and LUMO orbitals, and thus smaller 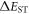 values. Unfortunately, a clear trade-off exists between the efficiency of the rISC mechanism and the electronic coupling between the ground and excited singlet states. While negligible orbital overlap leads to very small
values. Unfortunately, a clear trade-off exists between the efficiency of the rISC mechanism and the electronic coupling between the ground and excited singlet states. While negligible orbital overlap leads to very small 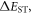 it also leads to low radiative rates and therefore reduced fluorescence yields
it also leads to low radiative rates and therefore reduced fluorescence yields 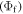 [32]. On the other hand, weak donors and acceptors induce less significant HOMO and LUMO localisation, which results in a relatively larger singlet–triplet energy gap, thus decreasing the TADF contribution. Further research is, therefore, necessary to find structures with well-balanced
[32]. On the other hand, weak donors and acceptors induce less significant HOMO and LUMO localisation, which results in a relatively larger singlet–triplet energy gap, thus decreasing the TADF contribution. Further research is, therefore, necessary to find structures with well-balanced  and
and  values. Strong TADF contribution with relatively high fluorescence yields is, in principle, also achievable in molecules using a combination of weak donor, coupled with a stronger electron-acceptor unit. In these cases, several weak donors are often used to strengthen the donor character of the TADF molecule, while simultaneously maintaining relatively high fluorescence yield [6, 35].
values. Strong TADF contribution with relatively high fluorescence yields is, in principle, also achievable in molecules using a combination of weak donor, coupled with a stronger electron-acceptor unit. In these cases, several weak donors are often used to strengthen the donor character of the TADF molecule, while simultaneously maintaining relatively high fluorescence yield [6, 35].
Figure 5 shows a series of TADF emitters based on carbazolyl dicyanobenzene (CDCB), with carbazole as a donor and dicyanobenzene as the electron acceptor, where these strategies were implemented by the Adachi group [6]. The emission of CDCBs, peak emission wavelength and fluorescence yield, is strongly dependent on the number and positions of substitution in the dicianobenzene, ranging from 450 nm for 2CzPN to 580 nm to 4CzTPN-Ph, and OLEDs fabricated with these compounds gave efficiencies of 19% (green), 11% (orange) and 8% (sky-blue).
Figure 5. Molecular structures of CDCBs. Me, methyl; Ph, phenyl [6].
Download figure:
Standard image High-resolution imageMost frequently used combinations of electron donor and acceptor units in TADF compounds are derivatives of the molecules represented in figure 6 [18].
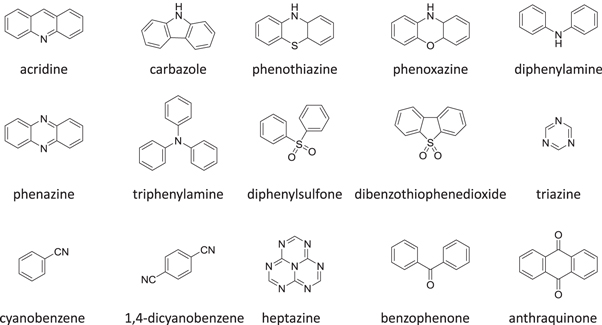
Figure 6. Molecular structures of most common electron donor and acceptor units used in TADF molecules.
Download figure:
Standard image High-resolution imageDespite the significant advances in recent years on the development of several TADF molecules, still further studies are necessary to design molecular structures with improved tuning of their emission and improved stability. The majority of TADF molecules have emission in the green region. However some blue emitters [20, 21, 36], and fewer red emitters [33, 37], have been reported recently, but clearly more structures are needed in these spectral regions. The design of blue TADF emitters is difficult because they require small 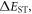 while keeping the emission in the blue. This requires using weak donors and acceptors, so the CT emission is not strongly shifted to lower energies. Red TADF emitters are particularly limited, because the excited states are strongly affected by non-radiative decay, in agreement with the energy gap law. This decay actively competes with the rISC mechanism and quenches the excited state population, leading to lower quantum yields. Further molecular design is thus necessary to achieve blue and red emitters with small
while keeping the emission in the blue. This requires using weak donors and acceptors, so the CT emission is not strongly shifted to lower energies. Red TADF emitters are particularly limited, because the excited states are strongly affected by non-radiative decay, in agreement with the energy gap law. This decay actively competes with the rISC mechanism and quenches the excited state population, leading to lower quantum yields. Further molecular design is thus necessary to achieve blue and red emitters with small 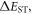 and strongly minimising non-radiative decay.
and strongly minimising non-radiative decay.
3. Fundamental understanding of the TADF mechanism
The complex photophysics involved on the TADF mechanism is best perceived when the luminescence from these emitters is followed using time-resolved methods. Following excitation with a fast laser pulse, the luminescence in TADF molecules shows a fast component, identified as prompt fluorescence (PF), due to the radiative decay of singlet excited states that were directly formed by excitation, according with the scheme in figure 1. This fast decay, usually occurring within a few nanoseconds, is followed by an emission tail due to the presence of DF. This occurs as a result of thermally activated reverse intersystem crossing from 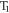 to
to 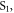 and is usually identified as TADF [18, 22, 38, 39].
and is usually identified as TADF [18, 22, 38, 39].
The fluorescence yield of TADF emitters 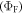 reflect the energy level diagram shown in figure 1. The equilibrium between singlet and triplet excited states due to intersystem crossing
reflect the energy level diagram shown in figure 1. The equilibrium between singlet and triplet excited states due to intersystem crossing 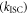 and reverse intersystem crossing
and reverse intersystem crossing 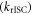 is a key parameter in the observation of TADF. Strong TADF is observed in molecules with relatively strong yield of triplet formation,
is a key parameter in the observation of TADF. Strong TADF is observed in molecules with relatively strong yield of triplet formation, 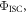 and where the yield of singlet states formed by reverse intersystem crossing is very high,
and where the yield of singlet states formed by reverse intersystem crossing is very high, 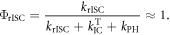 These conditions are met in compounds where the pathways for vibrational decay affecting the triplet excited state are suppressed, i.e.
These conditions are met in compounds where the pathways for vibrational decay affecting the triplet excited state are suppressed, i.e. 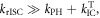 and the energy gap between the singlet and triplet states is small, usually less than 0.1 eV.
and the energy gap between the singlet and triplet states is small, usually less than 0.1 eV.
The total emission of a TADF emitter 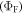 is described by equation (6), accounting for the recycling of singlet and triplet states.
is described by equation (6), accounting for the recycling of singlet and triplet states.
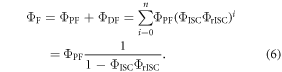
Equation (6) is of fundamental importance because it shows that a reverse intersystem crossing yield 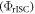 close to 100% is readily obtained, if the ratio between the DF and PF
close to 100% is readily obtained, if the ratio between the DF and PF 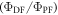 is around or above four [23]. This situation is obtained in most TADF emitters with a singlet–triplet energy splitting that is less than 150 meV, or even slightly above this. For
is around or above four [23]. This situation is obtained in most TADF emitters with a singlet–triplet energy splitting that is less than 150 meV, or even slightly above this. For 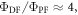 the product
the product 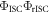 is already around 0.8, and as the triplet yield is limited by the fluorescence yield, as
is already around 0.8, and as the triplet yield is limited by the fluorescence yield, as 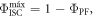
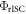 has to be for sure close to 1. The photophysical characterisation of TADF systems with
has to be for sure close to 1. The photophysical characterisation of TADF systems with 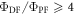 is thus very simplified, and in such cases, the triplet yield can be determined directly from equation (7), with very high accuracy.
is thus very simplified, and in such cases, the triplet yield can be determined directly from equation (7), with very high accuracy.
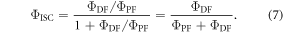
Following determination of 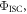 all the other relevant photophysical parameters are determined from relatively simple measurements, which are described in the following sections.
all the other relevant photophysical parameters are determined from relatively simple measurements, which are described in the following sections.
3.1. The PF yield,  and lifetime
and lifetime
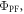
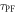
The PF yield, 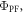 and lifetime,
and lifetime, 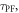 are determined in non-degassed solutions or solid films, and are both important parameters for the photophysical characterisation of TADF molecules. Knowing
are determined in non-degassed solutions or solid films, and are both important parameters for the photophysical characterisation of TADF molecules. Knowing 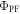 and
and 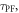 allows determination of the radiative rate constant
allows determination of the radiative rate constant 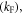 affecting the singlet excited state, from equation (8).
affecting the singlet excited state, from equation (8).
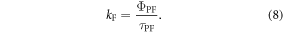
Efficient TADF emitters often have low fluorescence yield in the presence of oxygen. However, 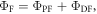 increases significantly upon degassing the sample, due to the DF contribution.
increases significantly upon degassing the sample, due to the DF contribution.
3.2. The delayed-to-PF ratio,

In strong TADF emitters, the triplet yield is determined directly from equation (7). Therefore, determining the 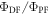 fluorescence ratio is of fundamental importance. There are two ways to determine
fluorescence ratio is of fundamental importance. There are two ways to determine 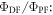 a first method collects the prompt and DF components, using degassed samples. Then in single time-resolved fluorescence decay, see figure 7(c) the prompt and delayed components are measured. In this case, the fluorescence decay is usually well fitted by the sum of two exponentials, one describing the PF and DF decays, see equation (9) [38].
a first method collects the prompt and DF components, using degassed samples. Then in single time-resolved fluorescence decay, see figure 7(c) the prompt and delayed components are measured. In this case, the fluorescence decay is usually well fitted by the sum of two exponentials, one describing the PF and DF decays, see equation (9) [38].
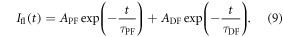
The 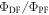 ratio is then easily determined from the integral of the PF and DF components, according with equation (10).
ratio is then easily determined from the integral of the PF and DF components, according with equation (10).
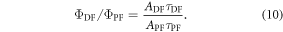
Figure 7(c) shows the fluorescence decay of DPTZ-DBTO2 in methylcyclohexane. DPTZ-DBTO2 is an efficient TADF emitter formed by two phenothiazine moieties, as the electron donor units, and dibenzothiophene-S, S-dioxide, as the acceptor, see figure 7(a). The contributions of prompt and DF decays are clearly visible in figure 7(c). Using the exponential amplitudes and decay times, 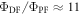 is determined from equation (10). Such high value for the DF/PF ratio clearly indicates that the triplet harvesting efficiency in DPTZ-DBTO2 is close to 100% [23].
is determined from equation (10). Such high value for the DF/PF ratio clearly indicates that the triplet harvesting efficiency in DPTZ-DBTO2 is close to 100% [23].

Figure 7. (a) Molecular structure of DPTZ-DBTO2; (b) steady-state fluorescence spectra of DPTZ-DBTO2, in MCH with and without oxygen; (c) fluorescence decay of DPTZ-DBTO2 in MCH at RT. Reproduced with permission from [23], John Wiley & Sons. CC BY 3.0.
Download figure:
Standard image High-resolution imageA second approach to determine 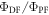 uses steady-state data and may be even more accurate than the first. Since the triplet excited state is strongly quenched by oxygen, the DF is practically fully suppressed in non-degassed solutions, or films, depending on the oxygen permeability of the host. Therefore, the integral of the steady-state fluorescence spectrum obtained in air-equilibrated conditions is proportional to
uses steady-state data and may be even more accurate than the first. Since the triplet excited state is strongly quenched by oxygen, the DF is practically fully suppressed in non-degassed solutions, or films, depending on the oxygen permeability of the host. Therefore, the integral of the steady-state fluorescence spectrum obtained in air-equilibrated conditions is proportional to 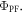 However, in degassed conditions, both the PF and DF contribute to the total emission. Therefore, the integral of the emission spectrum obtained in degassed conditions is proportional to the sum
However, in degassed conditions, both the PF and DF contribute to the total emission. Therefore, the integral of the emission spectrum obtained in degassed conditions is proportional to the sum 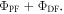 Since both PF and DF come from the same excited state and have the same spectrum, the proportionality constants affecting
Since both PF and DF come from the same excited state and have the same spectrum, the proportionality constants affecting 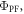 and
and 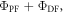 are exactly the same. The ratio of the integrated fluorescence spectra collected in degassed and non-degassed conditions, therefore, gives
are exactly the same. The ratio of the integrated fluorescence spectra collected in degassed and non-degassed conditions, therefore, gives 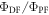 according with equation (11). For DPTZ-DBTO2, the data obtained in figure 7(b), gives
according with equation (11). For DPTZ-DBTO2, the data obtained in figure 7(b), gives 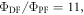 in excellent agreement with the previous determination from time-resolved data [23].
in excellent agreement with the previous determination from time-resolved data [23].
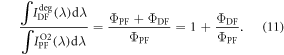
Following the determination of  the triplet formation yield is determined using equation (7). In the case of DPTZ-DBTO2,
the triplet formation yield is determined using equation (7). In the case of DPTZ-DBTO2, 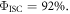 Using the fluorescence yield, and equations (8), (12) and (13), the intersystem crossing rate,
Using the fluorescence yield, and equations (8), (12) and (13), the intersystem crossing rate, 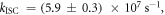 the radiative rate constant,
the radiative rate constant, 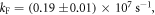 and the IC rate,
and the IC rate, 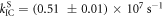 are all easily determined [23].
are all easily determined [23].
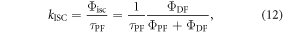
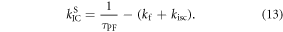
3.3. The rate constant for reverse intersystem crossing
Determining the rate of reverse intersystem crossing, 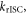 is obviously of extreme importance to the photophysical characterisation of TADF molecules. Fitting the fluorescence decay of a TADF emitter with equation (9), gives
is obviously of extreme importance to the photophysical characterisation of TADF molecules. Fitting the fluorescence decay of a TADF emitter with equation (9), gives 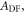
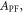
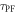 and
and 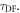 As
As 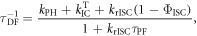 and since
and since 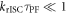 is always verified, the rate of reverse intersystem crossing
is always verified, the rate of reverse intersystem crossing 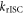 is determined according equations (14) or (15) [38].
is determined according equations (14) or (15) [38].
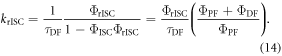
In the case of 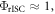 equation (14) simplifies to equation (15).
equation (14) simplifies to equation (15).
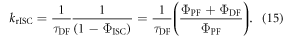
In the case of DPTZ-PTZO2, the rISC constant is determined as 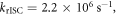 according with equation (15) [23].
according with equation (15) [23].
3.4. The temperature dependence of TADF
By measuring the fluorescence decay as a function of temperature and using equation (15), the temperature dependence of the reverse intersystem crossing rate is obtained. From here, and using equation (1), the energy barrier associated with the reverse intersystem crossing mechanism is determined from an Arrhenius type plot of 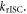 The energy barrier associated with TADF is often similar to the singlet–triplet energy gap, which is simply determined using the singlet and triplet energies obtained from fluorescence and phosphorescence spectra, when possible.
The energy barrier associated with TADF is often similar to the singlet–triplet energy gap, which is simply determined using the singlet and triplet energies obtained from fluorescence and phosphorescence spectra, when possible.
The temperature dependence of the DF is also fundamental to prove that the DF is due to a TADF mechanism. In the case of TADF the integral of the DF will increase when temperature increases, and this will be observed in time resolved data either in the form of decays or integrated DF. Figure 8, shows the temperature dependence of the fluorescence decay of PTZ-DBTO2, a phenothiazine-dibenzothiophene-S, S-dioxide, a D–A TADF emitter, in MCH. While the PF decay is unaffected by temperature, the DF shows a strong temperature variation, consistent with a thermally activated mechanism being responsible for the delayed emission [25].
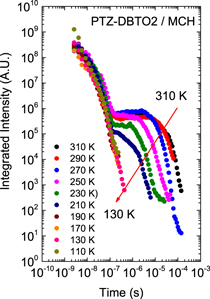
Figure 8. Fluorescence decay of PTZ-DBTO2 in MCH, as a function of temperature. Reproduced with permission from [25], John Wiley & Sons. © 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageMore complex approaches have to be used to determine 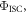 when the
when the 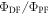 ratio is not large, e.g. below 3. In this case
ratio is not large, e.g. below 3. In this case 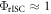 cannot be assumed, and fitting methods need to be applied in order to evaluate
cannot be assumed, and fitting methods need to be applied in order to evaluate 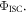 However, following the determination of
However, following the determination of 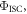 all the other relevant photophysical parameters are determined in the usual way. Methods to determine
all the other relevant photophysical parameters are determined in the usual way. Methods to determine 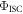 have been described in detail by Berberan-Santos and coworkers [38, 40, 41]. The most relevant expression is given in equation (16), where
have been described in detail by Berberan-Santos and coworkers [38, 40, 41]. The most relevant expression is given in equation (16), where 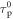 is the phosphorescence lifetime determined in the temperature range where rISC is not operative.
is the phosphorescence lifetime determined in the temperature range where rISC is not operative.
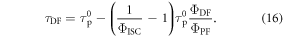
Using equation (16), 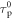 and
and 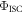 are determined from a linear plot of
are determined from a linear plot of 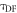 versus
versus 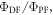 obtained at different temperatures [40]. The rate of reverse intersystem crossing is then obtained according with equation (17), which is equation (14) written in slightly different way.
obtained at different temperatures [40]. The rate of reverse intersystem crossing is then obtained according with equation (17), which is equation (14) written in slightly different way.

3.5. The dependence of TADF with excitation dose
Together with the temperature dependence, the variation of the DF integrated intensity with excitation dose is also an important criterion to establish the intramolecular origin of the DF, and it is critical to distinguish between the TADF mechanism and the DF that is observed as a result of TTA [39]. In the case of TTA, the DF intensity varies with excitation dose due to the competition between the rate for the monomolecular decay of triplet states, 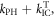 and the rate for collisional quenching of triplets,
and the rate for collisional quenching of triplets, 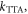 which is controlled by diffusion. When
which is controlled by diffusion. When 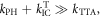 i.e. the triplets deactivate more quickly than they annihilate, the DF due to TTA shows a quadratic dependence on the excitation dose. However, when TTA dominates, usually at higher triplet concentration, this dependence turns to a linear regime [39, 42, 43]. In contrast, in pure TADF emitters, the mechanism that originates DF is entirely intramolecular; therefore, the TADF intensity varies linearly with excitation dose in the entire regime. This type of dependence is observed in figure 9 for DPTZ-DBTO2 in MCH [22, 23, 25, 39].
i.e. the triplets deactivate more quickly than they annihilate, the DF due to TTA shows a quadratic dependence on the excitation dose. However, when TTA dominates, usually at higher triplet concentration, this dependence turns to a linear regime [39, 42, 43]. In contrast, in pure TADF emitters, the mechanism that originates DF is entirely intramolecular; therefore, the TADF intensity varies linearly with excitation dose in the entire regime. This type of dependence is observed in figure 9 for DPTZ-DBTO2 in MCH [22, 23, 25, 39].
Figure 9. (a) Power dependence of the DPTZ-DBTO2 delayed fluorescence in MCH solution. (b) Linear fit of the DF intensity as a function of excitation power. The linear fit is obtained with strictly linear variation, e.g. gradient 1. Reproduced with permission from [23], John Wiley & Sons. CC BY 3.0.
Download figure:
Standard image High-resolution imageThere are situations, however, where a mixture of TADF and TTA have been observed [44]. This is revealed by power dependences of the emission integral that show slopes between 1 and 2. This situation occurs mainly in compounds where the rISC rate constant is not fast enough to completely deplete the triplet population before they annihilate trough triplet–triplet collisions (TTA), and is usually due to poor confinement of the triplet state by the host.
4. TADF molecules in solid Hosts
The application of TADF emitters has so far focused upon the field of OLEDs. This requires TADF emitters to be dispersed in solid hosts, which have strong impact on the photophysics of these compounds [45]. Therefore, the design and optimisation of TADF based OLEDs requires the photophysical characterisation of the TADF molecule in the host used in the device. In this section, we review briefly the fundamental requirements that a host material should fulfil, and then discuss host–guest interactions, which affect the photophysics of TADF molecules and have impact on device performance.
The first requirement when choosing a host material is its triplet energy. This should be higher than the energy of the triplet state of the emitter, so as to avoid unwanted quenching of the emitters triplet states by the host, and therefore suppression of the delayed emission. The situation is similar to the existing requirements when selecting hosts for OLEDs based on heavy metal complexes. Some of the most frequently used hosts are mCP, CBP, DPEPO, mCBP, TPBi, TCTA, 1, 5-DCN, and TAPC, see figure 10 for molecular structures [10, 46–54].

Figure 10. Molecular structures of organic host materials for TADF OLEDs.
Download figure:
Standard image High-resolution imageIn the case of TADF molecules, because their emission is broader, the energy difference between the triplet energy of the host and TADF molecule will also have to be larger [53, 54]. The objective is to achieve complete confinement in the emitter of singlet and triplet excitons created by charge recombination, thus avoid quenching of triplet states due to triplet–triplet energy transfer to the host and TTA. This requirement imposes a strong limitation on the choice of host materials for most blue emitters, for which hosts have to have triplet energies around or above 3.0 eV. A second requirement for a good host is the possibility to achieve host–guest energy transfer, which will allow excitons formed in the host material to be transferred to the TADF emitter. To fulfil this requirement the HOMO and LUMO frontier orbitals of the host should straddle those of the TADF emitter, and the host emission needs to show significant overlap with the absorption of the TADF emitter. Finally, host materials are required to have bipolar charge transport properties in order to facilitate exciton formation in the emitting layer and avoid exciton quenching at the electrodes.
One more important aspect when choosing a host for TADF emitters is related with the strong CT character of the excited state in TADF molecules, which induces strong local interactions between the excited state dipole moment of the emitter and the dipole moment of the host. In solution, such interactions lead to the observation of the usual bathochromic spectral shift observed in CT molecules with increasing solvent polarity. However, a similar effect also happens in the solid state when the emitters are doped in hosts of relatively high polarity, because these materials also stabilise the CT excited state due to local dipolar interactions, and thus tend to shift the emission peak to longer wavelengths, while in nonpolar host molecules the emission peak is not strongly affected.
There are, however, even more subtle effects in the solid state that affect the emission of TADF molecules, which require further investigation. In polar hosts the emission of TADF molecules often shows an unusual temporal spectral-shift, which is very intriguing, see figure 11 [23, 45]. In hosts with large dipole moment, the TADF emission occurring in the μs time range, shows an initial red-shift, which is followed by a pronounced blue-shift at longer times. This phenomenon has been tentatively interpreted as the result of local interactions between the dipole moment of the host and the excited state dipole moment of the TADF molecule [45]. The mechanism that has been proposed in [45] justifies the observation of this temporal red-to-blue spectral-shift, due to the fact that following the rapid decay of the prompt-fluorescence, the interaction with the host leads to an initial red-shift of the singlet and triplet CT states, 1CT and 3CT respectively. This shift occurs towards the low energy local triplet excited state of ππ* character, 3LE, which is not significantly affected by the host dipoles. Reverse internal conversion (rIC) from the 3LE to 3CT, followed by rISC from 3CT to 1CT then leads to TADF emission in the red. As the population of the CT states decays, the interaction between excited TADF molecules with the host also decreases, leading to an increasing randomisation of the dipole moment of the host molecules. The CT states in the TADF emitter then start to move away from the 3LE state, towards higher energies, and the emission starts to occur from increasing higher energies, leading to the observation of a spectral blue-shift in the DF spectrum. However, this explanation still requires further investigation. An alternative explanation for the temporal spectral shift observed in the TADF decay may be the presence of conformational heterogeneity in the TADF molecule [55], i.e. the presence of a distribution of CT states, where the most relaxed molecular geometries, with D–A relative orientation near orthogonality, emit at lower energies. It is known that the singlet–triplet energy splitting is strongly affected by the dihedral angle between the electron donor and acceptor units [23]. Therefore, the presence of slightly different conformers may lead to a distribution of 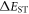 values, and thus to a distribution of intersystem crossing rates,
values, and thus to a distribution of intersystem crossing rates, 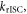 according to equation (1). In this scenario, the conformers with the smallest
according to equation (1). In this scenario, the conformers with the smallest 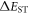 will emit at longer wavelengths, and have faster
will emit at longer wavelengths, and have faster 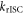 leading to a faster decay on the population of the triplet excited state, thus to fast decaying TADF. However, the less relaxed CT states will emit towards the blue, and will have relatively larger
leading to a faster decay on the population of the triplet excited state, thus to fast decaying TADF. However, the less relaxed CT states will emit towards the blue, and will have relatively larger 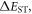 thus slower decaying TADF. The overall effect on the TADF emission would thus appear as a temporal spectral shift from red to blue. This also explains why such temporal shifts are observed in solid films, but not in solution [23].
thus slower decaying TADF. The overall effect on the TADF emission would thus appear as a temporal spectral shift from red to blue. This also explains why such temporal shifts are observed in solid films, but not in solution [23].

Figure 11. (a) Luminescence decay of DPTZ-DBTO2 in CBP as function of temperature. (b) Spectral shift, and luminescence decay of DPTZ-DBTO2 in CBP at RT. Reproduced with permission from [23], John Wiley & Sons. CC BY 3.0.
Download figure:
Standard image High-resolution imageDynamic effects observed in the TADF decay strongly contribute to the increasing complexity observed on the fluorescence decays of TADF molecules in solid films, and while in solution the fluorescence decays are usually well described by a sum of two exponentials, according with equation (9), in solid samples the fluorescence of TADF molecules often show complex decays that cannot be fitted by sums of two or even three exponentials. The formation of aggregates, and the presence of room-temperature phosphorescence, can also contribute to increasing complex luminescence decays from TADF molecules. The determination of photophysical parameters in solid samples is, therefore, much more difficult than in solution, and requires exhaustive studies.
In addition with the temporal spectral shifts described above, the formation of exciplex states between the TADF and the host molecules, also affect the emission from TADF emitters in the solid state. The formation of exciplex states involves electron transfer occurring between the TADF emitter and the host molecules, or between the D or A units and the host molecules. Since TADF molecules contain strong electron donors and acceptors, exciplex formation is facilitated [44, 56]. Unfortunately exciplex formation can lead to a decrease on the non-radiative decay rate, and certainly results on the observation of red-shifted emission, compared with those of isolated molecules [57]. The polarity of the host and the possibility of exciplex formation have thus to be carefully considered when selecting the components for TADF OLEDs.
5. Theory of TADF
Equation (1) casts the rate of reverse intersystem crossing (krISC) in terms of a simple Arrhenius equation, commonly adopted in the absence of a detailed knowledge of the vibrational density of states. This shows that a small energy gap between the lowest singlet and triplet states is most critical for determining the rate. However, krISC is more rigorously expressed using first order perturbation theory, namely Fermi's Golden rule. Within the Condon approximation this is expressed:
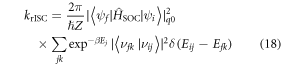
where 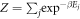 is a canonical partition function for vibrational motion in the initial electronic state, β is the inverse temperature and Ej is the energy of the vibrational level in the initial electronic state. The transition rate between two states of different spin multiplicity is mediated by the SOC matrix element [58]. This approach is effective for describing the excited state kinetics, provided that the motion of the electrons and the nuclei can be effectively decoupled and the coupling between the two states is small compared to their energy difference. If the latter is not true the validity of this approach, i.e. perturbation theory, becomes questionable, although it has still been used with some success [59]. Breakdown of the former (the Condon approximation) means the effect of vibrational motion on the electronic coupling elements has to be explicitly taken into account.
is a canonical partition function for vibrational motion in the initial electronic state, β is the inverse temperature and Ej is the energy of the vibrational level in the initial electronic state. The transition rate between two states of different spin multiplicity is mediated by the SOC matrix element [58]. This approach is effective for describing the excited state kinetics, provided that the motion of the electrons and the nuclei can be effectively decoupled and the coupling between the two states is small compared to their energy difference. If the latter is not true the validity of this approach, i.e. perturbation theory, becomes questionable, although it has still been used with some success [59]. Breakdown of the former (the Condon approximation) means the effect of vibrational motion on the electronic coupling elements has to be explicitly taken into account.
Due to the importance of CT states, it was initially assumed that the states involved in the TADF mechanism were the singlet and triplet CT, 1CT and 3CT, respectively. However, Lim and co-workers showed that SOC between these intramolecular CT states is formally zero [60]. This is because the SOC operator carries both the spin magnetic quantum number of the electron and its spatial angular momentum quantum number. Consequently, coupling between singlet and triplet states with the same spatial orbital occupation are formally zero. Therefore other electronic states should be involved in the rISC mechanism that supports TADF. This issue for describing the mechanism of rISC appeared to have been solved, as it was recently demonstrated that in D–A and D–A–D molecular TADF systems, two of the excited states involved in the rISC step could be independently tuned [23, 25, 61, 62]. These states must therefore be of different character and a 1CT and local excitonic triplet (3LE) pair appeared most likely. Indeed, SOC between these two sets of states will be allowed. However, Chen et al [63], and Marian [64], both used Fermi's Golden rule to calculate the 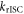 in a number of organic donor–acceptor CT complexes. In both cases they found that the rISC rates were unable to explain the high rates of rISC (106–8 s−1) reported experimentally.
in a number of organic donor–acceptor CT complexes. In both cases they found that the rISC rates were unable to explain the high rates of rISC (106–8 s−1) reported experimentally.
An alternative mechanism was recently proposed by Ogiwara et al [65], who used electron paramagnetic resonance (EPR) spectroscopy to probe the population of the 3LE and 3CT states. By fitting the transient experimental signals, they reported that complexes showing the largest rISC exhibited an EPR signal consistent with a mixture of both the 3LE and 3CT states. The authors used this to propose that efficient rISC not only includes the SOC pathway (3LE → 1CT), but also a hyperfine coupling (HFC) induced ISC pathway (3CT → 1CT). This conclusion is consistent with the proposal of Adachi and coworkers [66], who rationalised efficient rISC from 3LE → 1CT, as proceeding via rIC from 3LE to the 3CT and then using HFC induced ISC to cross to the 1CT. However, crucially the HFC constants are very small, usually in the range of 10−4 meV, and it therefore appears highly unlikely that such coupling accounts for efficient rISC.
Further insight into the mechanism for efficient rISC was recently presented by Ward et al [61], who found a huge difference in the rate of rISC in a range of D–A and D–A–D complexes exhibiting relatively similar ΔES1–T1. Indeed, in complexes, which included bulky substituents, designed to provide steric hindrance around the D–A dihedral angle, the TADF pathway was switched off totally and the complexes exhibited phosphorescent at room temperature, in the solid state. This indicates a dynamical aspect to the TADF mechanism, in that it appears to be promoted by molecular vibrations. This is consistent with the recent simulations of Marian, who proposed that efficient rISC is mediated by mixing the 1CT singlet state with an energetically close-lying 3LE state along a carbonyl stretching mode, which promotes spin-vibronic mixing between multiple excited states as being crucial to efficient rISC [64].
To address this dynamical aspect, which appears to be present in rISC, we have recently performed quantum dynamics simulations upon of the rISC process [67]. This is achieved using a D–A molecule composed of a phenothiazine donor and a dibenzothiophene-S, S-dioxide acceptor (PTZDBTO2), shown in figure 12(a) [67]. As also shown in [23, 25]. this dimer analogue and the D–A–D trimer both give identical photophysics and excellent OLED performance >19% EQE. Our model Hamiltonian, shown schematically in figure 12(b), and used during the excited state quantum dynamics simulations is composed of the most important electronic and vibrational degrees of freedom. A full description is provided in [67].

Figure 12. (a) Schematic representation of the donor–acceptor (D–A) molecule composed of a phenothiazine donor and a dibenzothiophene S, S dioxide acceptor (PTZ-DBTO2). (b) Schematic of the model Hamiltonian for PTZ-DBTO2 that incorporates the three most important excited states and the coupling between them. Reprinted with permission from [67]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageUsing quantum dynamics within the density matrix formulism to account for temperature, figure 13 shows the effect of the main Hamiltonian parameters, namely the vibronic coupling, SOC, HFC and energy gaps, on the calculated rate of rISC. In each case the simulations were initiated in the lowest triplet state, the 3LE state, and the rate determined as the change in population of the 1CT state during the 0.5 ns of the simulations.
Figure 13. (a) The relative populations of the 1CT state associated with reserve intersystem crossing after initially populating the 3LE state. Black: full model Hamiltonian containing vibronic, spin–orbit and hyperfine coupling, green: no hyperfine coupling, blue: no vibronic coupling, red: vibronic coupling increased by 10%, cyan: energy gap between 3LE and 3CT halved. (b) Population kinetics of the 3CT state during the rISC dynamics after initial population of the 3LE state. Figure reprinted with permission from [67]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageAs shown in figure 8, the rate of rISC is most sensitive to the vibronic coupling and SOC, with the hyperfine interaction having very little effect. The fact that vibronic coupling between the two triplet states (3LE and 3CT) has such a profound effect on the rate of rISC is somewhat surprising and means that efficient rISC cannot be described within the limits of first order perturbation theory. To explain this observation, i.e. the role of two coupling components, one must go beyond the first order effect of FGR and use a more general description including second order perturbation theory.
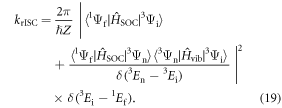
In the present case, a direct second-order coupling would require population transfer between the two CT states, via the HFI, that is, an initial 3LE state populates the 3CT via vibronic coupling, which decays into the 1CT, via the HFI. However, as already demonstrated, it plays an insignificant role. Consequently, in this case the rISC mechanism must occur via a 2-step mechanism.
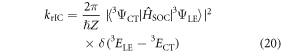
and
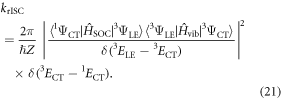
Firstly, the large vibronic coupling between the 3LE and 3CT states promotes, on a timescale much faster than the rISC, an equilibrium (or mixing) between the two states (figure 13(b)). This is called rIC. Obviously, the position of this equilibrium and therefore the amount of mixing between the two states depends both on the size of the vibronic coupling and the energy gap. Subsequently, the second-order term, couples the 3CT and the 1CT states, using the 3LE state as an intermediate. This latter second-order term is very efficient because of the good vibrational overlap between the almost degenerate initial and final states, 3CT and 1CT, respectively. Therefore, the two coupling terms driving this dynamics are the SOC and the vibronic coupling elements. This explains recent experimental results which demonstrated that steric hindrance of D–A dihedral angle switches the main pathway TADF to phosphorescence [61]. This steric hindrance is equivalent to removing the vibronic coupling term, which is shown herein to be strongest along modes exhibiting a distortion of the D–A dihedral angle.
The importance of the 3CT state as highlighted in figure 13(b) is also consistent with the time-resolved EPR study of Ogiwara et al [65], who, by fitting the transient experimental signals, reported that complexes showing the largest rISC exhibited an EPR signal consistent with a mixture of both the 3LE and 3CT states. The authors concluded that efficient rISC occurs simultaneously via the spin–orbit and hyperfine pathways. However, the EPR actually only probes the population of the 3LE and 3CT states and not the mechanism for its connection into the 1CT state. However, as shown in our present work, equilibrium between the 3LE and 3CT exists and depends on the strength of vibronic coupling. Therefore strong vibronic coupling increases the population of the 3CT state and provides efficient rISC, as observed in [65], via the second order mechanism we demonstrated in [67].
This 3 state model for efficient rISC [67], means that there are two important energy gaps not just one. As the states involved are of different character, it opens the opportunity to study the effect of the host environment on the dynamics of TADF. This was recently achieved using a combination of photoinduced absorption and quantum dynamics [68], confirming the spin-vibronic mechanism proposed in [67]. By exploiting the temperature dependent polarity of the PEO host (figure 14(a)), the charge-transfer states were brought into energetic resonance with the 3LE state, and a significant increase in TADF was observed. These results allowed three distinct regimes of TADF to be categorised, TADF I (CT > LE), TADF II (CT = LE), and TADF III (CT < LE). These observations were simulated using quantum dynamics (figure 14(b)) and the model Hamiltonian outlined above. This correctly predicts the resonant behaviour of TADF as a function of temperature, confirming the proposed model for efficient TADF.

Figure 14. (a) The temperature dependence of the intensity (black line) and CT onset energy (purple circles). The change in CT onset energy plateaus below the Tg, representative of the PEO film becoming rigid. The black dashed line represents the energy of the 3LE at 2.58 eV, with the peak in intensity apparent as the CT energy crosses resonance. The error bars are indicative of the error in the fit of 0.01 eV for all points, the same error is expected on the 3LE energy. (b) Relative rate of reverse intersystem crossing as a function of temperature for the D–A complex PTZ-DPTO2. The rates were extracted from the population of the 1CT state at 0.5 ns of dynamics simulations initiated from the lowest triplet state. Figure reproduced from [68]. CC BY 3.0.
Download figure:
Standard image High-resolution image6. TADF in macromolecules and intermolecular exciplex systems
6.1. TADF in polymers and dendrimers
TADF based OLEDs are usually fabricated by vacuum deposition. However, despite solution-processing methods, such as spin-coating, being more suitable for deposition over large areas and also cheaper, making solution processed OLEDs very attractive, the performance of these devices are still inferior when compared to vacuum deposition devices [69]. Unfortunately, small molecules are often not appropriate for film deposition directly from solution, and tend to form films of poor quality; there are also difficulties to fabricate multilayer devices, which also contribute to generally weaker performance of these devices. Therefore, the advantages offered by solution processing methods, such as spin-coating, spray-on and ink-jet printing, allowing rapid deposition over large area at room temperature, and on flexible substrates [70], are still in general unavailable for TADF devices. Despite some significant progress during recent years [69, 71–73], further work is still required to create efficient ways to promote TADF in large molecules, such as polymers, and dendrimers which are more suitable for solution processed devices. However, the observation of TADF in oligomers, polymers and dendrimers is challenging, because IC is more difficult to be minimised in macromolecules, and also because TTA might be more efficient in large molecules. Both processes rapidly quench the triplet population and, therefore, compete with reverse intersystem crossing. Hosts can still be used in the case of large molecules to help confining the triplet states in the TADF emitter, thus avoiding the effect of TTA. Small TADF molecules are dispersed in hosts with high triplet energy levels exactly for this reason. Unfortunately, intramolecular TTA can be operative in macromolecules, and thus cannot be avoided by simple host confinement. This method may therefore not be effective in the case of large molecules.
As already mentioned, hosts frequently influence the dynamics of the excited state, affecting both photoluminescence (PL) and electroluminescence (EL) properties of TADF emitters, and causing large variations in the emission yield and lifetime due to the formation of exciplexes [44], and also due to the presence of heterogeneities in the host–guest molecular geometries [45]. Strong variations on the TADF contribution are often observed in different hosts due to the effect of the dielectric medium, and host–guest systems are susceptible to suffer phase separation due to the differences between the molecular structures of the constituent molecules, which may result in unstable luminescence. Achieving efficient TADF in neat films, i.e. without using host–guest systems, is thus of major interest and has only very recently been reported [25, 74–77].
TADF in non-doped films, made of dendrimers and fabricated from solution have been recently reported by Albrecht et al [77]. The authors reported three different dendritic structures with s-triazine core and carbazole dendrons, GnTAZ, with n = 2, 3, 4, where n is the generation number, see figure 15. The three generations were all highly soluble in organic solvents, in toluene and THF for example, and showing fluorescence quantum yields close to 100% in nitrogen saturated solution. In the presence of oxygen, the fluorescence quantum yield decreased significantly in all cases, but the difference between the fluorescence quantum yield measured in nitrogen and oxygen saturated solutions increased with the generation number. For n = 2, the ratio between DF and PF, 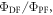 is around 0.23, for n = 3,
is around 0.23, for n = 3, 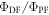 is 0.69, and for n = 4,
is 0.69, and for n = 4, 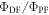 is around 13, indicating that the TADF contribution is larger in the higher-generation dendrimers. In neat film however, the fluorescence quantum yield in nitrogen atmosphere decreased with increasing generation number, from 52% for n = 2 to just 8.5% for n = 4. This effect is attributed to emission quenching due to intermolecular interactions, such as excimer formation, which are more prevalent in the higher-generation dendrimers [77]. OLED devices incorporating these dendrimers as spin-coated emitting layers gave EQE of up to 3.4%. More recently, Yang et al [78], reported dendrimer based solution processed OLEDs with EQE around 10% at 1000 Cd m−2. Despite these promising results, optimised dendrimer structures are still necessary to improve device performances, maintaining strong fluorescence yield in solid pristine films, and thus improving device efficiency.
is around 13, indicating that the TADF contribution is larger in the higher-generation dendrimers. In neat film however, the fluorescence quantum yield in nitrogen atmosphere decreased with increasing generation number, from 52% for n = 2 to just 8.5% for n = 4. This effect is attributed to emission quenching due to intermolecular interactions, such as excimer formation, which are more prevalent in the higher-generation dendrimers [77]. OLED devices incorporating these dendrimers as spin-coated emitting layers gave EQE of up to 3.4%. More recently, Yang et al [78], reported dendrimer based solution processed OLEDs with EQE around 10% at 1000 Cd m−2. Despite these promising results, optimised dendrimer structures are still necessary to improve device performances, maintaining strong fluorescence yield in solid pristine films, and thus improving device efficiency.

Figure 15. Molecular structure of a GnTAZ, dendrimer with n = 3 [77].
Download figure:
Standard image High-resolution imagePolymeric structures showing efficient TADF in neat films, have also been reported recently [25, 74, 76, 79, 80]. Slightly different architectures were used, but all containing spacer groups with higher triplet level, in order to confine the triplet states in the TADF unit, either within the polymer backbone or with the TADF group used as a pendant, in order to limit TTA. Figure 16 shows the molecular structure and the fluorescence decay of a TADF polymer based on PTZ-DBTO2 as the TADF unit, used as a pendant group and confined by a large band-gap spacer [25].
Figure 16. Molecular structure and fluorescence decay of the PTZ-DBTO2 polymer neat film as a function of temperature. Reproduced from [25], John Wiley & Sons. © 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageThe time resolved fluorescence decay in the pristine film of the PTZ-DBTO2 polymer shows a clear delayed component, due to TADF, following the decay of the PF. Therefore, the rate of reverse intersystem crossing from the lower energy triplet state is clearly able to compete with the rate of TTA even in the neat film of the PTZ-DBTO2 polymer.
Despite the progress made in the synthesis of TADF large molecules, and while the photophysics clearly shows the presence of efficient triplet harvesting via the TADF mechanism, solution process devices made of polymers still show consistently low efficiencies, with EQE below 5%. Part of the problem is due to the method of fabrication of these devices rather than due to the material itself. Solution methods are unable to allow deposition of consecutive layers, which makes the device optimisation difficult. The development of TADF solution processing materials that will allow fabrication of two or three layer devices, exploring orthogonal solvents or other methods, are therefore, an area of high interest in the development of TADF materials for solution processing.
6.2. TADF in intermolecular exciplexes
From equation (5), it is possible to envisage an alternative way in which the exchange energy can be minimised. If considering that the two electrons in the excited state are separated by a large distance, then the term in the exchange operator goes to zero and J also becomes very small. This situation can be achieved when a CT occurs between two different molecules forming an intermolecular excited state complex, also known as an exciplex.
Exciplexes, are, therefore, intermolecular charge-transfer states formed under photo- or electrical excitation by the interaction of electron donor (D) and electron acceptor (A) molecules. Frederichs and Staerk [81], have confirmed experimentally the assertion made by Weller that thermally assisted ISC from a triplet to singlet states in the exciplex manifold can occur. They showed also that certain exciplexes have very small exchange energies (<0.1 eV), demonstrating clear E-type emission (TADF) from an exciplex in solution [82], and highlighting the importance of electronically coupling the exciplex emissive state to the D ground state to achieve high luminescence yields. These solution studies also demonstrated the role of the environment polarity on stabilising the degree of charge separation in the exciplex.
Exciplex excited states are formed from a linear combination of the possible excited states of the D–A system, i.e. CT ∣D+A−〉CT and locally excited states, ∣D*A〉Loc and ∣DA*〉Loc, see equation (22). However, the radical ion pair is usually only stabilised for highly polar environments, which tends to give poor luminescence yields because of the weak coupling between the excited and ground states. In the solid-state, the environment is usually only weakly or moderately polar, and less stabilisation is thus achieved, giving rise to more excitonic-like ∣DA*〉Loc exciplex, rather than the full ion pair ∣D+A−〉CT. This has the benefit of enhancing both the ground state coupling and the luminescence yields. Hence, exciplex states in the solid state are good candidates for efficient triplet harvesting via TADF in OLEDs. The exciplex emission maximum is related to the ionisation potential of the donor (ID) and electron affinity of the acceptor (AA), stabilised by the electron–hole coulomb potential energy (EC), see equation (23) [83].
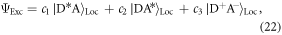
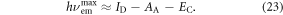
Importantly for OLEDs, direct comparison between exciplex and intramolecular CT systems seems obvious, however two different factors that control SOC need to be considered: (i) the overlap of the wavefunctions of the two electrons in the exciplex state and (ii) the electronic coupling between them, which falls off as 1/r3 [58, 84]. One way to view SOC is considering the spin interaction of one of the two electron's angular momentum in the magnetic field of the other orbiting electron, which quantum mechanically follows the exchange interaction between the two electrons and falls off very quickly with increasing electron separation, typically at 1.5 nm separation the SOC rate <100 s−1 [85]. Therefore, the exact orientation of the D and A molecules in an exciplex does not affect SOC rates, as it does in intramolecular D–A systems. This is because the exchange interaction is already very small, as the electron separation distance is easily larger than 1 nm. SOC in exciplexes is thus totally dominated by the spatial electron separation term, which for an intermolecular exciplex system is often in the range 2–3 nm, and can be controlled by the external electric field [86]. SOC in exciplexes, therefore, occurs mostly by hyperfine interaction between 1CT and 3CT, irrespective of relative orientation of D and A in an exciplex, as this effect is totally overwhelmed by the large electron separation.
There is literature dating back to the early days of OLED research discussing the pros, but mostly the cons, of exciplexes in the context of OLEDs [87]. This early work focused on exciplexes formed unintentionally at the interface between a transport layer and the emitting layer, usually seen only in electroluminescence (EL) and not in photoluminescence (PL). The first report of interfacial exciplex emission was in 1998 by Itano et al [88], and then in a blended exciplex device by Cocchi et al [89]. The devices in the latter work were inefficient as they incorporated the emitter molecules in polycarbonate matrix and the exciplex had low photoluminescence quantum yield ca 0.17. The authors clearly described exciplex evolution from a tightly bound ∣DA〉* exciplex to an ionic ∣D+A−〉* ion pair, and the effect of Coulomb relaxation which yields large red shifts, thereby explaining the previously observed 'electroplex'.
Kalinowski et al [90], and Palilis et al [91], were the first to report true D and A blend devices, using an exciplex system of high PLQY, ca 0.62, between a triarylamine hole transporter (the D unit) and a highly fluorescent (PLQY ca 0.85) silole-based emitter and electron transporter (the A unit). OLED devices with EQE of 3.4% were reported, which at the time was excellent. These results clearly show that it is possible to engineer exciplexes with strong ground state coupling and thus high luminescence efficiency. It is important to emphasise that, in principle, exciplexes can have vanishingly small exchange energies [92]. In the OLED context, this was first highlighted by Cocchi et al [89, 93], who discussed the possibilities of electrophosphorescence from exciplexes. However, their system (donor TPD : acceptor BCP in polycarbonate matrix) has a rather large singlet–triplet gap, ΔES–T ca 0.4 eV. The first report of an exciplex-based device giving E-type exciplex emission was by Goushi et al in 2012 [94]. The donor molecule was a triarylamine (m-MTDATA) and the acceptor a triarylborane derivative. OLEDs with EQE of 5.4% where realised from an exciplex system having a PLQY of only 0.26, indicating that far more than 25% singlets were being generated in the device. Subsequently Goushi reported a device giving up to 10% EQE, 47 lm W−1 for green emission [95].
A very important reason why exciplex systems are so interesting in OLEDs, is that they can be used in very simple device structures with very low working voltages, ca 2.5 V. This was first reported by Morteani et al [96], who demonstrated that electron and holes are directly injected into the exciplex HOMO and LUMO levels giving origin to a low drive voltage. This is a critical finding and is vitally important for high luminance efficacy lighting and good compatibility with CMOS backplanes in mobile devices. The high EQE and luminance power efficiency derive from efficient electron–hole capture directly at the exciplex. Therefore, there are no voltage drops associated with charge injection and transport through additional layers, and the usual necessity of forcing the electron and hole onto a single molecular emitter site is overcome. Thus, TADF exciplex devices have many potential advantages over phosphorescence based devices, notably a very simple device structure (two materials in three layers) and very high power efficiency. However, not all exciplex systems showing enhanced OLED performance, i.e. above 25% singlet generation, do so through TADF. As it was shown by Jankus et al for many exciplex systems one or both of the local triplet states, of the D and/or A, lies at lower energy with respect to the CT states. In this case, the delayed emission often arises as a result of TTA, not TADF. This indicates that the low lying triplet state acts as a quencher for CT emission, but that the triplet population is very high giving rise to efficient TTA [97].
A vanishing singlet triplet energy splitting, ∆EST, is in principle relatively easy to be engineered in exciplex systems, because the large spatial separation of electron and hole in these complexes ensures negligible exchange integral, according with equation (5). However, the number of available exciplex emitters showing a true TADF mechanism is still limited and initial strategies for designing good TADF exciplexes seem too simplistic. For example, the first 'design' rule for an exciplex to yield TADF, i.e. that the CT state of the exciplex should be positioned below the local triplets of the D and A molecules, derived from the above observation of Jankus, is very powerful but does not explain anything about the rISC mechanism and how this can be maximised. In order to expand on this, the photophysics of a series of exciplexes is discussed below, from which a more detailed and better understanding of TADF in exciplex systems is envisaged, and discussed in light of the D–A intramolecular CT systems already given above.
Figure 17(a) shows the chemical structure of four well known hole and electron OLED transport materials [98–101]; m-MTDATA, TPBi, TPD and OXD-7, with their normalised absorption (dash lines) and emission spectra (full lines) in figures 17(b)–(d). Three exciplex blends were made from these molecules: m-MTDATA:TPBi; TPD:TPBi; and TPD:OXD-7 [102]. The corresponding absorption and emission spectra of these exciplexes are also shown in figures 17(b)–(d). The films were excited at 337 or 355 nm, at room temperature.
In common with most exciplex systems, the absorption spectra of the blends show features that are nearly identical to the combination of the absorption of their pristine D and A units. This result shows that the formation of a new ground state transition does not occur in these blends. However, their emission spectra are broader and significantly red shifted when compared to both the D and A, confirming the formation of new excited state in the blend films, i.e. the exciplex formation.

Figure 17. (a) Chemical structure of m-MTDATA, TPBi, TPD and OXD-7, (b) normalised photoluminescence (PL) and absorption spectra of m-MTDATA:TPBi, (c) TPD:TPBi and (d) TPD:OXD-7, as well as their pristine acceptor and donor molecules. Reprinted with permission from [102]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageTo investigate the exciplex excited state, we again turn to time resolved emission. Figure 18 shows the temperature dependent decay curves of m-MTDATA:TPBi from the early prompt emission, collected with time delay (TD) of 1.1 ns, to the end of the DF emission (TD = 89 ms). The curves were obtained with 355 nm excitation. The decay curves show first a rapid decay, associated with the prompt emission, and later a long lived emission, associated to the DF. The DF can be analysed in two different time regions, A and B. Initially, region A shows a temperature dependence typical of the TADF mechanism, i.e. increased intensities at higher temperatures, consistent with a thermal activated mechanism. However, region B shows the opposite behaviour, the intensity is higher for low temperatures. This behaviour can be understood by analyzing the dependence of the rISC rate with temperature [39]. At high temperature, the rISC rate is maximised, according with equation (1). This leads to a short TADF lifetime, i.e. giving rise to a faster decay time of the delayed component. On the other hand, at low temperature, both the rISC and the IC rates that affect the triplet excited state are minimised and the triplet state will live longer, therefore, leading to long-lived emission decay, as observed. Thus, the decay lifetime increases with decreasing temperature, as shown in figure 18(a).
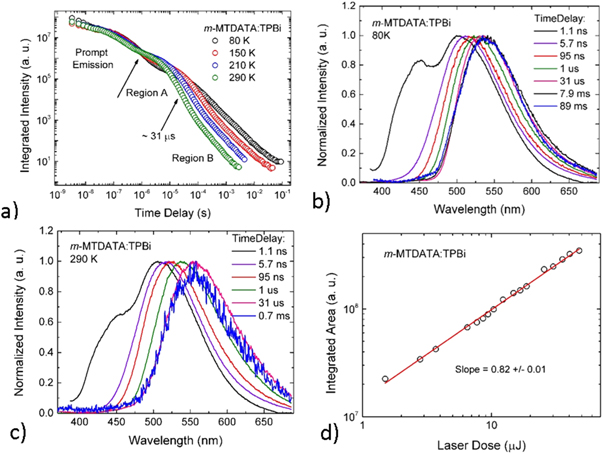
Figure 18. (a) Time resolved fluorescence decay curves at different temperatures. (b) Time resolved normalised emission spectra at 80 K (c) at 290 K (d) Integrated area as a function of the laser excitation (337 nm) of m-MTDATA:TPBi blend. Reprinted with permission from [102]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageFigure 18(b) shows the time-resolved emission spectra of the m-MTDATA:TPBi in the entire region of the study at 80 K. At earlier times, TD = 1.1 ns, a first peak at 425 nm and the 1CT state emission are observed. The first peak is associated to a vestige of the donor emission (1LED), m-MTDATA. As the time delay increases, this peak emission disappears, showing that the process of electron transfer from the donor to the acceptor molecules has finished over about 4.6 ns, giving a rate for the electron transfer process estimated at ca 2 × 108 s−1. During the prompt emission and in region A, a continuous red shift of the emission is observed, which is associated with the energetic relaxation of the CT state, or via dispersion of CT decay times from CT states of different energy, i.e. the lower the CT energy, the less coupled it is to the ground state and so the longer the radiative lifetime. Around TD = 31 μs, the 1CT state stabilises and the same emission is observed until TD = 89 ms. The same analyses is made at 290 K, the temperature at which TADF mechanism is maximized, in figure 18(c). The main difference from those spectra measured at 80 K is that the 1CT is slightly further red shifted at longer time delays. This shows that the 1CT state suffers more relaxation at higher temperatures, due to increased vibrational energy.
As can be seen in figures 18(b) and (c), no phosphorescence emission was detected from the m-MTDATA:TPBi blend. Most probably, all the excitons in the triplet states are efficiently converted back to 1CT due to the small energy splitting between the singlet and triplet states. The m-MTDATA:TPBi blend shows a 1CT energy level of (2.64 ± 0.02) eV, which was determined by the onset of the PL emission at 290 K. From the on-set of the phosphorescence of the pristine donor and acceptor molecules the triplet energies of the D and A units, 3LED, and 3LEA, were determined at (2.65 ± 0.02) eV and (2.66 ± 0.02) eV, respectively. This leads to very small values of ∆EST, (−0.01 ± 0.03) eV and (−0.02 ± 0.03) eV in both cases, which maximises the ISC/rISC processes in this blend. The negative values meaning that the 3LE is located above the 1CT [102].
Finally, in figure 18(d), the intensity of the DF emission in region A (TD = 2 μs, Ti = 5 μs) was measured as a function of the laser excitation dose to certify that the DF was due to a TADF mechanism and not to TTA. A linear gradient of 0.82 ± 0.02 was found (figure 18(d)), confirming the thermally activated mechanism as opposed to TTA. Generally, TADF complexes show a slope close to 1 at low and high excitation doses while TTA complexes show a slope close to 2 at low excitation doses turning to slope close to 1 at high excitation doses [39, 43].
Turning now to the TPD:TPBi blend. The decay curves from the early prompt emission (TD = 1.1 ns) to the end of the PH emission (TD = 89 ms), at different temperatures (excitation at 355 nm), are shown in figure 19(a). Again, we see an initial rapid decay, associated with the decay of the prompt emission, and a long lived emission, associated with the decay of DF. Also, a very long lived emission is observed at low temperatures, see figure 19(b), which is associated with the phosphorescence emission of the D unit, see discussion below. There is no strong temperature dependence from 80 to 210 K, but a slight increase of the intensity of the assigned DF region is observed at higher temperatures. However, it is clear that at low temperature, the emission lives longer time than at high temperature.

Figure 19. (a) Time resolved fluorescence decay curves at different temperatures. (b) Time resolved normalised emission spectra at 80 K (c) at 290 K (d) Integrated area as a function of the laser excitation (337 nm) of TPD:TPBi blend. Reprinted with permission from [102]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageFigure 19(b) shows the time-resolved emission spectra in the entire interval at 80 K. Initially, at early times, the 1LED emission of the D molecule, TPD, is observed, before the 1CT emission grows in. At TD = 5.7 ns pure 1CT emission is observed with onset at (3.05 ± 0.02) eV. The rate of electron transfer in the TPD:TPBi was thus estimated at ca 2 × 108 s−1. After 5.7 ns only 1CT emission is observed, and no high energy shoulder coming from 1LED emission is seen after this. However, at 1.1 ms a contribution from the 1LED emission is observed again. This delayed emission from 1LED must come from TTA as this is the only mechanism that can regenerate the 1LED state. Given that films are made by co-evaporation it is very unlikely that there are large regions of completely segregated D, hence the 1LED emission most likely comes from 1LED states formed by TTA, i.e. due to 3LED states that are able to collide with each other and annihilate. This process creates 1LED states that are then able to decay radiatively before electron transfer to the 1CT manifold occurs.
At later times, phosphorescence (PH) starts to compete with the 1CT emission, and both emissions are observed at TD = 1.1 ms. The PH is the dominant emission at the longest measurement times, TD = 89 ms, but we still can detect a vestige of 1CT emission around 450 nm. The phosphorescence emission is well structured and show two peaks, at 523 and 564 nm, and a shoulder, at 615 nm, which matches the local triplet state phosphorescence of the D, TPD (3LED has onset at 2.44 ± 0.02) eV). The PH of the TPBi acceptor molecules, also emits in this wavelength region (onset at 3LEA = (2.66 ± 0.02) eV), but has different peak positions [102]. Thus, the energy splitting between 1CT and 3LED was found to be ∆EST = (0.61 ± 0.03) eV, and between 1CT and 3LEA, ∆EST = (0.39 ± 0.03) eV. Both ∆EST are large and TADF is unlikely in this blend. The emission spectra collected at 290 K, figure 19(c), show that the phosphorescence emission from the 3LED is very weak at this temperature, and just a shoulder is observed around 550 nm. This clearly indicates that the TTA mechanism is dominating at high temperatures. To confirm this, the integrated area of the emission spectrum, collected in the DF region (TD = 2 μs and Ti = 20 μs), was measured as a function of the laser excitation dose. The intensity dependence shows a slope of 1.60 ± 0.03 at low excitation dose (<11 μJ), which turns to slop of 1.08 ± 0.02 at high excitation doses, see figure 19(d). This behaviour strongly indicates a dominant TTA mechanism [39, 43].
Turning now to the TPD:OXD-7 blend. Figure 20(a) shows the decay curves of TPD:OXD-7 collected as a function of temperature. There is a clear separation in this blend, between the prompt emission and the DF emission around TD = 2 μs. The prompt emission does not show temperature dependence, exhibiting the same intensity from 80 K to room temperature, whilst, the DF and PH regions show clear temperature dependence. The DF intensity is stronger at higher temperatures, in accordance with a TADF mechanism, while the PH shows higher intensity at low temperatures, as expected, with reduction in the non-radiative decay channels.
Figure 20. (a) Time resolved fluorescence decay curves at different temperatures. (b) Time resolved normalised emission spectra at 80 K (c) at 290 K (d) Integrated area as a function of the laser excitation (337 nm) of TPD:OXD-7. Reprinted with permission from [102]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageThe time resolved emission spectra collected at 80 K, see figure 20(b), show that at earlier times, two shoulders, at 403 and 426 nm, are observed. These match those of the local singlet state (1LED) of the D molecule, TPD. As the time delay increases, the emission spectrum shifts to longer wavelengths, moving from 1LED to 1CT emission, showing clearly that the process of electron transfer from the D to the A molecules lasts over about 2 ns, and a rate of electron transfer of kET ∼ 5 × 108 s−1, is estimated. This is significantly faster than those estimated in the m-MTDATA:TPBi and TPD:TPBi blends. Until ca TD = 124.7 ns the 1CT emission is observed to relax and further red shift, giving finally an onset of (2.95 ± 0.02) eV. At later times, the 1CT fluorescence starts to compete with phosphorescence emission, and both emissions are observed, for example at TD = 0.8 ms. However, the phosphorescence dominates at TD = 89 ms, but still a vestige of 1CT emission around 475 nm is detected. The PH emission is well structured and shows two peaks, at 526 and 569 nm, and a shoulder at 615 nm. This triplet emission originates from the localised triplet state of the donor, TPD. The 3LED has an onset energy at (2.44 ± 0.02) eV. Thus, the 1CT and the 1LED (the lowest energy excited states) were identified in the TPD:OXD-7 blend, and the energy splitting between these two states was found to be ∆EST = (0.51 ± 0.03) eV. The ∆EST between 1CT and 3LED is relatively large and is unlikely for TADF to be efficient. However, the triplet of the acceptor OXD-7 molecule, 3LEA, was identified to have an onset energy at (2.72 ± 0.02) eV. Thus, the energy splitting between 1CT and 3LEA is ∆EST = (0.23 ± 0.03) eV and rISC involving these two states would have much higher probability. Therefore, there could well be a competition between TTA, (process involving the 1LED and 3LED states) and TADF (process involving the 1CT and 3LEA states), depending on the relative lifetime of each state. Figure 20(c) shows the normalised emission spectra at different time delays, collected at 290 K. The contribution of the donor emission at TD = 1.1 ns is higher at 80 K than at 290 K, because at low temperature the IC is minimised. Also, the 1CT is slightly red shifted at longer time delays, since at higher temperatures the local host–guest dipolar interactions are able to lead to the formation of a more relaxed 1CT state.
The integrated area of the emission spectrum, collected over the DF region (TD = 5 μs and Ti = 50 μs), was again analysed as a function of the laser excitation dose, see figure 20(d). This time, the intensity dependence of the DF emission shows a slope of 1.33 ± 0.03 at low excitation dose, turning to a linear dependence with slope of 1.00 ± 0.02, at high excitation doses. This suggests that the DF of this exciplex has strong contribution of TTA, but TADF might have some contribution to the DF, otherwise the behaviour at low excitation doses would show a slope much closer to 2. Most probably, the TADF is also able to contribute to the DF because the TTA requires triplet excitons to have bimolecular interactions, which require diffusion. This might be a sufficiently slow process that allows some fraction of triplet states to be directly upconverted using thermal energy, i.e. through rISC, giving origin to TADF. Therefore, the DF mechanism in TPD:OXD-7 is a mixture of TADF and TTA, which are competing processes in this exciplex, and controlled by the relative energy positions of the excited states.
From the analysis of these three exciplex systems, and other cases in the literature, it is possible to conclude that the difference between 1CT and 3LE states, from the D and A molecules, plays a key role in which pairs of molecules are likely to yield TADF. The energy diagram in figure 21, represents the decay pathways available for exciplexes to decay, in general. Green dashed arrows represent the non-radiative transitions and green full ones the radiative transitions. Upon optical excitation of the donor (D) or acceptor (A) molecules, a population of 1CT states is formed, relatively slowly, kET ∼ 108 s−1, by electron or hole transfer. At early times, we detect the transitions 1CT → S0 and 1LED → S0. In the cases discussed here, the acceptor molecules absorb very weakly at 355 nm, thus the transition 1LEA → S0 is not detected. However, even when the A is excited, It is also likely that non-radiative transitions from 1LEA → 1LED will occur as potentially would 1LEA → 1CT, which contribute to quenching emission from the 1LEA state.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
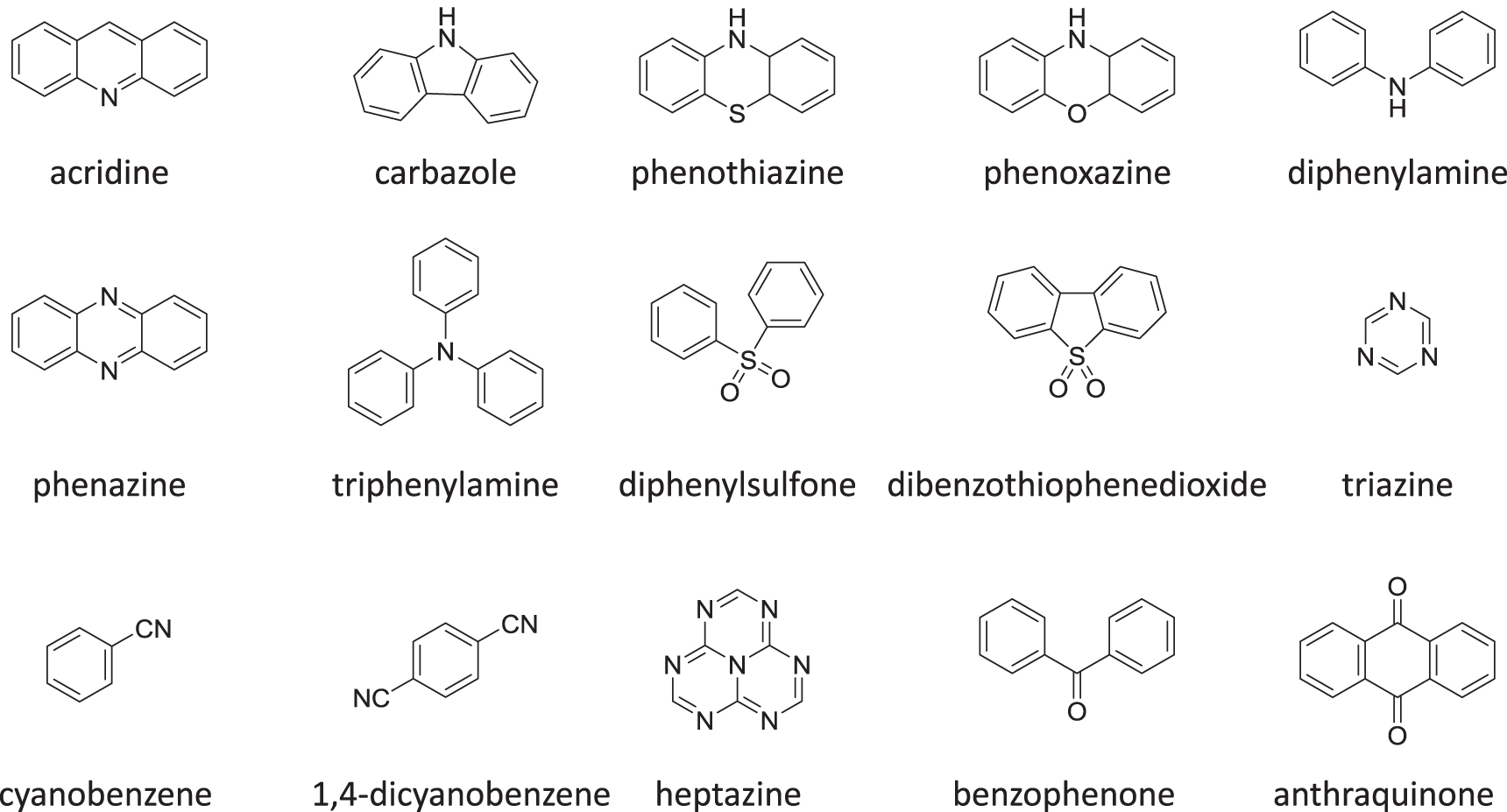{kind=link}
{kind=link}
{kind=link}
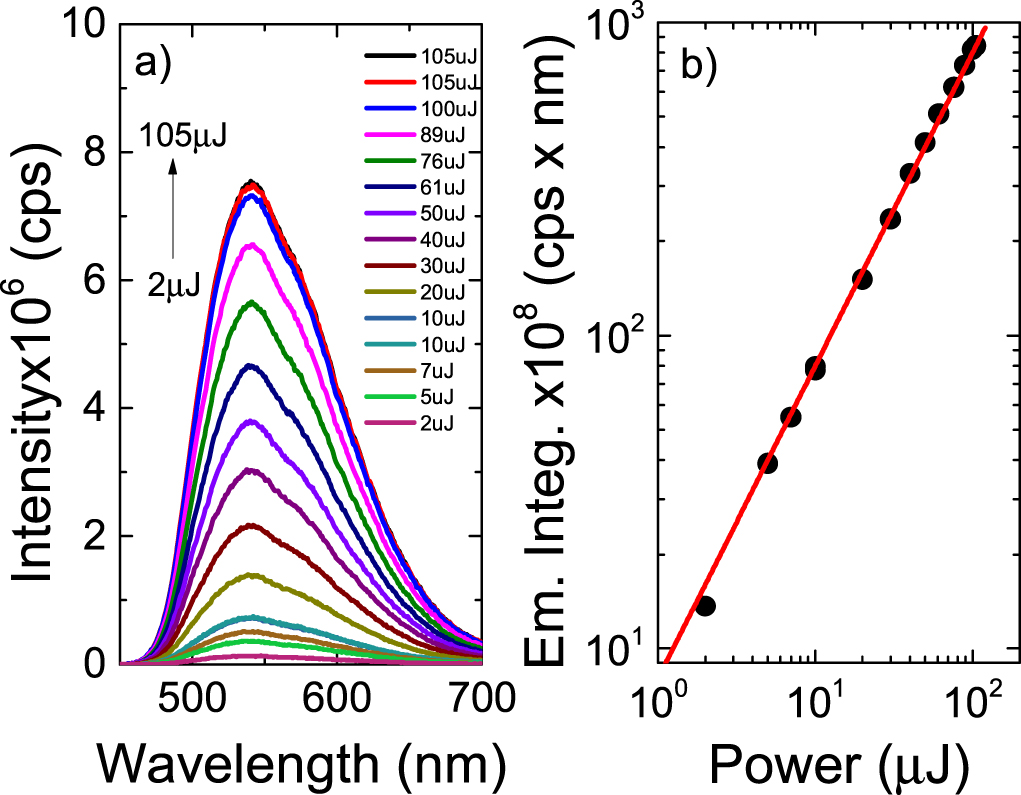{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
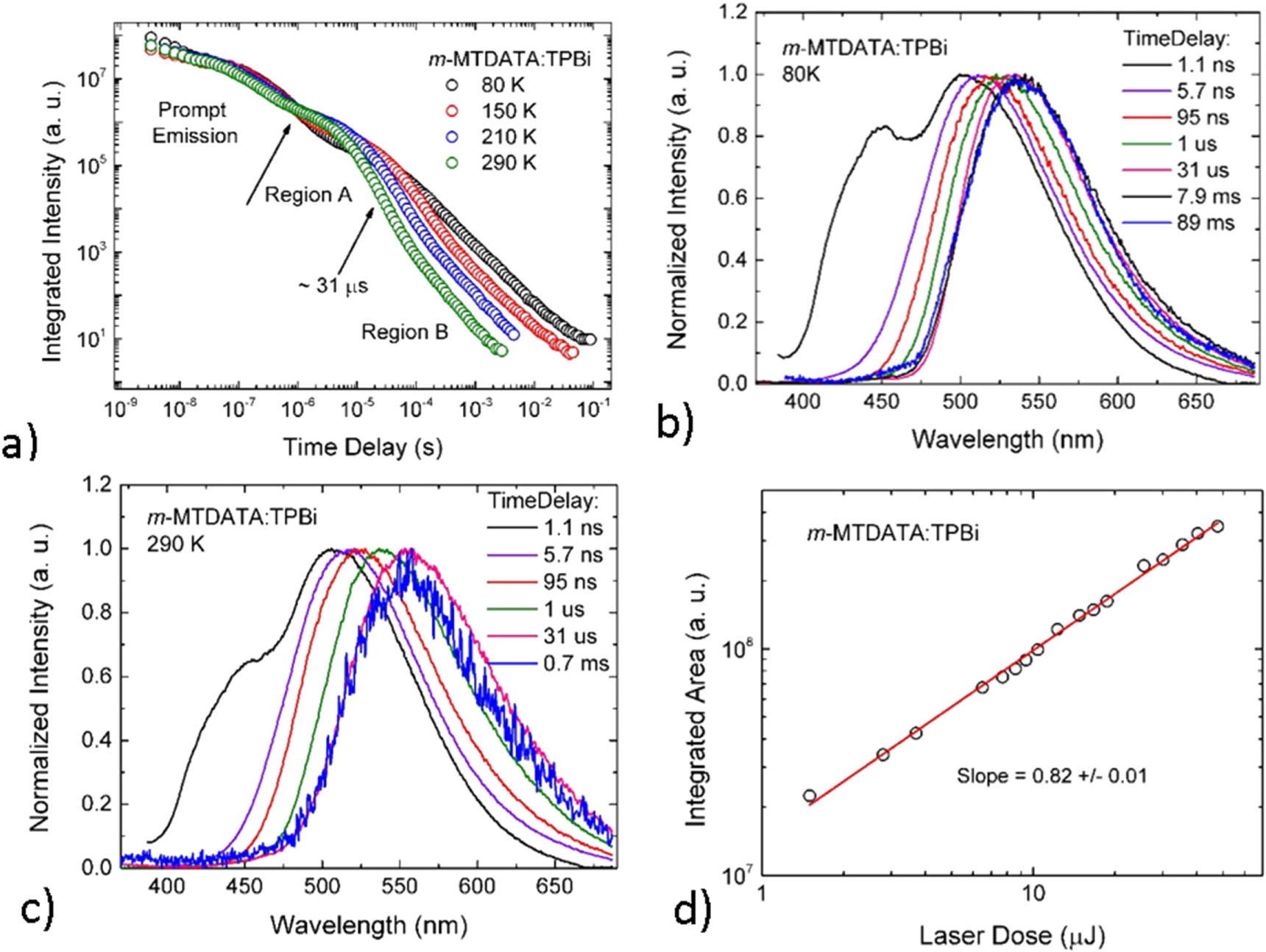{kind=link}
{kind=link}
{kind=link}
Figure 21. Energy diagram showing all the radiative and non-radiative available pathways of decay in an exciplex blend. Reproduced with permission from [102]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
As intersystem crossing between 1CT and 3CT states potentially can only occur by HFC, i.e. when the energy difference between the singlet and triplet CT states is very small, of order 10−3 meV, this channel for intersystem crossing is extremely inefficient compared to that between the 1CT state and a local excited triplet state (3LE) [60]. Therefore, the transition 1CT ↔ 3CT was assigned as a forbidden process in the diagram. The energy splitting between 1CT ↔ 3LED and 1CT ↔ 3LEA are obviously dependent on which molecules are chosen as donor and acceptor. However, when these energy gaps are small, excitons in the localised triplet levels gain enough thermal energy to match the 1CT level and cross to the upper level, considering that rISC/ISC processes are adiabatic transitions. Hence, the transition 1CT → S0 is detected also at longer times and assigned as delayed fluorescence (TADF). If however the energy splitting between 1CT ↔ 3LED and 1CT ↔ 3LEA are large, TADF is unlikely to occur. In this case, another mechanism, TTA, may be active, occurring between 1LEA ↔ 3LEA, 1LED ↔ 3LED, or 3LEA↔, 1LED, converting two triplet excitons into one singlet exciton in the 1LE state and one in the ground state, thus giving origin to DF.
If TTA occurs in the acceptor molecules, the exciton that is converted to the 1LEA state can decay back to 3LEA, relax to 1LED or alternatively to 1CT, from where it can then decay radiatively to the ground state. Finally, at very late times and at low temperature, we detect the emission from the transition 3LED → S0, assigned as phosphorescence. The transition 3LEA → S0 is unlikely to be observed because the Dexter energy transfer from 3LEA to 3LED may be active. Therefore, from our observations it seems clear that in exciplex systems, as with D–A intramolecular CT systems, ISC and rISC are mediated by the same, or very similar second order mechanism, which involves CT states and triplet excited states localised in the D or A molecules.
In summary, as in the three exciplexes discussed here, the DA separation must be about the same, at around 3–4 nm, thus the energy separation between the 1CT and 3CT states is expected to be very small in all cases. Therefore, the differences observed in the contribution of DF due to TADF among the three blends, must be explained as in the case of intramolecular D–A molecules, i.e. it is the energy gap between the CT manifold and the local triplet states that control rISC. It is clear that TADF and TTA compete with each other. However, when the energy gap between the CT singlet state, 1CT, and one of the localised triplet states, 3LED or 3LEA, is small, then TADF is the dominant mechanism that originates DF. Thus, in order to obtain efficient pure TADF from an exciplex it is very important to avoid the presence of local triplet states, from the D or A molecules, having an energy offset larger than ca 0.1 eV relatively to the 1CT state. This is in order to avoid quenching of TADF. Therefore, to achieve efficient thermally activated reverse intersystem crossing it is of fundamental importance that one or both of the local triplets is in near resonance with the 1CT state to facilitate rISC, as we find in D–A intramolecular systems. From this, it would not be a good strategy to design a system where both local triplets are well above the 1CT and 3CT states, as here rISC between 1CT and 3CT would be very slow and in devices polaron excited state quenching may become very efficient. However, as yet no exciplex system has been found that fulfils this latter criterion.
7. Conclusions
This topical review summarises the recent progress in the photophysics of TADF molecules, and introduces current methods to characterise these molecular systems. The global interest on research addressing the TADF mechanism in various molecular systems is reflected by the exponential increasing number of publications and citations, since the first papers reporting the application of TADF molecules in OLEDs were published in 2009, in a complex of tin(IV) chloride and coproporphyrin III [103], and in 2010, in copper complexes [10], and in 2011 in pure organic molecules [104].
The TADF mechanism represents a unique photophysical process originated from a series of molecules containing electron donor (D) and electron acceptor (A) units, or that result from the intermolecular interaction of D and A molecules, in the case of exciplexes, which allows the up conversion of low energy triplet states, usually non emissive at room temperature, into high energy and strongly emissive singlet states. The emergence of the TADF phenomenon initiated a conceptual revolution in the area of OLEDs. In OLEDs, three quarters of the charge recombination events result in triplet excitons that are usually non-emissive. This is a major loss mechanism, which demanded the application of metal organic complexes in order to be able to harvest the entire population of excitons to be utilised in the light output. Such heavy-metal complexes have been very successful, but are often chemically unstable, particularly in the blue emission region, and may create environmental and economic difficulties if used in large scale industry applications, such as lighting. In contrast, TADF molecules are free of heavy metals, and are still able to achieve 100% internal electroluminescence luminous efficiency. This was a major revolution in the field of OLEDs.
The rationale of molecular design, the photophysics and the advantages of using TADF fluorophores in the area of OLEDs have been introduced in this article. The current research momentum in TADF molecules is still for application in OLEDs. However, despite the understanding of the photophysics of these systems is still at their infancy, innovative applications in the areas of sensing and biological-imaging are already envisaged. Fundamental mechanistic understanding especially by applying advanced photophysical techniques will certainly appear in the forthcoming years, which will motivate innovative applications. Further investigation of TADF is anticipated to address the current limitations of TADF chromophores by emphasising the following research directions: (a) improve the optoelectronic performance of these chromophores; (b) achieve enhanced TADF efficiency in the blue and red regions; (c) design and synthesise novel TADF chromophores, in order to achieve water solubility, and high selectivity for analytes, so these materials can be exploited in bio-imaging and sensing applications; (d) explore the possibilities offered by the interaction of the TADF mechanism with magnetic fields in the understanding of the TADF mechanism and to support novel applications. This will require theory work, design and synthesis of novel molecules, and extensive photophysical characterisation, working together to full understand the complexities involved in the TADF mechanism, so novel applications can be anticipated.
Acknowledgments
The authors thank the support of EPSRC, grants EP/L02621X/1 and EP/N028511/1.