
Abstract
In the quest to decipher the chain of life from molecules to cells, the biological and biophysical questions being asked increasingly demand techniques that are capable of identifying specific biomolecules in their native environment, and can measure biomolecular interactions quantitatively, at the smallest possible scale in space and time, without perturbing the system under observation. The interaction of light with biomolecules offers a wealth of phenomena and tools that can be exploited to drive this progress. This Roadmap is written collectively by prominent researchers and encompasses selected aspects of bio-nano-photonics, spanning from the development of optical micro/nano-spectroscopy technologies for quantitative bioimaging and biosensing to the fundamental understanding of light–matter interaction phenomena with biomolecules at the nanoscale. It will be of interest to a wide cross-disciplinary audience in the physical sciences and life sciences.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Since its first invention in the 17th century, the optical microscope is still the only practical means of examining living cells and tissues with high specificity and spatio-temporal resolution. The evolution of this technology has been highlighted by landmark discoveries and inventions, including the Nobel Prize in Chemistry in 2014 for the development of super-resolution fluorescence microscopy beyond the Abbe diffraction limit. Still, many challenges remain, e.g. toward achieving biomolecular specificity label-free, developing quantitative imaging and sensing modalities, and reaching single molecule detection.
Progress toward biomolecular specificity label-free has been brought by the development of coherent Raman scattering (CRS) microscopy techniques [1]. In Raman scattering, the inelastic interaction of light with vibrating chemical bonds produces light scattered at a different frequency compared to the incident light. The frequency shift is equal to the frequency of the vibration, which in turn depends on the type of chemical bond. Spontaneous Raman scattering, however, is a very weak process, owing to the small Raman scattering cross-sections of vibrating modes in biomolecules. This limitation is overcome in CRS whereby two laser fields with different colours are used to drive molecular vibrations coherently and resonantly, via the beat term at the laser frequency difference. Raman scattered light from identical vibrational modes which are coherently driven constructively interferes, generating a signal that can be acquired at high speed to imaging living cells and tissues. Notably, CRS does not suffer from photobleaching and is amenable to quantitative analysis.
Ultrafast pump-probe spectroscopy techniques, including two-dimensional (2D) methods [2], offer a powerful way to increase biomolecular specificity and at the same time address fundamental questions into the importance of ultrafast dynamics and quantum coherences in biology. Measuring phenomena such as electronic and vibrational quantum coherent oscillations in biomolecules, albeit being technically challenging, could bring the highest degree of biomolecular specificity, alongside fascinating new discoveries into the fundamental machinery of Nature.
Reaching single molecule sensitivity and spatial resolution at the nanoscale also remains a major challenge in bioimaging and biosensing, especially without the aid of fluorescent labels. Significant progress in this area has been brought by the local field enhancement effect occurring in the vicinity of metallic (plasmonics) nanostructures [3], merging the fields of bio-photonics with nano-plasmonics.
This Roadmap article aims to provide a concise yet authoritative overview on the present and future of bio-nano-photonics, spanning from the development of optical micro/nano-spectroscopy technologies for quantitative bioimaging and biosensing to the fundamental understanding of light–matter interaction phenomena with biomolecules at the nanoscale.
Section 1 discusses recent advances in the development of nanoantenna platforms to achieve single-molecule biosensing, owing to the strong light field confinement and enhancement effects from localized surface plasmon resonances with these antennas. Present status and future challenges are reviewed, particularly regarding the design and fabrication of plasmonic nanoantennas, and their integration with optical micro-spectroscopy platforms, both fluorescence-based as well as label-free.
Sections 2 and 3 address CRS microscopy applied to bioimaging, both as a label-free chemically-specific technique, and in combination with recent advances in the development of vibrational labels. Section 2 specifically discusses label-free biomolecular specificity, sensitivity, and in-depth imaging. These aspects are considered alongside the corresponding requirements in CRS hardware and instrumentation development. Section 3 reviews the field of vibrational labelling to increase biomolecular specificity, via the use of chemical bonds which vibrate at wavenumbers in the so-called 'Raman silent region'. Besides labelling by replacing hydrogen with deuterium, the emerging field of designing new small molecules acting as vibrational tags by the insertion of triple bonds, such as alkynes and nitriles, is discussed. Notably, in contrast to fluorophores, vibrational labels do not photobleach; they also exhibit sharp Raman resonances amenable to multiplexing.
Section 4 considers the polarisation of light as an additional tool to provide valuable information on the symmetry and organisation of biomolecules, with high sensitivity and at a scale below the resolution limit of a conventional optical microscope. Polarization-sensitive imaging is reviewed in the context of linear and nonlinear (multiphoton) microscopy techniques, including CRS. Technological advances and challenges are discussed, in particular concerning the speed of polarized imaging when observing living specimens, and the ability to measure molecular orientations in 3D.
Sections 5 and 6 consider the application space in the life sciences. Section 5 specifically addresses how the development of various optical microscopy technologies has progressed our understanding of the transport and trafficking of biomolecules inside cells, which is critical for eukaryotic cell's physiology and function. Opportunities, as well as challenges, in this area include elucidating the role of intracellular trafficking in health and disease, and exploiting these pathways to deliver drugs to distinct and targeted locations inside cells. Section 6 discusses recent advances in the optical biopsies of tissues for disease diagnostics by means of multiphoton microscopy, to complement and potentially replace traditional histopathology. Combining different label-free imaging modalities offers a very powerful platform in this area, with the potential to expand into in-vivo or intravital imaging. Challenges in image analysis are highlighted, alongside emerging opportunities in machine learning algorithms, to drive a shift from the subjective to the objective interpretation of quantitative datasets.
Sections 7 and 8 tackle the challenge of addressing single molecules. Section 7 discusses the micro-spectroscopy investigation of single emitters at cryogenic temperatures. By cooling single molecules well below room temperature, their thermal motion and interaction with the fluctuating environment is slowed down significantly. Hence, it becomes possible to investigate fundamental properties of single molecules as quantum emitters in the coherent light–matter interaction regime, before they are perturbed. The section reviews the present status and future directions in terms of phenomena observed with various emitters (from organic molecules to semiconductor quantum dots), and the range of spectroscopic instrumentation and techniques utilized in this low-temperature regime. Section 8 addresses the study of single molecules at ambient temperature, which is key to biological applications. At room temperature, many processes, such as electron dephasing and vibrational dynamics, occur at an ultrafast time scale, in the femtosecond to picosecond regime, and require the development of novel measurement schemes. Section 8 considers the limitations of conventional pump-probe methods, and discusses alternative strategies exploiting the use of stimulated absorption and/or stimulated emission. Significant challenges in these experiments are signal-to-noise ratio and photon budgets, requiring further development into ingenious excitation and detection schemes.
Sections 9 and 10 discuss ultrafast 2D spectroscopy methods applied to organic molecules, to unravel fundamental insights into the correlations of characteristic modes and the importance of quantum coherences in biology. Section 9 specifically addresses 2D electronic spectroscopy (2DES), and how this technique has been applied to tackle the highly debated topic of electronic coherences in light-harvesting complexes and their role in controlling excitation energy transfer pathways. Challenges and limitations of 2DES include the considerable experimental complexity and the lack of spatial resolution resulting in ensemble-averaged information. The latest efforts toward implementing high spatial resolution schemes are considered, as these would pave the way to the grand goal of performing 2DES in single photosynthetic complexes at room temperature. Section 10 focuses on ultrafast two-dimensional mid-infrared (2DIR) spectroscopy, a technique that allows probing correlations of characteristic vibrational modes in the 'fingerprint' spectral window of biomolecules to retrieve their chemical structure. This section considers the opportunity of combining 2DIR with recent advances in mid-IR plasmonics, to achieve unprecedented sensitivity and spatio-temporal resolution towards the goal of single molecule spectroscopy.
Section 11 reviews from a theoretical standpoint our understanding of photoinduced charge separation processes and electron transfer phenomena, which are at the heart of natural functions such as photosynthesis, vision, and DNA damage repair. The ongoing scientific effort to achieve a fully quantum and microscopic description of these phenomena is summarised. Challenges in the implementation of theoretical methods are discussed, including how to properly describe the coupling between electron and nuclear motion in a non-adiabatic picture, and the role of quantum coherences.
Finally, section 12 addresses the present status and future directions in the development of laser sources for biophotonics. Requirements such as pulse duration, pulse energy, wavelength tunability and repetition rate are reviewed, and recent advances in the development of solid-state and fibre lasers are highlighted. Considerations around the need to simplify the complexity and shrink the size of laser systems are also discussed, important for the translation of laser sources into biomedical applications.
Overall, this Roadmap aims to provide a meaningful snapshot of important selected topics in bio-nano-photonics. The field is much broader hence this selection is not intended to be exhaustive but wishes to create a constructive picture of some of the major advances for the scientific community.
Acknowledgments
This Roadmap embraces the research vision and ambition of the European Training Network MUSIQ, funded by the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
1. Nano-plasmonics for bioimaging and biosensing
Ediz Herkert1, Nicole Slesiona2, Martina Elisena Recchia2, Thomas Deckert3 and Maria F Garcia-Parajo1,4
1 ICFO-Institut de Ciencies Fotoniques, The Barcelona Institute of Science and Technology, Barcelona, Spain
2 School of Biosciences, Cardiff University, Museum Avenue, CF10 3AX Cardiff, United Kingdom
3 Department of Physics and Materials Science, University of Luxembourg, 162a avenue de la Faïencerie, L-1511 Luxembourg, Luxembourg
4 ICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
Status
Life scientists seek to understand how nanoscale molecular interactions in living cells influence biological function. Given the small scales involved, these interactions cannot be resolved with conventional light microscopy, requiring approaches that surpass the diffraction limit of light.
Nanostructures that exhibit localized surface plasmon resonances provide huge electromagnetic field enhancements on the nanoscale. These properties have enabled fluorescent enhancements of weakly fluorescent molecules and enhanced label-free vibrational spectroscopy. A broad range of applications aimed at probing the molecular electronic and vibrational states in biochemical environments with single-molecule sensitivity have already been documented. Amongst others, resonant nanoantennas allowed detection of individual fluorescent molecules at micromolar concentrations [4], surface-enhanced infrared absorption (SEIRA) [5] and surface-enhanced Raman spectroscopy (SERS) [6] with enhancement factors up to 105 [5], and 109, respectively [6] (figure 1(a)).
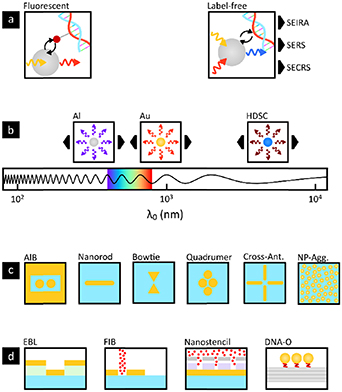
Figure 1. State-of-art nanoantenna platforms for bioimaging and biosensing. (a) Plasmonic nanoantennas enhance the fluorescence of individual molecules attached to the biomolecules of interest (left), or enable enhanced label-free detection of biomolecules via different spectroscopic approaches (right). (b) Different nanoantenna materials such as aluminium, Gold and highly doped semiconductors support resonances at distinct spectral regions, from the ultraviolet, visible, near-infrared to mid-infrared. (c) The plasmonics community has reported a broad variety of nanoantenna shapes that provide specific benefits for the selected spectroscopic approach, i.e. fluorescence, SEIRA, SERS, SECRS, etc. Designs include dimer antenna-in-box, nanorods, bowtie configurations, quadrumers, aggregates, etc. (d) Nanofabrication approaches used to produce arrays of antennas on single chips. The optimal nanofabrication depends on the requirements regarding biocompatibility, scalability, cost, and precision. The pictograms depict only a subset of spectroscopic approaches, materials, nanoantenna shapes, and fabrication methods. (SEIRA: surface-enhanced infrared absorption, SERS: surface-enhanced Raman spectroscopy, SECRS: surface-enhanced coherent Raman spectroscopy, HDSC: highly-doped semiconductor, AIB: antenna-in-box, Cross-Ant.: cross-antennas, NP-Agg: nanoparticle aggregate, EBL: electron-beam lithography, FIB: focused-ion-beam, DNA-O: DNA-origami).
Download figure:
Standard image High-resolution imageRecently, great scientific effort is devoted towards establishing nanostructures suitable for in-vivo enhanced fluorescence monitoring [4] and surface-enhanced pump-probe schemes to deliver molecular bond information with ultimate sensitivity [7]. Moreover, antennas based on highly-doped semiconductors (HDSCs) have been proposed for third harmonic generation in the mid-infrared [8] since they offer a tunable bulk plasma frequency through doping and high third-order susceptibility. This makes them promising candidates for surface-enhanced ultrafast pump-probe vibrational spectroscopy, providing previously inaccessible information about the target molecule and its biological environment.
Many of these breakthroughs have resulted from the optimization of nanostructure designs, providing strongly confined resonances at the desired spectral region (figure 1(b)) and using different geometries depending on the application (figure 1(c)) [4, 7–9]. Similarly, a wealth of nanofabrication technologies have been explored, including traditional lithography approaches and more recently, nanostencil [10] or DNA-origamis [11] (figure 1(d)). In particular, the possibility of using DNA as modular breadboard to self-assemble nanostructures opens up a widely unexplored space of new plasmon-based designs that can be further extended to HDSC-based plasmonic devices.
Further advances in the field promise to provide biocompatible and cost-efficient tools to interrogate individual molecular transitions in living biological systems. Nevertheless, there are still numerous challenges to be addressed before these devices can be readily integrated as reliable tools for life scientists.
Current and future challenges
Nanoantenna platforms have proven to outperform conventional biosensing approaches. However, most of the proposed designs are still in a conceptual phase and have not reached the maturity for reliable in-vivo applications. In the case of fluorescence, planarized gold-antennas-in-box platforms have shown great potential for ultrasensitive, nanoscale fluorescence detection in living cells [4]. Yet, broadband operation for multicolour applications will require materials with resonances in the short-wavelength visible. In this respect, aluminium is a promising candidate, but the achievable enhancement factors will stay below that of Gold and its biocompatibility remains to be asserted. Moreover, schemes that provide multiplexed fluorescence readout from thousands of antennas simultaneously are confronted with the lower temporal response of state-of-art camera detectors. Plasmonic super-resolution fluorescence imaging also holds great promise. Unfortunately, coupling of dipole emitters with plasmonic nanoparticles induces emitter mis-localizations of up to 30 nm [12] that might be challenging to compensate for.
In the case of label-free vibrational spectroscopy, considerable effort has been devoted to designing nanostructures with strong resonances to enable single-molecule SEIRA and SERS. Strategies to embed these nanostructures into biocompatible platforms are still awaiting. Although surface-enhanced coherent Raman spectroscopy (SECRS) promises higher sensitivity as compared to SERS [7], its application for in-vivo monitoring is currently pending.
Future challenges will also likely involve merging nanoantenna platforms with state-of-the-art spectroscopic approaches. Two-dimensional (2D) spectroscopy for instance, could deliver unprecedented spatiotemporal details about the structure and environment of the target molecule when combined with nanoantennas. However, the ultrafast and broadband nature of these spectroscopic techniques places huge demands on the nanoantenna´s optical properties and design. In general, nanofabrication methods like DNA-origamis or nanostencil enable low-cost and large-scale nanostructure arrays [10, 11] but they are still restricted in terms of design freedom and material selection. DNA-origamis also enable to dynamically reconfigure the optical properties of nanoantenna arrays and allow inclusion of analyte binding sites. However, their full integrability into biosensing platforms is yet unproven. Finally, HDSCs provide an alternative for active plasmonics since they can be switched dynamically from transparent to metallic by optical pumping. However, the high peak intensities needed are incompatible with most biological samples.
Advances in science and technology to meet challenges
Plasmon-enhanced-fluorescence and -label-free spectroscopy, outperform their diffraction-limited counterparts, but standard spectroscopy is also developing at a fast pace. To match these advances, plasmonics must adapt to the spectroscopic requirements while preserving biocompatibility. The investigation of ultrathin layers is therefore crucial to decouple the high spectroscopic demands from the requirements of biocompatibility and thus tackle both challenges independently. Atomic layer deposition of dielectrics like Alumina holds great potential to physically separate the nanoantennas from the biological environment without losing their electromagnetic interaction. Moreover, planarization strategies [4] could be used to expose the sample to the highest region of nanoantenna confinement and to minimize curvature effects that may influence measurements in living cells.
Since most of the related literature employs gold nanoantennas, alternative antenna designs with other materials that support tunable resonances outside the near-infrared will be required. This will enable multicolour fluorescence and vibrational spectroscopy in the visible and mid-infrared-region. Nanoantennas based on aluminium, dielectrics, or HDSCs are known to provide resonances in these spectral regions but further optimization of the sensor design and fabrication are needed to compete with Gold-based platforms in terms of enhancement and confinement. Figure 2 shows as example how to convert nanoantenna arrays into truly biocompatible sensing platforms.
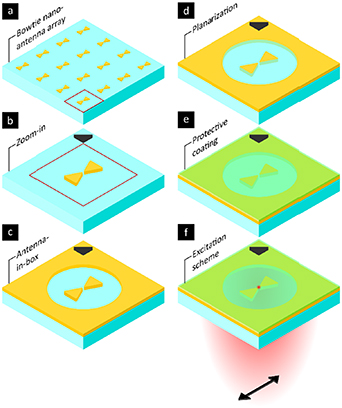
Figure 2. Exemplary scheme of how to convert nanoantenna arrays into biocompatible sensing platforms. (a), (b) Bowtie nanoantennas are often used since the localized near-field allows to strongly couple to emitters close to the gap region. (c) The antenna-in-box design increases the effective signal-to-background of nanoantennas by reducing surrounding background from fluorescent molecules outside the gap region. (d) Planarization of the nanoantennas is required if the curvature of the nanostructures can impose undesired curvature effects on the probed biological specimen. (e) Using protective ultrathin coatings allows to decouple biocompatibility demands and requirements on the spectral tunability and enhancement of the deployed nanoantenna material. (f) Using suitable excitation schemes is crucial to probe the desired properties of the biological system at the nanoscale with ultimate sensitivity and minimal photodamage.
Download figure:
Standard image High-resolution imageMany of these goals are already within reach, so that plasmon-enhanced biosensors will eventually mature into inexpensive and user-friendly devices, making them available to non-experts. Only then, will they be broadly applied to relevant problems in life sciences that cannot be addressed with conventional methods. This requires a holistic mindset that considers the cost and portability of the light source, nanoantenna platform, and detector. Recently, Tittl et al [13] introduced pixelated dielectric metasurfaces to reveal molecular absorption signatures in the mid-infrared without the need for spectrometers. These pioneering results pave the way for low-cost spectrometer-free approaches in different spectral regions. High-throughput nanofabrication methods will also play a key role in the transition towards highly sensitive, cost-efficient biosensors. In particular, DNA-origamis offer design and active reconfiguration possibilities that cannot be achieved with conventional top–down fabrication methods and thus promise completely new biosensor concepts at low cost.
Concluding remarks
The strong field confinement and enhancement of nanoantennas are key properties that yielded exciting perspectives for single-molecule fluorescence and label-free spectroscopy. Yet, successful commercialization of nanoantenna platforms is still pending due to high demands on nanofabrication and biocompatibility. New nanofabrication techniques promise the expected breakthrough and applicability at low cost. Moreover, alternative materials and coupled plasmonic systems that support tunable resonances at multiple wavelengths could be exploited. Aluminium, gold, and HDSCs cover the full spectral range from ultraviolet to mid-infrared, while oligomer nanoantennas provide tunability of field enhancement, confinement, and resonance frequency for multicolour spectroscopy. With this knowledge at hand, the implementation of spectroscopic techniques like 2D spectroscopy and SECRS for single-molecule sensitive in-vivo applications is foreseeable. Ultrafast surface-enhanced spectroscopy will make it possible to study functional groups, conformational changes, or coupling effects of single molecules far beyond current capabilities, enabling to better understand their role in health and disease.
Acknowledgments
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992. Further support has been provided by the European Commission H2020 Program under grant agreement ERC Adv788546 (NANO-MEMEC), Government of Spain (Severo Ochoa CEX2019-000910S and FIS2017-89560R), Fundació CELLEX, Fundació Mir-Puig and the Generalitat de Catalunya through the CERCA program and AGAUR (Grant No. 2017 SGR 1000).
2. Coherent Raman scattering for label free bioimaging
Eric Michele Fantuzzi1, Andrea Pruccoli2, Imaiyan Chitra Ragupathy3, Dominykas Gudavičius4,5 , Martina Elisena Recchia6 and Hervé Rigneault1
1 Aix Marseille Univ, CNRS, Centrale Marseille, Institut Fresnel, Marseille, France
2 Department Chemie, Universität Konstanz, D-78457, Germany
3 Leica Microsystems CMS GmbH, Am Friedensplatz 3, 68165 Mannheim, Germany
4 Light Conversion, Keramiku st. 2B LT-10233 Vilnius, Lithuania
5 Cardiff University, School of Physics and Astronomy, The Parade, CF24 3AA Cardiff, United Kingdom
6 Cardiff University School of Biosciences, Museum Avenue, CF10 3AX Cardiff, United Kingdom
Status
Coherent Raman scattering (CRS) encompasses stimulated Raman scattering (SRS) and coherent anti-Stokes Raman scattering (CARS). Both SRS and CARS processes have been discovered shortly after the first laser was demonstrated as they require intense light fields to activate multiphoton processes. The main feature of the SRS and CARS processes is to be resonant with molecular vibrational energy levels and therefore both can probe chemical bonds present in matter without requiring any label or staining. Initially used to perform nonlinear spectroscopy, CRS became a striking label free biomaging techniques after CARS and later SRS were implemented with solid state lasers and combined with laser scanning microscopes. Contrary to spontaneous Raman that requires long (ms) integration time per pixel, CRS can image cells, model organisms and living tissues (figure 3) with μs pixel dwell time allowing video rate images of specific chemical bonds such as found in lipid and proteins. Since then, the field of coherent Raman imaging developed rapidly to reach today a level of maturity that brings the technology towards the application markets [14, 15].

Figure 3. CARS image of mouse spinal cord reveals the organization of myelin sheets (magenta) and is combined with fluorescence (CFP) to reveal the axons (blue).
Download figure:
Standard image High-resolution imageCurrent and future challenges
Although the CRS speed imaging can be easily pushed down to 1 μs pixel dwell time [14] when imaging highly concentrated samples such as bulk lipid the sensitivity of CRS becomes an issue when dealing with more diluted samples. This is especially true for SRS that requires to modulate one beam at MHz frequencies and demodulate the other beam to catch the weak (10−4, 10−6) stimulated Raman gain or loss signals [16]. For this, besides low noise electronic and high speed lock-in amplifier detection, the shot noise limited nature of the optical source is the crucial parameter [14]. Shot noise limited SRS laser systems for frequencies above few MHz can be easily achieved using bulk crystal laser oscillators coupled to optical parametric oscillators (OPO) to generate the two beams necessary to activate CRS [14] but fibre lasers are more problematic due to the remaining amplified spontaneous emission noise inherent to the fibre gain media. Recent effort to perform SRS with fibre lasers systems have used balanced detection schemes that sets the sensitivity limit to at best 3 dB above the shot noise limit [17]. These efforts currently limit the detection sensitivity of CRS to the mM concentration range which is often not enough for applications where the targeted molecules are more diluted.
Besides molecular sensitivity, molecular specificity is also a practical issue when dealing with biological samples. This is because bio-molecules are mainly constituted with the same chemical bonds and the relevant vibrational signature associated to a studied bio-process is often buried in the congested fingerprint (700 cm−1, 1800 cm−1) vibrational spectra. So far CRS demonstrations have mostly targeted lipids and few reports only exist on probing the much more informative fingerprint region [14].
Finally, the in depth imaging CRS ability and its ultimate spatial resolution are still limitations for current bioimaging applications. Imaging biological tissues in depth is challenging because of inhomogeneity that scatter and absorb light. Similar to multiphoton fluorescence microscopy CRS is limited to few hundreds of microns penetration depth depending the tissue whereas its spatial resolution (∼1 μm) is diffraction limited and directly bounded by the numerical aperture of the used objective lenses [18].
Advances in science and technology to meet challenges
The detection sensitivity can be further enhanced using optical or molecular strategies.
From the optical side, the sensitivity limit is often related to the CRS background. This is especially true in CARS which is associated to an inherent non resonant background [14] that hampers its sensitivity. On the contrary SRS has less non-resonant background but it is also affected by nonlinear spurious effects such as thermal lensing, two-photon absorption and cross phase modulation [14], these artefacts limit the SRS sensitivity. Using dedicated schemes with three or two beams [14], it is possible now to cancel and/or evaluate SRS artefacts to produce background free SRS images whose molecular sensitivity is limited by the shot noise limit. This pushes up the sensitivity to hundreds of μM concentration in bio samples.
From the molecular side SRS sensitivity has been dramatically improved to the μM range using electronic pre-resonances. When one of the SRS excitation laser is spectrally close to the absorption band of the targeted molecule, the SRS four wave mixing process becomes pre-resonant with the molecular electronic states and resonant with the molecular vibrational states, this boost up the overall SRS sensitivity by 3 orders of magnitudes to the few μM range [19]. Of course, this strategy implies to work with specific molecules that have been designed to be used as Raman tags with dedicated electronic absorption in the NIR and chemical groups that cannot be found in natural biosamples such as C≡N (nitrile) or C≡C (alkyne). More recently the combination of nitrile Raman tags conjugated to dedicated fluorophore has enable the vibrational detection of single fluorescence molecule through their vibrational signature [20].
Although the use of fluorescence provides superb specificity, this is not compatible with applications where fluorescent tags cannot be used or are too big to penetrate through complex media. This is for instance the case for transcutaneous active molecule penetration in skin that requires to use tiny Raman tags. For this application carbon–deuterium bonds (CD) can be used and conjugated to active biomolecule to be further followed when the drugs penetrate into tissues. Since C–D vibrational bonds vibrates at 2100 cm−1, it does not overlap with other chemical bonds present in tissue and provides a superior sensitivity.
To image tissues in depth, strategies have to be developed to overcome absorption and scattering. The latter can be circumvented when using the latest advances in wave front shaping that enable to create a focused spot through a scattering medium, whose transmission matrix has been pre-measured. Extending this concept to nonlinear optics it has been recently demonstrated CARS imaging over tens of micrometres field of view through thick mice spinal-cord tissues where initially no signal was measurable due to scattering [21].
The second more straightforward approach consists in developing endoscopes that can penetrate deep in tissues to activate the CRS processes, as reported recently using scanning tip endoscopes combined with hollow core fibres that have the ability to deliver ultra-short pulses together with collecting the CARS signals [22].
Beside the above mentioned innovations, CRS contrast mechanisms have abilities that cannot be performed using other optical approaches. This is for instance the case for CARS that can access molecular orientation distribution to a level of precision that cannot be achieved with other linear optical contrasts, leading to high speed polarization CARS imaging with application in myelin organization within the context of neurodegenerative diseases [23] (figure 3).
Concluding remarks
Besides the scientific innovations reported above, SRS has been pushed recently to the medical field as being able to provide images of freshly excised tissues with histological quality [14, 15, 22] (figure 4). This step forwards combined with artificial intelligence should bring a novel diagnostic tool directly in the operatory room to help in decision making and cancer tissue removal. This also proves that CRS bioimaging has reached a mature stage 20 years after its first demonstration. However, there is still a lot to explore and to expect from multispectral detection [14], multi-focus and future wide field [24] CRS implementations that should keep the field at the forefront of label free imaging.

Figure 4. Stimulated Raman histology: Imaging the CH2 and CH3 chemical bonds using SRS allows to generate a colourful stimulated Raman histology (SRH) image that reproduce the classical hematoxylin and eosin (H&E) histology image used as the gold standard in the hospital. SRH image (a) and classical H&E image (b). Adapted from [15]. Reproduced from [15]. CC BY 4.0.
Download figure:
Standard image High-resolution imageAcknowledgment
This work was supported by the European Union through the H2020-EU MSCA-ITN-2018 MUSIQ program.
3. Non-linear Raman microscopy with vibrational labels
Jan Majer1, Andrea Pruccoli2, Imaiyan Chitra Ragupathy2 and Andreas Zumbusch2
1 GSK Medicines Research Centre, Stevenage, United Kingdom
2 Department of Chemistry, University of Konstanz, Konstanz, Germany
Status
Without a doubt, fluorescence microscopy is one of the most important imaging techniques in the biological sciences. Yet, fluorescence excitation is always accompanied by photobleaching which limits the achievable observation times. In addition, fluorescence techniques require the labelling of the samples with comparatively bulky fluorophores. These drawbacks have provided a motivation for the development of complementary imaging methods. Raman microscopy is especially well suited as an alternative approach since it allows the generation of contrast based on the vibrational spectra of the sample molecules. Even small molecules possess a large number of distinctive vibrational bands which allows their spectral identification. This abundance of spectral lines, however, poses problems for spectral separation in a biological environment, as for example in a living cell, where myriads of different molecules are present in the excitation volume. This results in dense spectra in which individual bands are difficult to assign. Therefore, recently an increasing number of reports have been published, which describe the use of vibrational labels, i.e. small chemical groups which are covalently bound to a target molecule. The labels are chosen such that their vibrational frequencies lie in the so-called Raman silent region. In this spectral area ranging from approximately 1900 cm−1–2700 cm−1, only a few very specific vibrational resonances are found. In microscopy, vibrational labels of this type have first been employed with spontaneous Raman scattering [25], but they are equally well suited for non-linear Raman microscopy techniques such as coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) microscopy [26]. Compared to spontaneous Raman scattering, the latter methods have the advantages of being robust against sample autofluorescence and of offering much faster image acquisition [27]. The combination of novel methods such as SRS microscopy with new approaches for vibrational labelling therefore holds great promise for non-invasive long-term imaging in the biological sciences [28–30].
Current and future challenges
The most important vibrational labelling groups with vibrational resonance in the Raman silent region are either deuteriated groups in which the C–D stretch vibration is found at 2070–2210 cm−1 or groups with triple bonds such as alkynes, nitriles, or organic azides which possess resonances at 2100–2260 cm−1, 2210–2260 cm−1, and 2080–2120 cm−1, respectively (figure 5). For either type of compound, a major bottleneck for current research is the low availability of appropriately modified small molecules. While a number of deuteriated compounds are commercially available, most of these molecules are of little biological interest. Use of deuteriated compounds as starting products for the chemical synthesis of more relevant molecules, however, is often impeded by their high price. No matter which type of modification is employed, tests for the biocompatibility of the modified compounds are prerequisite for their application in imaging experiments. Important aspects with this respect are cytotoxicity, efficiency of enzymatic processing of the compounds in competition with natural substrates, and the chemical stability in a typical biological environment. Information of this type is rarely available.

Figure 5. Typical spectral ranges of the resonances for vibrational labels in the Raman silent region.
Download figure:
Standard image High-resolution imageThe more suitably labelled small molecules become available, the more aspects for their future application in non-linear Raman experiments become relevant. As can be seen above, all relevant markers have their vibrational resonances in a small spectroscopic window. Since many potential applications would benefit from the simultaneous detection of several compounds, it is therefore desirable to provide modifications with different vibrational resonances allowing the spectral distinction between several different vibrational labels in one sample. Ideally, these modifications would also offer readouts for cellular parameter such as pH value, ionic strength, or membrane potentials.
With respect to spectroscopy, not only spectral resolution, but also detection sensitivity will become more important in the future. To date, spontaneous and non-linear Raman imaging applications were mainly successful when the target molecules are present in comparatively high local concentrations of 1 mM and above. Despite decades of intense research, this still puts severe limits on the applicability of all Raman imaging techniques.
Advances in science and technology to meet challenges
Concerning the synthesis of alkyne modified small molecules, a lot of progress has recently been achieved due to the surge of applications of copper(I)-catalyzed azide-alkyne cycloadditions for bioorthogonal fluorescent labelling [31]. This resulted in intense efforts for the synthesis of a broad variety of alkyne containing molecules, some of which are now also commercially available. For deuteriated and nitrile containing compounds, other sources might be helpful, especially with respect to investigating pharmaceutical compounds. It has been pointed out that many bioactive compounds already contain an active group with a resonance in the Raman silent region. In addition, pharmaceutical research often relies on mass spectrometry data for which deuterated compounds are synthesized. As pointed out above, the latter would be equally valuable for Raman imaging.
Apart from using these existing sources, also the specific synthesis of Raman labelled molecules has made important progress. With respect to the possibility of doing multispectral Raman experiments, impressive results on the use of isotopic labelling of alkyne and nitrile groups have been achieved recently [19]. In these experiments, also an important approach for increasing the sensitivity of Raman imaging based on the exploitation of electronic pre-resonance conditions has been demonstrated. So far, most pre-resonance Raman imaging work was done using near-infrared excitation which in turn necessitates long wavelength absorbing chromophores as reporter molecules. However, the availability of laser systems with synchronized dual emission in the visible [32] should give access to smaller chromophoric systems or even the exploitation of resonance enhancement of small molecules themselves. First steps in this direction have recently been published [33]. As a side effect, the authors also demonstrated a significant improvement of the spatial resolution using short wavelength excitation.
Concluding remarks
The possibility to generate molecule specific contrast without the need for labelling is often seen as the biggest advantage of spontaneous or non-linear Raman microscopy techniques. Yet, recent work shows that the use of vibrational labels offers exciting possibilities for imaging especially of biological samples. The sacrifice of working label-free is outweighed by the higher selectivity and sensitivity that can be achieved. In addition, in contrast to fluorophores, vibrational labels do not photobleach. These advantages currently motivate intense work on both the chemical synthesis of suitable vibrational labels, but also on Raman methodology itself. With the first results emerging from these efforts, we expect that especially SRS microscopy will increasingly become a method of choice mainly for long term imaging studies of small molecules in a biological context.
Acknowledgments
The project leading to this work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 812992.
4. Exploiting vector-fields in optical microscopy for biology
Eleanor Munger1, Martina Elisena Recchia2, Dominykas Gudavičius3,4 and Sophie Brasselet1
1 Aix Marseille Univ, CNRS, Centrale Marseille, Institut Fresnel, France
2 School of Biosciences, Cardiff University, Museum Avenue, CF10 3AX Cardiff, United Kingdom
3 School of Physics and Astronomy, Cardiff University, The Parade, CF24 3AA Cardiff, United Kingdom
4 Light Conversion, Keramiku st. 2B LT-10233 Vilnius, Lithuania
Status
Optical microscopy imaging offers tremendous possibilities to explore questions related to biological phenomena or in nanosciences with high resolution and dynamics. While microscopy provides morphological observations, it misses important information about how molecules, proteins, or even atoms in a nano-object are organized which requires a read-out of their orientation. Light polarization offers a way to access this information, for instance molecules respond to light excitation most efficiently if its input electric field oscillates along the bound electrons' motion direction. A strong dependence to light polarization can be found in many optical processes: one- and multi-photon fluorescence, but also nonlinear coherent processes such as second harmonic generation (SHG), third harmonic generation (THG), and the coherent Raman processes coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) [34].
Among the successful investigations of molecular organizations using polarized imaging, polarized-coherent Raman processes are particularly interesting for their chemical selectivity to molecular vibrational bonds. Polarized CARS has permitted to probe the heterogeneities of nanoscale organization of C–H bonds in lipid structures in myelin sheaths, and their sensitivity to neuro-degenerative diseases in Krabbe disease [35] and multiple sclerosis models [23] (figure 6). Polarized SHG which probes electronic molecular polarizabilities, has brought considerable insight into collagen assemblies for medical read-outs [36], while polarized THG has revealed organization of lipid phase changes at the interfaces of lipid droplets.

Figure 6. Polarized nonlinear microscopy based on sequential tuning of the incident linear polarizations (from [37] with permission). (A) Polarized resolved CARS in the myelin sheath of neurons in fixed spinal cord tissues allows the fine read-out of lipids molecular order. The sticks represent the averaged direction of lipids within the focal volume (phi 2) while their colour denotes the local orientational order of lipids (S2). (B) Polarized resolved SHG in metal nanoparticles of complex shapes. The stick colour (right image) denotes the degree of anisotropy of the local response, which is not visible from a pure SHG intensity image (left image). Reprinted with permission from [37]. Copyright (2017) American Chemical Society.
Download figure:
Standard image High-resolution imageThese methods have been pushed to their ultimate nanoscale resolution, capitalizing on their sensitivity to near-field properties of nanostructures. Sensitivity to local surface effects and plasmon mode symmetry has permitted to evidence the spatial and vectorial properties of local nonlinear radiation sources in metal nanoparticles down to 45 nm size of various symmetry [37] (figure 6), as well as in dielectric nanoparticles of 150 nm sizes. Interestingly, the versatile manipulation of incident light polarizations under high numerical apertures (NAs) has allowed coupling 3D geometries in vertically aligned semiconductor GaAs nanowires, thanks to the manipulation of radial or azimuthal incident polarizations [38]. Despite the considerable added information, these methods are however not yet entirely applicable as a standard for live measurements in complex biological tissues.
Current and future challenges
Live imaging of samples with complex morphology offers both medically useful and complex challenges today to the field of polarized nonlinear imaging. (a) Most of the current approaches using polarized nonlinear imaging use sequential imaging of multiple incident polarization states, which considerably slows down the measurement to the order of the minute time scale. Increasing speed in polarized imaging requires faster modulation devices, which electro-optic modulators can provide. Accessing high speed imaging is in particular required for in vivo measurements, which involve both real time sample motion and live molecular processes to be observed. (b) Another important challenge is the capacity to measure orientations in 3D, and not only projections in the sample plane. Polarization-sensitive imaging generally relies on the manipulation of the excitation polarization state in the transverse plane of light propagation. Molecular assemblies however often expand in 3D both in space and in orientation, in particular in complex biological tissues such as instance neuronal tracks in the mouse brain or spinal cord. Similarly, nano-objects of investigation in nano-optics exhibit 3D structures of complex shapes, which require coupling of light along the longitudinal direction of polarization. Controlling polarization in 2D not only leads to biased information when interpreting molecular or nanoscale organizations, it also considerably decreases the coupling efficiency between incident polarization and the observed structure. Achieving polarized control in 3D requires creating longitudinal polarization components, such as using radial/azimuthal polarization under high NA focussing. (c) When light propagates in-depth in tissues, not only the focus of a microscope objective degrades due to both sample aberrations and pure scattering, but also light polarization is affected. On the one hand, anisotropic samples introduce strong birefringence effects, yet light scattering introduces progressively a strong depolarization of light due to random directions taken by propagation paths in the tissue. While birefringence can be anticipated and partially corrected for via post-processing treatments, depolarization is a random process which cannot be numerically corrected for. Recent progresses in adaptive optics and wavefront shaping offer however interesting tools to manipulate light in complex environments.
Advances in science and technology to meet challenges
Technological advances in imaging are constantly progressing and likewise, polarization resolved imaging benefits from these advances. To increase the speed of polarized imaging, fast polarization modulations up to MHz rates have been obtained with the use of electro-optics modulators (EOM). Combined with lock-in detection, second time scale polarized measurements have been reached in polarized CARS imaging [39]. A similar approach in THG polarized imaging based on line-to-line switching of polarization uses an EOM and detects crystalline particles in zebrafish embryos [40]. This scheme was also recently applied by the same group to dynamic SHG imaging of collagen in skin samples. With adapted electronics and demodulation processes, sub-second time scales should be envisioned. To be able to induce 3D light-molecular coupling and access the third dimension of molecular orientations, novel schemes have been developed based on the manipulation of spatial patterns in phase, amplitude and polarization at the Fourier plane of a high NA focussing objective. Such polarized wavefront shaping, with the use of specific phase plates or spatial light modulators, offer new opportunities for versatile vectorial fields focussing in 3D (figure 7). Spatial phase shaping of a Hermite Gaussian beam was for instance applied for efficient SHG generation in semiconductors 3D nanowires beyond pure radial/azimuthal polarized beams [38]. Another approach has employed specific mirrors' configuration that recombine different phase shifted sectors of a beam in the Fourier plane of the objective [40]. While the characterization of such fields is still an open challenge, they show a high potential accessible via current technologies. At last, it has been shown that recovering a polarized focus can be achieved using wavefront shaping through highly scattering medium, opening to polarized control in-depth in tissues via coherent shaping [41] (figure 7). These advances offer novel directions towards a full control of vectorial beams in complex environments. Such control is moreover a unique opportunity to increase the resolution capabilities of polarized-resolved imaging [42]. Using a combination of these important recent inputs should permit a real step forward for live tissue polarized imaging in the near future.
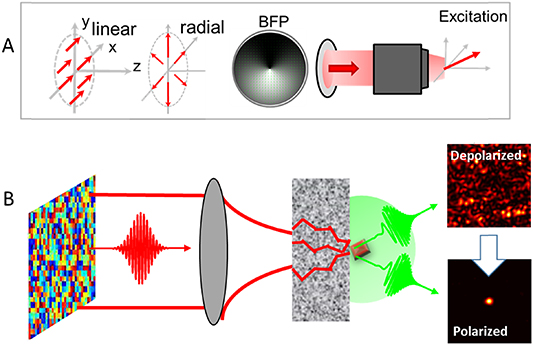
Figure 7. (A) Use of complex focused beams such as combination of radial/azimuthal beams allow the manipulation of 3D vectorial excitations at the focus of a high numerical aperture objective. BFP: back focal plane of the focussing objective. (B) Coherent wavefront shaping in scattering media allows recovery of polarization states of light.
Download figure:
Standard image High-resolution imageConcluding remarks
Optical polarized imaging is rapidly developing thanks to conceptual and technological advances, expanding over many optical contrasts, from fluorescence and super resolution imaging to vibrational nonlinear coherent microscopy. A major asset of polarized microscopy is to be able to reveal molecular organization at a scale below the resolution limit of optical microscopes, which is valuable in many fields from nanosciences and fundamental biology to medical applications. While the developed tools are today demonstrating their potential, they still show important challenges to overcome, in order to explore the full complexity of biological tissues in 3D. Recent concepts and technological steps will nevertheless ultimately permit to include polarization as a routine and complementary tool in microscopy imaging, applicable from living cells to in vivo investigations in small animals. Combined with other modalities, for instance hyperspectral coherent Raman imaging, we expect the approach to expand over new information such as molecular-targeted structural microscopy, in which both chemical/vibrational signatures and molecular orientations can be imaged.
Acknowledgments
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
5. Tracking intracellular trafficking in live cells with optical microscopy
Nicole Slesiona1, Ediz Herkert2, Jan Majer3, Arwyn T Jones4 and Peter Watson1
1 School of Biosciences, Cardiff University, Museum Avenue, CF10 3AX Cardiff, United Kingdom
2 Institut de Ciencies Fotoniques (ICFO), The Barcelona Institute of Science and Technology, Barcelona, Spain
3 GSK, GSK Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY Hertfordshire,, United Kingdom
4 School of Pharmacy and Pharmaceutical Sciences, Cardiff University, Redwood Building, CF10 3NB Cardiff, Wales
Status
The correct transportation of biological components between distinct intracellular compartments is critical for eukaryotic cell physiology and function. Intracellular trafficking involves the packaging of cargo into membrane bound compartments that bud from and fuse with each other and travel on well-defined pathways to specific destinations [43, 44].
Fluorescence based optical microscopy has been the cornerstone of intracellular trafficking research as it allows researchers to simultaneously study the cargo being moved, and the cellular machinery responsible for transport. This has resulted in the characterisation of a multitude of pathways (e.g. endocytosis, exocytosis, secretory transport) regulated by distinct classes of proteins and lipids. Traditionally, fast timelapse fluorescence microscopy (widefield, spinning disk or confocal) has been employed to visualise membrane trafficking in live cells. Specialised imaging techniques allow detailed analysis of trafficking pathways in various situations: total internal reflection fluorescence (TIRF) illumination allows events at the plasma membrane to be visualised with high contrast by restricting the excitation field to a few hundred nanometres beyond the coverslip; light sheet fluorescence microscopy (LSFM) and lattice lightsheet microscopy allows the gentle visualisation of trafficking events in large multicellular structures; the application of live cell super-resolution techniques such as stimulated emission depletion (STED) microscopy enables the imaging of trafficking events at spatial resolution in the tens of nanometres (for an overview see figure 8).

Figure 8. A wide range of optical microscopy techniques can be utilised to study intracellular trafficking, with the challenge often being knowing which technique to utilise. Understanding how to balance signal to noise, spatial and temporal resolution, and sample health is critical in live cell imaging of vesicular transport. IO—illumination objective, CO—collection objective, IO/CO—illumination and collection objective, F—fluorophore, F*—visualised excited fluorophore, MNP—metallic nanoparticle, M—molecular vibrations.
Download figure:
Standard image High-resolution imageAn understanding of intracellular trafficking can elucidate its role in health and disease (reviewed in [44]) but also holds potential for us to exploit these pathways to deliver therapeutics such as nucleotides and proteins to cells [43]. Increasing our knowledge of intracellular trafficking allows us to understand the intracellular pharmacokinetics of molecules when they enter the cell and assists the development of novel drug delivery systems delivering cargo to distinct locations such as the nucleus. Effective trafficking and delivery of large biological molecules such as plasmids encoding therapeutic proteins, siRNA/mRNA, peptides and functional proteins remains a significant clinical challenge despite their therapeutic potential and advances in analysis of their cellular dynamics [45]. The key behind developing more successful therapeutics will be not only an increased understanding of the cell's endogenous trafficking pathways at a higher spatial and temporal resolution, but also knowledge of the vesicular luminal environment.
Current and future challenges
Most fluorescence-based microscopy solutions attempt to balance the need to obtain sufficient signal to noise with resolution and acquisition speed: these components are intrinsically linked, and an attempt to optimise one will usually detrimentally affect the others. With live cell microscopy, additional constraints are imposed as sample health must be of paramount importance to prevent the researcher simply reporting the effects of photodamage on the tested model. This leads to the development of a 'pyramid of frustration' [46] where optical conditions cannot be fully optimised and compromises must be made in order to maintain sample health.
Fluorescence imaging introduces inherent challenges for tracking intracellular trafficking in live cells. To understand the temporal dynamics and interplay of cargo and machinery, multiplexed imaging techniques are crucial: however, fluorophores have broad overlapping spectra that restrict the number of individual components that can be visualised. The presence of a fluorescent moiety can dramatically affect the dynamics and trafficking properties of the molecule/protein under test [47]. Typical super-resolution fluorescence techniques have lateral/axial spatial resolution in the tens/hundreds of nm: still too low to resolve or enable an understanding of the 3D geometry of many trafficking compartments. As spatial resolution improves, the use of fluorescent proteins as localisation tags will becomes less effective as their similar size to their target (3 nm) will results in an inherent uncertainty as to where the attached protein of interest resides in relation to the observed fluorescent signal. As a solution, small fluorescent tags that incorporate directly into proteins are being developed (e.g. by the inclusion of fluorescent non-natural amino acids).
In order to improve delivery of therapeutic cargo through endocytic pathways, additional information is required about cargo experience during transportation within a vesicle in real time (figure 9)—its spatiotemporal dynamics (A) and orientation (B), what proteins/molecules is it interacting with (C), what is the status of its local environment (e.g. pH, ion concentration, chemical composition)(D) and how it is responding to that environment (E). We are clearly moving away from a model where fluorescence imaging alone is able to answer these questions researchers wish to pose, and with the increased use of nanoparticles as therapeutics, we require novel and innovative imaging solutions with which to visualise them.
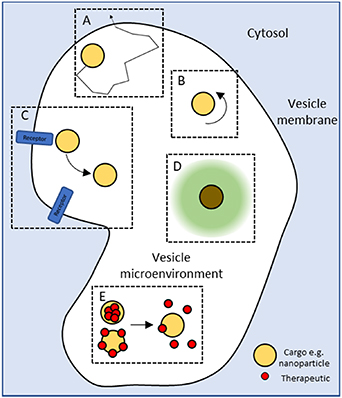
Figure 9. There are still challenges to our understanding of how cargo behaves within an intracellular trafficking compartment—its spatiotemporal dynamics (A) and orientation (B), what proteins/molecules is it interacting with (C), what is the status of its local environment (e.g. pH, ion concentration, chemical composition) (D) and how it is responding to that environment (E). The increased use of nanoparticles within the field presents and exciting opportunity to develop novel imaging techniques capable of addressing these challenges.
Download figure:
Standard image High-resolution imageAdvances in science and technology to meet challenges
MINFLUX nanoscopy utilises concepts from both single molecule localisation microscopy and the donut beam principles of STED, and has demonstrated the localisation of proteins which play key roles in receptor rearrangement [48] with a localisation precision estimated to be 2–3 nm in all three dimensions in live cells. Temporal resolution across increased scan volumes are still the major challenge with images requiring minutes to be produced. Parallelisation of acquisition (multiple scan points) and optimisation of acquisition are now the targets of development to allow the required fast timelapse imaging at nm resolution in live cells required to understand the dynamics of membrane trafficking. Orientation of nanoparticle cargo and their dynamics within vesicles can be studied using parallax-quantified differential interference contrast microscopy [49], dual colour dual particle tracking [50] and four-wave-mixing microscopy [51, 52]. Although spatial (10 s of nm) and temporal (ms) resolution of these techniques is excellent, the sensitivity of the systems still require relatively large (compared to proteins) gold spheres, nanorods or fluorescently labelled latex beads. Advances are needed that can detect NPs an order of magnitude smaller to truly understand the conditions encountered by cargo within an intracellular vesicle. Metallic nanoparticles provide an excellent opportunity to exploit Raman based imaging for sensing applications due to the local field enhancement effect. Surface-enhanced Raman spectroscopy has the potential to chemically define the local environment of a nanoparticle, with a sensitivity enhancement of 1010–1015-fold, pushing the technique to the single molecule detection range (see section 1 in this roadmap). This would potentially allow for sensing of the intravesicle environment, detect cargo-receptor interactions and any changes/release of conjugated molecules. A significant challenge will be the design of nanoparticles able to limit non-specific binding of biological material that form a corona [53], which would prevent local detection of chemical changes. The use of 'Raman tags' such as polyynes or dyes bearing conjugated and isotopically-edited triple bonds, allow advanced multiplex imaging due to their narrow spectral bandwidth. Ten-colour imaging combining Raman active and fluorescence-based organelle probes has been demonstrated in live cells [54, 55]. Invariably sensitivity is the limiting factor of the expanded use of these probes, with the potential for electronic pre-resonant Stimulated Raman Scattering of polyynes one potential solution that could then allow (a) detection of components at low concentrations and (b) the application of super resolution based techniques [55] (Detailed in section 2 in this roadmap).
Concluding remarks
Understanding the complex and dynamic environment within intracellular vesicles remains a formidable challenge for cell biologists. Optical imaging still has a major role to play, however it is clear that the development of techniques capable of reporting on local environmental conditions alongside spatial location will be key to unravelling the cellular mechanisms that are critical for therapeutic delivery and efficacy.
Acknowledgments
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992. We thank the EPSRC (EP/J021334/1) for funding.
6. Label-free multi-modal multiphoton imaging of the tissue and tumour microenvironment
Stephen A Boppart1
University of Illinois at Urbana-Champaign, Illinois, United States of America
Status
Our understanding of cancer and the role that the tumour microenvironment has on carcinogenesis was greatly advanced by seminal papers that described the Hallmarks of Cancer [56]. Historically, advances in science and our understanding of fundamental principles has often been driven by advances in technology that enable new measurements and new data to better elucidate mechanisms and processes. New optical imaging technologies have enabled scientists to visualize bioprocesses at the molecular and cellular scale, and have helped address these fundamental questions. Optical imaging techniques have benefitted immensely with the complementary use of targeted dyes and stains to site-specifically label molecules and cells of interest. However, this ability has often come at the cost of having to visualize largely only ex vivo tissue specimens. Performing in vivo or intravital imaging with exogenous dyes, nanoparticles, or genetically-expressed proteins often present perturbative conditions that at best alter biological function, or at worst, induce toxic by-products and cell death. Therefore, there has been an increasing interest in leveraging the endogenous optical properties and signatures from living biomolecules, cellular structures, or metabolic and dynamic processes that reveal natural behaviour, and potentially offer new label-free biomarkers indicative of disease and pathological processes [57–60].
The current gold-standard for the microscopic evaluation of disease in tissue is histopathology, most commonly by staining thin tissue sections with hematoxylin and eosin (H&E), and in recent decades, with immunohistochemistry (IHC) for more molecularly-specific labelling. This historical process, however, is both time- and labour-intensive, and can only be performed on dead and processed tissues specimens that have been removed from the living organism. While H&E-stained histology preserves structural and architectural features of cells and tissues for diagnosis, it forsakes many of the dynamic, molecular, and metabolic features that undoubtedly could contribute valuable information to elucidate bioprocesses in pathogenesis as well as new biomarkers for disease diagnosis. Label-free multi-modal multiphoton imaging of either fresh, respiring, ex vivo tissues, or even in vivo optical biopsies (figure 10), therefore offer new-found potential for both biological discovery and clinical applications [61, 62].
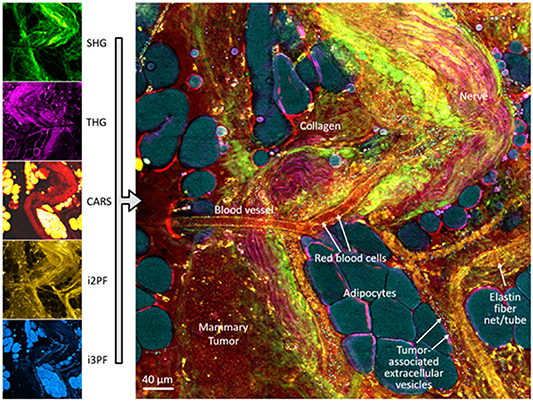
Figure 10. In vivo multimodal multiphoton imaging of the living tumour and tumour microenvironment in a rat mammary tumour model of human disease. Simultaneous acquisition of multimodal channels enables non-perturbative visualization of the collagen scaffold (SHG, green), refractive index interfaces (THG, magenta), lipid and protein content (CARS, yellow/red), distribution of FAD (intrinsic two-photon excited autofluorescence, yellow/brown), and distribution of NAD(P)H (intrinsic three-photon excited autofluorescence, blue). Reprinted with permission and modified from [61]. From [61]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageCurrent and future challenges
One current major challenge in the detection and diagnosis of disease is that we rely largely on the visual detection of patterns in stained tissue sections that pathologists learn, through years of training and specialization, to associate with various diseases and disease processes. While this has served the medical and biological professions well, these steps do not extract the full amount of information available from the tissue and cells, information that is both complementary and additive for biological discovery and medical decision making. Additionally, the time and labour required to process tissue for histology can limit rapid diagnosis for immediate interventions, such as during surgical procedures. To address this challenge, future technologies need to perform optical biopsies in living in situ environments, or in freshly resected but still respiring tissues. The iconic microscope slide can be replaced by a 'living slide' or microfluidic chamber in which freshly biopsied tissues can be maintained or cultured to remain viable for hours after removal, allowing sufficient time to acquire new data on cell and tissue metabolism and dynamics, or preserving cell constituents such as extracellular vesicles that can be used as biomarkers of disease and disease aggressiveness [63].
A second major challenge is maximizing the amount of information that can be extracted from tissue without the introduction of dyes or stains that would subsequently alter viability and the bioprocesses of interest. The wide range of dyes and the targets they can label provide a high level of insight into tissue and cell composition, which is currently unmatched by the more limited number of label-free optical signatures. However, multimodal multiphoton imaging, particularly if multiple channels are collected simultaneously in both time and space, enable perfect co-registration for assessment of redox ratios for metabolic activity, as well as new computational approaches that exploit the mathematical relationships that exist between the physical mechanisms of each of the nonlinear laser-tissue interactions.
A third major challenge is how to both expand on the pattern recognition of stained tissue sections that have historically driven the field of pathology for diagnosis, as well as integrate the complex multi-dimensional multimodal data that is becoming increasingly more accessible. Here lies the value of artificial intelligence (AI) and machine-learning (ML) and deep-learning (DL) algorithms that with training, can be used to recognize complex patterns that often evade the human eye and brain. AI algorithms will also drive a shift from the subjective interpretation of microscopic image-based data, often coming from several pathologists reviewing the same slides to reach a consensus, to the objective interpretation of images based on training sets with known ground-truths and hundreds of thousands of images with known patient prognoses and disease outcomes.
Advances in science and technology to meet challenges
There is an increasing focus on developing label-free multiphoton imaging technologies to address the current and future challenges raised. Without the need for dyes, nanoparticles, probes, or labels, real-time in vivo optical biopsies are possible, and can be more directly integrated into human clinical studies without the need for and delay of lengthy drug-approvals and certifications. The acquisition of a relatively limited number of label-free optical signatures can be addressed by the development of multimodal imaging systems that can, within a single shot, excite and collect multiple co-registered optical signatures that can be directly utilized by AI/ML/DL algorithms for automated classification, disease detection, and diagnosis (figure 11) [62, 64].

Figure 11. Classification of human breast cancer by a deep neural network. (Top row) Multimodal multiphoton composite images of four channels (THG, 3P-NADH, SHG, 2P-FAD) for two cancer and two normal label-free images. (Middle row) Corresponding classification results, identifying regions of cancer, lipids, and non-cancer areas. (Bottom row) AUC statistics for different model architectures and tile sizes, along with representative examples of false positive and negative images. Scale bar represents 200 μm. Figure reprinted with permission from [64]. Reproduced from [64]. CC BY 4.0.
Download figure:
Standard image High-resolution imageAdvances in optical science and source technology is required, both to generate new wavelengths and ultrafast pulse properties including spatial, spectral, and temporal pulse shaping, as well as new laser and light source platforms that are compact, portable, robust, and reliably stable even under the harsher clinical environments, compared to the controlled optics lab. Such laser sources and portable systems will enable label-free nonlinear optical imaging at the point-of-procedure, such as in the operating room or needle-biopsy procedures to assess freshly resected tissue specimens for the presence of tumour, for alterations of the tumour microenvironment, or for the presence of new biomarkers including extracellular vesicles [65].
New AI/ML/DL algorithms will also learn pattern relationships between the imaging modalities themselves, so that the collection of image data by one modality alone may predict the image or contrast features of others. Whereas IHC is now used to stain specific molecular receptors for their presence, density, and spatial distribution, and correlate these to disease presence and severity, it should be possible to predict the presence and spatial distribution of these molecules based on the multimodal multiphoton image features that are collected from fresh unstained tissue for stain-free slide-free molecular histopathology.
Finally, there must be concomitant adoption and integration of this new high-dimensional multimodal image-based information derived from these nonlinear tissue interactions. While H&E- and IHC-stained histopathology serve as the gold-standard, they do not reveal or even predict the new image features and biomarkers obtained from many of the nonlinear imaging modalities. The fresh or in vivo tissue and tumour microenvironment contain a wealth of new data and information that has often been missed by our gold standards and overlooked.
Concluding remarks
The convergence of advances and new technologies in optical science and engineering, in label-free multimodal multiphoton imaging systems, and in AI algorithms that feed on high-dimensional image-based information, will undoubtedly lead to a deepening of our biological understanding of carcinogenesis, and new cancer detection, diagnosis, and monitoring paradigms. The ever expanding and increasingly available computational power will not only drive more sophisticated and smart AI or augmented intelligence systems in medicine, but will also enable the integration of data from genomics, from bioassays and biosensing, and from physical exam, laboratory, and radiological data sources. Collectively, the convergence of these revolutionary changes will challenge the gold-standard microscopic assessment of disease with complementary or even replacement imaging technologies and techniques that will ultimately serve to improve the diagnosis of disease and the human condition.
Acknowledgments
I would like to thank all of the researchers from the Biophotonics Imaging Laboratory, the Centre for Optical Molecular Imaging, and our clinical partners who have contributed to this direction of research. This research was supported in part by grants from the National Institutes of Health (1R01CA213149 and 1R01CA241618). Additional information can be found at http://biophotonics.illinois.edu.
7. Cryo-micro-spectroscopy of single molecules and quantum dots
Vikramdeep Singh1, Saurabh Borkar2, F E Quintela Rodriguez3 and Wolfgang Langbein1
1 School of Physics and Astronomy, Cardiff University, The Parade, CF24 3AA Cardiff, United Kingdom
2 ICFO-Institut de Ciencies Fotoniques, The Barcelona Institute of Science and Technology, Barcelona, Spain
3 Dipartimento FIM—Università di Modena e Reggio Emilia, Italy
Status
Interrogating single quantum emitters is enabling to pose a range of unique and unprecedented questions, and to develop novel applications of such emitters in optical quantum technology (QT) as single photon sources. The local interaction of such emitters with the environment is sometimes a nuisance, for example when striving to create on-demand indistinguishable photons, and at other times a resource, providing information about the local environment with the emitter serving as nanometre-sized probe.
While much work has been dedicated to single molecule studies at ambient temperature, most prominently in the life sciences to investigate the structure of cells—via single molecule super-resolution techniques, the fast thermal motion and the strong interaction with the thermally fluctuating environment limits the detail with which single molecules can be investigated, as there is not enough observation time before the molecule is affected by the environment.
At cryogenic temperatures, the dynamics and interaction processes slow down enormously, thus much more detailed observation is possible before the emitter is perturbed. Individual molecule dynamics can be unraveled for testing of theoretical molecular quantum electrodynamics, microscopic theories of coupling, and understanding of inherent spectral inhomogeneity of such a system in complex environments, which is not accessible in ensemble measurements. Furthermore, single emitters trapped in a solid host matrix are shielded from environmental fluctuations, including from encountering reactive species such as oxygen, which is enhancing photo-stability.
First reports of single molecule spectroscopy at cryogenic temperatures [66, 67] were followed by investigations of the influence of the environment, such as vibrational decay [68], and hosting local two-level systems influencing the zero-phonon line shape [69]. Using single molecules as two-level systems for QT has motivated the investigation of their non-linear properties [70]. Highly sensitive energy tuning via external electric fields in novel host-guest system, enabled by a symmetry breaking of host, is promising for probing weak electric fields [71]. Placing such molecules into a narrow-linewidth and small optical cavity, their radiative rate was enhanced by a Purcell factor of 40 [72], resulting in a system at the onset of strong coupling. Similar, yet more stable and electrically tunable systems made of semiconductors with embedded quantum dots as emitters, even reached deep strong coupling [73].
The photo- and mechanical stability at low temperatures allows for very high resolution in localization microscopy. A resolution of 0.5 nm has been reached at 4 K [74], and used to investigate 3D protein structure, exploiting multiple fluorophore labelling.
To increase the spontaneous emission rate of fluorophores, and thus their observability and photostability, one can exploit the Purcell effect. This requires a modification of the local photon density of states (LDOS), which can be achieved by some form of antenna structure close to the emitter of interest. What is 'close' depends on the principle used by the antenna. For resonators with a small spectral linewidth, such as dielectric cavities (see sketch Fig. 12(b)), as in [72], the size of the enhancement region (the mode volume) can be larger than the wavelength, as this type of antenna uses a concentration of the LDOS by redistribution in spectral domain. Therefore, they can be applied without affecting the nanometre scale surrounding of the emitter. The small spectral width requires spectrally narrow emission lines, which can be achieved at cryogenic temperatures. Antennas which are concentrating the LDOS by redistribution in space, such as plasmonic structures [75, 76] (sketch Fig. 12(c)), can be broadband, and thus effective also for room temperature emission. However, the emitter must be situated at sub-wavelength distance to the antenna, which limits the experimental scenarios where it is applicable. The higher the Purcell factor, the closer the antenna—factors of 1000 can be reached at distances below 10 nm.
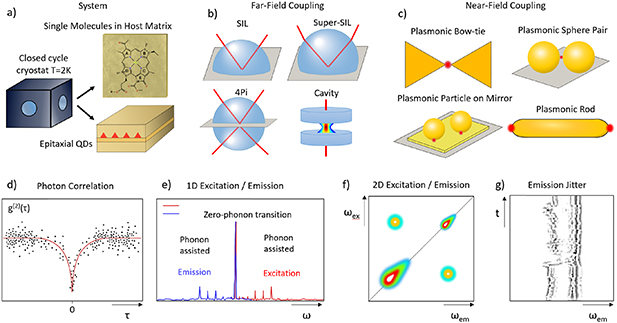
Figure 12. Sketches of the various methods discussed in the text.
Download figure:
Standard image High-resolution imageIndependent of Purcell enhancements, maximizing collection and detection efficiencies is important to improve measurement speed and provide feasibility. Typical approaches today (sketch Fig. 12(b)) use high numerical aperture (NA) objectives for collection, paired with suited solid immersion lenses of hemisphere or Weierstrass type. Additionally, so-called 4Pi collection, using opposing objectives, can be applied, to reach collection efficiencies above 50%.
Presently used detectors can be imaging arrays, or single channel detectors. Imaging arrays mostly use silicon technology, sensitive in the 400–1000 nm wavelength range. CCDs (charge-coupled devices) , CMOS (complementary metal–oxide–semiconductor) , and EM-CCD (electron multiplying CCD) arrays are available, with detection quantum efficiencies of typically 50%–90%. Deep Peltier cooling to −100 °C reduces dark current, and 1e per hour per pixel can be reached as well as a read noise of 1e. Electron multiplication allows for photon counting with these cameras, providing negligible read noise. For time-resolved detection, photomultipliers or avalanche photodiodes can be used in photon counting mode, with time resolutions of around 100 ps. Superconducting nanowire detectors achieve faster time-resolutions in the 10 ps range and can be sensitive at longer wavelength, but need cooling to liquid helium temperatures.
Spectroscopy techniques—the early experiments [66, 67] used tunable single frequency lasers to achieve a spectral resolution in the sub-µeV range suited for single molecules at low temperatures, and this is still a method of choice. Either excitation spectroscopy (sketch Fig. 12(e)), detecting the emission, or pump-probe spectroscopy, detecting the non-linear response of the molecules, have been employed. The emission can be spectrally resolved using a dispersive spectrometer and an imaging array [77], allowing to measure many molecules at once. Fourier transform techniques can also be used to achieve spectral resolution, either in excitation [78, 79], or in emission [80]. These can be combined with photon correlation techniques (sketch Fig. 12(d)) to measure lifetimes, photon anti-bunching, or photon indistinguishability.
Multidimensional coherent spectroscopy (sketch Fig. 12(f)) allows to investigate internal coherences, dephasing and density lifetimes of multiexcitonic states [81]. To enable to use such spectroscopy, heterodyne spectral interferometry [82] can be employed, which allows to distinguish the non-linear coherent response of different orders and detects the spectrally resolved signal in amplitude and phase. It has been used to detect coherent coupling [83], the quantum strong coupling between a single emitter and a cavity [84], and cavity induced coherent coupling between single emitters [85]. More recently, improved phase referencing was used [86] to identify coherent coupling between single emitters via different coherent pathways.
Current and future challenges
The paramount challenges in cryo-micro-spectroscopy of single emitters are:
- (a)Cryo-environment: Mounting and positioning the sample at low temperatures, with efficient optical excitation and collection, is a technical challenge. The simplest setup is to place the collection optics at room temperature, and only cool the sample in a vacuum environment [80, 86]. This limits the numerical aperture (NA) and thus the collection efficiency. Short distances to room temperature elements affect the sample temperature due to radiation heating for temperatures below 50 K. Additionally, spatial drift between sample and collection optics is an issue. Using helium bath cryostats, and inserting the collection optics (aspheres or simple multi-element objectives) at low temperatures avoids temperature and NA limitations [66, 67, 69, 70, 82], and allows to use superfluid helium to reach 1.4 K. It also suppresses image drift, and dedicated reflective optics have been developed [87]. However, these cryostats require a local liquid helium supply and are laborious to handle.
- (b)Background: Since the typical observation volume even at the optical diffraction limit of visible light is around a femto-litre, that is 109 nm3, and single molecules are in the size range of 1 nm, selecting a single molecule is challenging. The simplest way is to reduce their density so that there is only a single molecule per volume, which is the approach typically used. Additionally, at low temperature, the sharp spectral response combined with the inhomogeneous distribution [68, 77] due to the varying local environments allows to use spectral selectivity, given by the ratio of inhomogeneous to homogeneous broadening, typically 102–105. Background is a problem even if subtracted since its shot noise remains for incoherent detection. For the linear coherent response, scattering by dielectric disorder in the surrounding provides a background. It is therefore an art on its own to embed the emitters into a 'clean' environment. For some studies, the density of emitters is not adjustable, as they form part of the physics to be investigated, for example when investigating coherent coupling between molecules in light-harvesting complexes (LHCs).
- (c)Photostability: To obtain useful information, much more than one photon probing the single emitter is required. The measurements are taken as time average, during which the emitter needs to be stable. One instability is photochemistry since the optical excitation provides sufficient energy to trigger a chemical reaction destroying the emitter. Another instability are long term fluctuations of the emitter and its local environment, such as charging. While this spectral diffusion (sketch Fig. 12(g)) can be a subject of investigation in its own right, it leads to fluctuations of the spectral position, for example by the electric field of the charges, or even the spectral structure, by charging of the emitter, or conformational changes of the molecule.
Advances in science and technology to meet challenges
The challenges identified are addressed by
- (a)Cryo-environment: In the last decade, closed cycle cryostats (sketch Fig. 12(a)) have become available [88, 89] with low vibrations, and temperatures down to 2 K, suited for microscopy. They can be operated continuously without user intervention and make cryo-microscopy more accessible. Piezoelectric positioning at low temperatures enables sample alignment.
- (b)Background: Apart from the development of 'cleaner' samples, which is ongoing and very specific to the particular system investigated, there is the option to reduce the probe volume. Plasmonic antennas (sketch Fig. 12(c)) can reduce probed volumes by some 7 orders of magnitude, down to 10 nm3 [76], which is effectively eliminating the background from other molecules. Background by the antenna itself is still present. Furthermore, fabricating and positioning such antennas is an ongoing challenge. A relatively simple way to produce such antennas which can embed single emitters is the use of colloidal gold nanospheres, with a self-assembled molecular spacer to another nanosphere [90], or a flat gold layer [91]. More work is needed here to make such constructs mainstream and reproducible, but they are promising due to the chemical stability of gold, combined with the availability of single-crystalline spheres and rods [92] forming stable flat facets.
- (c)Photostability. How we can get more photons interacting with a given emitter within its stability time? Using engineered emitters and environment is solving this issue, as seen for semiconductor quantum dots [73]. Otherwise, for a given emitter, such as a biological molecule of interest, there are two options. Using the Purcell effect, one can increases the emission rate of photons, and accordingly the number of photons extracted within a given timescale. Plasmonic structures can provide Purcell factors of 102–106, making single emitter spectroscopy easy. However, the faster emission can affect the very process one wants to study, such as the energy transfer in LHCs. The second option, much less explored, is to use the concept of quantum non-demolition (QND) measurements [93]. The sharp linewidths at low temperatures allow to probe the molecular transitions off-resonantly, reducing the amount of excitation of the molecule per information extracted [72].
Non-linear multi-dimensional optical methods to study coherence pathways presently require real excitations of the system. QND approaches to such methods are conceivable, lending concepts from adiabatic transfer. Another approach to non-linear spectroscopy with reduced real excitation is the use of correlated photons in the excitation [94], which can enhance non-linear pathways for a given excitation budget.
Concluding remarks
Cryo-micro-spectroscopy of single emitters is poised to expand its scope over the next decade to become a wide-spread method to investigate single quantum systems. Apart from the technological applications for triggered indistinguishable photon sources and single photon switches, it lends itself to the investigation of biological systems with very high spatial resolution, exploring correlations and quantum effects.
Acknowledgments
The authors acknowledge funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
8. Ultrafast molecular dynamics of single molecules at room temperature
Saurabh Borkar1, Vasilis Petropoulos2, Vikramdeep Singh3, Frank E. Quintela Rodriguez4 and Niek F. van Hulst1,5
1 ICFO—Institut de Ciencies Fotoniques, The Barcelona Institute of Science and Technology, Barcelona, Spain
2 IFN-CNR, Dipartimento di Fisica, Politecnico di Milano, Piazza Leonardo da Vinci 32, I-20133 Milano, Italy
3 School of Physics and Astronomy, Cardiff University, The Parade, CF24 3AA Cardiff, United Kingdom
4 Dipartimento FIM—Università di Modena e Reggio Emilia, Italy
5 ICREA—Institució Catalana de Recerca i Estudis Avançats, Pg. Lluís Companys 23, Barcelona, Spain
Status
Light matter interaction at the nano-scale is being studied by various groups to address spectroscopic response at dimensions below the diffraction limit or even at the level of individual nanoparticles or molecules. The field of single molecule detection has experienced tremendous developments after the first detection by optical means by the groups of Moerner and Orrit [66, 67] in the early 90s. Ever since the demonstration by Orrit et al [67] of detection of a single molecule using background-free fluorescence detection under cryogenic conditions, the detection based on fluorescence has dominated. Cryogenic conditions provide high photo-stability and large absorption cross-section, allowing the recording of both high-resolution excitation and emission spectra [95] of individual molecules over long observation times. Yet, the study of molecules at ambient room temperature is crucial to the vast majority of chemistry and biological applications. The higher temperature is accompanied by broadened absorption lines, reduction in absorption cross section and photo-stability. As a result molecules bleach abruptly after ∼106 photo-cycles, making sensitive fluorescence based detection much more demanding. Many ultrafast processes, like electronic dephasing, excitation energy transfer and vibrational dynamics, all occur on a femto- to picosecond timescale, and in practise are all faster than the fluorescence lifetime. As a result, the conventional fluorescence based single molecule detection is not compatible with modern ultrafast pump-probe spectroscopy, preventing ultrafast processes to be uncovered at the level of an individual molecule. Moreover the spontaneous emission is incoherent, again unsuited for most ultrafast nonlinear approaches. Thus alternative pump-probe schemes, exploiting the use of stimulated transitions, either in absorption or emission, are needed to carry ultrafast spectroscopy to the realm of single molecules (figure 13).

Figure 13. Conceptual scheme for ultrafast spectroscopy on a single molecule. The molecule is excited by the pump pulse: stimulated absorption. The molecule is de-excited by the probe pulse: providing stimulated emission, ground state depletion or excited state absorption. The excited state population change is read-out by fluorescence (depletion). Alternatively the minute change in probe pulse intensity is detected (stimulated emission). Excitation or de-excitation spectroscopic data can be acquired by Fourier spectroscopy of time delayed broad band pump or probe pulses to acquire 2D excitation-emission (depletion) spectra.
Download figure:
Standard image High-resolution imageCurrent and future challenges
The principle of conventional pump-probe spectroscopy is based on the use of short pulses to optically pump a molecule to a particular excited state, followed by another delayed pulse which probes the evolution of created population (figure 14(a)). A molecule in an excited state can be stimulated to the ground state by stimulated emission (SE), resulting in emission of an extra identical photon, causing in increase of the probe beam intensity. Alternatively, ground state depletion, when both pump and probe are resonant with the absorption band of molecule, will also increase the probe beam intensity. Finally, any excited state absorption will decrease the probe signal. The detection of probe signal changes in transient pump-probe spectroscopy essentially relies on absorption contrast. Unfortunately, the absorption cross-section of a single molecule at room temperature is smaller than 1 N m2. Thus, even for illumination with a diffraction limited spot, the presence of a single molecule will change the probe intensity by less than 10−6, below the noise level of conventional detection. The Sandoghdar group presented an ultrasensitive balanced photodetector technique to detect the 10−6 contrast of a single molecule in absorption at room temperature [96]. The Orrit group showed that the single molecule's absorption detection can be enhanced by photothermal contrast [97] or that the subtle index change by the presence of a molecule can be perceived using the surface plasmon resonance of a gold nanorod [98]. Yet, despite these ultrasensitive, non-fluorescence, detection advances, the resulting signal/noise is too low for any ultrafast experimental application. Raman scattering seems an attractive alternative since it is a fast process, largely background-free, and unlike fluorescence all molecules do show Raman response. Unfortunately, the cross section for Raman scattering is yet another 1012 times smaller and single molecule Raman detection requires huge enhancements by metal nanoparticles, with the molecules in tiny nanogaps.
Figure 14. Pushing detection of single emitters to the ultrafast coherent and non-linear regime. (a) Schematic of transient pump-probe spectroscopy signal contributions: stimulated emission (SE), ground state bleach (GSB) and excited state absorption (ESA); (b) Spectroscopy scheme for CdSe-core/CdS-shell quantum dots: two photon excitation is followed by stimulated emission and photoluminescence detection. (c) Confocal photoluminescence image of individual rod-in-rod quantum dots, and (d) corresponding stimulated emission image showing the tiny (∼10−5) modulated probe pulse contrast for a positive time delay after the pump pulse. Mind the subtle differences between (c) and (d). (e) Non-linear pump-probe spectroscopy can track the electronic excited state vibrational decay of a molecule. (f) Fluorescence of single molecule, excited by a pump pulse, is depleted by a probe pulse, depending on the pump-probe time delay. The depleted fluorescence reveals the fs-ps development of the excited state de-excitation spectrum, when depleted with a phase-locked de-excitation probe pulse-pair. Thus the excited state vibronic dynamics for a single fluorescent molecule is retrieved. Adapted from [99, 100] with author´s permission. From [99]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageThus, Stokes-shifted fluorescence detection is still unbeaten as the most candid way to enable background free efficient detection of an individual molecule. One can only visualize world of fluorophores with sufficiently high quantum efficiency. Moreover, the photobleaching probability is ∼10−6 per photocycle at room temperature, resulting in only few seconds for observation. Altogether this results in limited fluorescence photon counts, incoherent and with a slow ns fluorescence lifetime, not ideal for fs-ps dynamics. Still the single molecule fluorescence is a good observable to read-out the excited state population, while the ultrafast dynamics can be explored on the (stimulated) absorption side. Indeed the first ultrafast responses of single molecules were reported by excitation with multiple delayed fs pulses, while relying on slow fluorescence photon counting detection. Using a delayed pulse pair the ultrafast effect on the saturation, and competition between the stimulated transitions, absorption and emission, was observed on a single molecule [98]. Using ultra-short broad-band pulses, spanning the full molecular absorption band, the presence of vibronic sidebands was observed in temporal beats in the time delay response [101]. However, exciting in the linear regime, the delayed pulse scheme comes down to a time domain Fourier interferometry probing of the excitation spectrum. Thus, while effective to probe excitation spectra of single molecules at room temperature, no ultrafast information is gained [102]. In fact, by scanning the narrowband excitation wavelength derived from a white-light continuum the excitation spectrum is equally obtained, yet incomplete due to blinking and bleaching of the molecule [103]. The ultrafast broadband scan is more robust against blinking as the full spectral information is collected all the time, while resolution builds up with delay scan [104].
Advances in science and technology to meet challenges
Detection of ultrafast single molecule interaction and response remains a challenge. Linear single-photon excitation is not sufficient. Schemes that involve multi-photon interaction, saturation or coherent detection are needed. At the same time the limited photo-stability puts severe limits on the non-linear schemes. Also excitation/detection of single molecules relies on tight focussing with very high numerical aperture objectives, introducing many mm of glass with strong dispersion. Compensating second and third order dispersion can be accomplished by using pulse compressors. Use of a reflective objective avoids the dispersion all together. Finally, for efficient ultrafast single molecule spectroscopy a proper spatio-temporal alignment is required, with pump and probe coinciding both in time and space. Given these challenges, advances in fs-ps single molecule research have so far been very limited.
Ultrafast dynamics of molecules mainly involves the excited electronic state. In a pump-probe experiment the excited is populated by stimulated absorption and depleted by SE. The competition between the stimulated photon interactions (absorption and emission) reveals the ultrafast response. Thus the SE needs to be part of the single molecule detection scheme. Beyond detection in absorption and fluorescence, one can try to detect directly the SE. Every molecule, also non-fluorescent molecules, exhibits SE and moreover the response is coherent. Yet the SE photon provides only a weak contrast on top of the stimulation probe pulse, at best ∼10−6, similar to the contrast absorption. Alternatively one can detect the SE through the depletion of the fluorescence. The fluorescence detection is more effective, yet the stimulating depletion probe pulse will rapidly lead to photo-dissociation. Here we review examples of both approaches.
SE detection
Direct SE detection of singles was recently demonstrated by Piatkowski et al on colloidal CdSe/CdS rod-in-rod quantum dots, not molecules [99]. Quantum dots have a larger absorption cross section, providing contrast of ∼10−5. The experiment involves exciting the quantum dot using a modulated pump pulse. The excited electron–hole decays to the lowest excited state, injecting a second delayed probe pulse leads to charge recombination and the quantum dot is stimulated back to the valence band, emitting a stimulated photon. A lock-in amplifier is used to measure the modulation enhanced probe signal. These photons contain information about excited state population and relaxation dynamics. The photoluminescence is detected simultaneously with the SE. This scheme is coherent, independent of quantum efficiency, has a femtosecond temporal resolution and can map the relaxation dynamics of any excited state. Figures 14(c) and (d) show the simultaneously recorded photoluminescence and SE (modulated signal) images of single quantum dots, respectively. The modulated response is only detected for positive pump-probe time delay. Note that some particles show only luminescence, or only SE contrast [99]. The next step forward would be to extend this approach to organic single molecules instead of semiconductor quantum dots.
Non-linear approach
Alternatively, the depleted fluorescence is detected, providing an excited state transient de-excitation spectroscopy, as demonstrated by Liebel et al on individual dibenzoterrylene (DBT) molecules embedded in a crystalline anthracene matrix [100]. This method is based on three-pulse scheme with fluorescence detection, where the first pump pulse provides the excitation, while a delayed phase-locked pulse-pair exerts the de-excitation (figure 14(e)). The transient change in fluorescence as a function of pump-probe delay shows the depletion at positive time delay (figure 14(f)). The spectral development can be retrieved by Fourier transform spectroscopy using a phase-locked de-excitation pulse-pair. Figure 14(f) shows Fourier spectral response with increasing fs-ps time delay. Fourier transformation reveals the ultrafast spectral evolution of the individual DBT molecule [100]. As a road ahead, the next step would be to extend this technique of three pulses to four pulses, ultimately towards single molecule 2D electronic spectroscopy. This should unravel the energy transfer processes and the role of quantum coherence further at the level of single molecules.
Concluding remarks
We presented an overview of recent advances in detection of single molecule ultrafast dynamics. At a fundamental level, these techniques provide a platform to investigate the molecular dynamic under the conditions which previously seemed to be unattainable. With SE, molecular processes can be probed directly and instantaneously without relying on the slow detection via fluorescence. This comes at a price of signal-to-background ratios of around 10−7 that needs to be compensated. The nonlinear approach of transient absorption addresses both ground and excited electronic states by spectral shaping. Tracing of femtosecond evolution of excited electronic state spectra of molecules reveals their non-linear ultrafast response in an effective three-pulse scheme.
Clearly, signal/noise and photon budget are challenging the progress in these ultrafast experiments, requiring even more ingenious detection schemes to move ahead.
Acknowledgments
S B thanks L Bolzonello, P Woźniak and Matz Liebel for stimulating discussion. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992 (ITN project MUSIQ).
N F v H acknowledges the financial support by the European Commission (ERC Advanced Grant 670949-LightNet), the Spanish Ministry (PGC2018-096875-B-I00 and CEX2019-000910S), the Generalitat de Catalunya (CERCA program and AGAUR 2017SGR1369), Fundacioé Privada Cellex and Fundacioé Privada Mir-Puig.
9. Two-dimensional electronic spectroscopy of light-harvesting systems
Vasilis Petropoulos1, Vikramdeep Singh2, F E Quintela Rodriguez3, Saurabh Borkar4, Margherita Maiuri1 and Giulio Cerullo1
1 IFN-CNR, Dipartimento di Fisica, Politecnico di Milano, Piazza Leonardo da Vinci 32, I-20133 Milano, Italy
2 School of Physics and Astronomy, Cardiff University, The Parade, CF24 3AA Cardiff, United Kingdom
3 Dipartimento FIM—Università di Modena e Reggio Emilia, Italy
4 Institut de Ciencies Fotoniques, The Barcelona Institute of Science and Technology, Barcelona, Spain
Status
Ultrafast multidimensional spectroscopy uses sequences of ultrashort laser pulses to reveal multivariable correlation functions of matter [105]. In two-dimensional (2D) spectroscopy the system under study interacts with three distinct and time-delayed ultrashort pulses and the generated third-order nonlinear polarization emits a signal which is fully recorded in amplitude and phase by optical heterodyning with a fourth phase-locked pulse, the so-called local oscillator (LO). Pulse 1 drives a macroscopic linear polarization in the system, then a second phase-locked pulse, delayed by the coherence time t1, puts the system in a population state. After evolution along the waiting time t2, pulse 3 reinstates the system to a coherent state, generating the third-order nonlinear polarization P(3)(t1,t2,t3) which follows pulse 3 with a delay t3 and emits the nonlinear signal E(3)(t1,t2,t3), retrieved by spectral interference with the LO (figure 15(a)). For a fixed value of t2 and by performing Fourier transforms with respect to t1 and t3, one obtains the 2D map S(ω1,t2,ω3), where ω1(ω3) is the excitation(detection) frequency.

Figure 15. (a) Pulse sequence used in a 2D optical experiment. (b), (c) Experimental configurations used for 2D spectroscopy: fully non-collinear 'boxcar' geometry (b) and partially collinear pump-probe geometry (c). (d) 2DES maps at t2 = 40 fs for the FMO complex for AP (0°, 0°, 0°, 0°) (left) and DC (45°, −45°, 90°, 0°) (right) polarized conditions. Adapted from [106]. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, Nature Chemistry, [106], Copyright © 2018, The Author(s).
Download figure:
Standard image High-resolution image2D spectroscopy was first developed in the infrared, targeting vibrational transitions (2DIR) [107]. The technical challenge of generating phase-locked pulses at short visible wavelengths has delayed the advent of 2D electronic spectroscopy (2DES). 2D spectroscopy resolves the signal both in the detection frequency ω3, as in conventional transient absorption (TA) spectroscopy, and in the excitation frequency ω1, while simultaneously maximizing temporal and spectral resolution [108]. In a 2D map, the diagonal peaks at t2 = 0 allow the determination of the homogeneous and inhomogeneous broadening, given by their anti-diagonal and diagonal widths, respectively. At later times, system-bath interactions lead to energy gap fluctuations that broaden the anti-diagonal width, a process called spectral diffusion. The cross peaks at t2 = 0 characterize the couplings between different transitions of the system. By tracing cross peaks as a function of t2, one can monitor in real time correlations between excitation and detection frequencies due to energy/charge transfer pathways [107].
The two main experimental configurations used to perform 2DES are the fully non-collinear 'boxcar' geometry (figure 15(b)) and the partially collinear (pump-probe) geometry (figure 15(c)) [109]. The major advantages of the boxcar geometry are the background-free signal detection, the possibility to discriminate rephasing and non-rephasing signals and the ability to control the polarizations of the individual pulses. Its major drawbacks are the experimental complexity and the requirement of interferometric stability of two pairs of pulses (pulse 1 with pulse 2 and pulse 3 with the LO). On the other hand, the partially collinear geometry is easier to implement, as it is a natural extension of TA in which the pump pulse is replaced by a phase-locked collinear pulse pair. However, it suffers from the congestion of desired and undesired signals emitted in the same direction. The rapid progress of pulse shaping technologies has allowed the isolation of the desired signals, making 2D spectroscopy widely accessible [110].
Following the milestone demonstration of the mapping of excitonic structure and energy transfer pathways in the Fenna-Mathew-Olson (FMO) complex [111], 2DES established itself as a powerful tool in photosynthesis research. It has been used to track energy transfer pathways in light harvesting (LH) complexes [112], reveal dark excited states in carotenoids [113] and address long-standing questions regarding quantum coherences [114, 115] and photoprotection mechanisms [116] in LH systems.
Current and future challenges
The advent of 2DES spectroscopy has provoked hot debates in the photosynthetic community concerning the presence of electronic coherences in LH complexes and their role in controlling excitation energy transfer (EET) pathways. Coherences are defined as the off-diagonal elements in the density matrix in the basis of system eigenstates. Such elements denote that the system is in a linear superposition of different eigenstates [115]. In photosynthetic complexes the excitation is delocalized over a small number of pigments and is described in terms of excitons [117]. Coherences in the excitonic eigenstates of the system are nonstationary states manifested as damped oscillations in the time domain, with frequencies corresponding to the energy differences between the involved eigenstates.
During the last decade, the observation of long-lived oscillating signals, the so-called quantum beats (QBs), in the cross peaks of room-temperature 2DES maps in photosynthetic systems, attracted considerable interest in view of their role in EET mechanisms. Initially these signals were assigned to electronic coherences, in contrast to the expected short electronic dephasing times caused by the system-bath interactions, and it was speculated that these coherences may support efficient EET via a wavelike motion rather than an incoherent hopping from site to site [118, 119]. The proposed mechanism, which would allow the survival of long-lived electronic coherences, relied on correlated motions of the protein matrix enclosing the chromophores [120]. An alternative explanation for the observed QBs assigned them to impulsively excited vibrational coherences of the chromophores, at frequencies matching the differences between excitonic eigenstates energies.
To address this problem, Thyrhaug et al recently performed polarization-controlled 2DES in the FMO complex at cryogenic temperatures [106]. Figure 15(d) presents the 2DES maps for all-parallel (AP) and double-crossed (DC) polarizations. The AP maps display intense diagonal signals corresponding to the excitonic contributions, while the DC maps are dominated by cross peaks, revealing correlations between transitions. Simultaneous analysis of the entire set of 2DES maps can lead to characteristic patterns [115] that enable one to distinguish among vibrational, electronic and vibronic (mixed electronic and vibrational) coherences. The data unambiguously show that the long-lived coherences are of vibrational origin, while the electronic coherences are short-lived and dephase within 240 fs. Similar results have been obtained in a recent room temperature 2DES study on FMO, showing dephasing of excitonic coherences on the 60 fs timescale [114]. These studies point to a negligible role of electronic coherences in photosynthetic light harvesting. The future challenge of polarized-2DES is to identify vibronic coherences in photosynthetic complexes that may affect the EET mechanism, as suggested in recent studies [121, 122].
Advances in science and technology to meet challenges
One of the limitations of conventional 2DES is that the signals represent an ensemble-averaged response, lacking spatial resolution. The combination of 2DES with confocal microscopy is highly promising for the understanding of natural and artificial photosynthetic complexes, but it has been so far unachievable due to the difficulties in separating the different nonlinear signals and to photobleaching of biological samples [110]. Nowadays, the ability to generate a train of collinear phase-locked pulses and control their relative phases via phase cycling or phase modulation techniques, could overcome such limitations. An alternative approach to include spatial resolution in 2DES experiments comes from fluorescence-detected 2DES (F-2DES) [123, 124], which is more sensitive and requires less pump power.
F-2DES uses a fourth pulse to convert the third-order coherence to a population state, which is measurable via spontaneous emission. Instead of directly measuring the emitted nonlinear signal, as in 2DES, in F-2DES the information on the third-order nonlinear response is encoded in the incoherent fluorescence signal. Pioneering work by Ogilvie et al performed spatially-resolved F-2DES on a mixture of photosynthetic bacteria [125], which are known to exhibit variations in electronic structure according to the light exposure during their growth. Using phase modulation to generate a sequence of four phase-locked pulses in a fully collinear configuration [123], F-2DES was combined with microscopy and used to measure a 2D map for every spot of the fluorescence image (figure 16(a)). By measuring as a reference the F-2DES maps of unmixed photosynthetic bacteria, under low light (LL) and high light (HL) growth conditions (figure 16(b)), the authors were able to identify the spatial concentrations of the LL and HL bacteria in a heterogeneous sample with sensitivity orders of magnitude better than conventional spatially-averaged electronic spectroscopies.
Figure 16. (a) Scheme of the collinear four-pulse sequence used for F-2DES coupled to a scanning microscope, which allows synchronous recording of F-2DES maps and fluorescence images. (b) F-2DES maps at t2 = 0 fs, obtained for unmixed photosynthetic bacteria under LL and HL growth conditions, showing different relative amplitudes of the peaks. Adapted from [125]. Reproduced from [125]. CC BY 4.0.
Download figure:
Standard image High-resolution imageFurther resolution enhancement of such setups, by using plasmonic nanoantennas that allow sub-diffraction-limited focussing of electromagnetic fields [126], could pave the way to real-time monitoring of conformational changes that govern photoprotection and charge separation processes in natural and artificial photosynthetic complexes. This would bring us a step closer to the grand goal to implement 2DES of a single photosynthetic complex [81].
Concluding remarks
The past decade has witnessed a series of ground-breaking technological developments in the field of 2DES, such as novel phase-locking techniques, extended wavelength tunability, and the combination of temporal, spectral and spatial resolution. The photosynthetic community can use it as a powerful tool to address long-standing questions in photosynthesis such as: (a) the quenching mechanisms involved in photoprotection; (b) the coupling regime between chromophores enabling high efficiency EET; (c) the relationship of EET with the protein structure and the pigment arrangement.
Acknowledgments
This project is funded by the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
10. 2D mid-IR/vibrational micro-spectroscopy of organic molecules
Thomas Deckert1, Vasilis Petropoulos2 and Daniele Brida1
1 Department of Physics and Materials Science, University of Luxembourg, 162a avenue de la Faïencerie, L-1511 Luxembourg, Luxembourg
2 IFN-CNR, Dipartimento di Fisica, Politecnico di Milano, Piazza Leonardo da Vinci 32, I-20133 Milano, Italy
Status
Ultrafast two-dimensional mid-infrared (2DIR) spectroscopy is an advanced technique that allows probing correlations of characteristic vibrational modes in the 'fingerprint' spectral window (3–20 µm) of biomolecules to retrieve the specimen chemical structure, its biological function and interactions within its environment, while providing an insight on fundamental dynamics on ultrafast timescales. Contrary, the field of plasmonics in the mid infrared is just emerging, stemming from visible and near-infrared research, and is quickly becoming a crucial addition to study biomolecules through the field enhancement provided by plasmonic nanostructures. Merging these two fields will provide vastly increased sensitivity to probe specimen in very diluted amounts towards the single-molecule limit, with unprecedented spatiotemporal resolution in the ability to access ultrafast inter- or intramolecular dynamics.
2DIR spectroscopy exploits the spectral interference of two phase-locked pump pulses to resolve the excitation frequency in addition to the detection frequency to resolve correlations between different resonances. Furthermore, well developed theory on 2DIR allows for direct comparison of experiment and computational models. Exemplary, Kratochvil et al resolved the mechanism of ion permeation across cell membranes with ultrafast 2DIR spectroscopy and comparison to computational models, enhancing the experiment sensitivity through isotope labelling [127]. A complementary extension to 2DIR is 2D electronic-vibrational spectroscopy, which resolves electronic-vibrational coupling through visible excitation and mid-infrared (mid-IR) detection, probing a strongly off-diagonal window in the frequency map.
Only recently, plasmonic nanoantennas based on heavily doped germanium (Ge) were introduced for the mid-IR spectral window. The advantages of this novel plasmonic platform include: (a) excellent material quality due to single-crystalline epitaxial growth, (b) compatibility with the microelectronic industry for fabrication, (c) tunability of the plasma response either by doping, or transiently through ultrafast electrical/optical excitation, (d) the absence of interband transition losses [128–131]. Experiments with various molecules already show signal enhancement factors of two orders of magnitude [128]. Furthermore, Fischer et al optically resolved single Ge antennas through third harmonic generation between 3 and 5 µm, which can serve as nonlinear source for single antenna plasmonic sensing [8] and constitutes an excellent starting point for mid-IR near-field microscopy in combination with ultrafast multidimensional experiments [131].
Current and future challenges
Given the recent advances in mid-IR plasmonics based on heavily doped germanium and the possibility to implement state-of-the-art 2DIR spectroscopy, the consequent next challenge is the combination of the two. The result will be the development of a characterization technique capable to retrieve bond-specific structural information through vibrational couplings and its dynamic conformational changes on time scales ranging from femtoseconds to hours on biomolecules in very diluted amounts. In fact, biomolecules and their dynamic processes are strongly linked to the complex environments in which they are found. Disorder and low concentrations are typical in realistic conditions. Crucially, studying phenomena like the real-time detection of protein conformation and misfolding through amide-bond signatures is fundamental to understand their role in major pathologies, for example in Alzheimer's and Parkinson's diseases. In this context, challenging single-molecule resolution is highly desirable [132]. To push towards this limit, the additional field confinement and signal enhancement of plasmonic antennas is essential.
Figure 17 schematically shows the principle of a combined measurement, where the spectroscopic signal is emitted from biomolecules in the antenna hotspot after a three-pulse sequence consisting of two mutually coherent pump pulses and one probe pulse. Implementing a setup for this measurement is the ultimate goal in the field of 2D mid-IR/vibrational micro-spectroscopy and has the potential to monitor chemical reaction dynamics. This goal requires major effort from an experimental point of view with a high degree of complexity. The four elements that must be combined are: (a) generation of short and carrier-envelope stable pulses in the fingerprint region, (b) multidimensional spectroscopy scheme, (c) microscope capable of handling ultrashort pulses and focus them onto an individual plasmonic antenna, and (d) detection of the mid-IR probe in amplitude and phase after interaction with the sample. Additionally, high sensitivity is crucial to measure faint spectroscopic signals, which limit the amount of dilution towards the goal of single molecule spectroscopy. Consequently, designing and optimizing highly sensitive detection schemes for 2DIR spectroscopy is as important as increasing the signal enhancement factors via plasmonics.

Figure 17. 2D mid-IR spectroscopy experiment performed on molecules with different possible geometries sketched in the insets. The heavily doped germanium (Ge) double-rod antenna on a silicon (Si) substrate provides additional field enhancement. The signal is detected by means of electro-optical sampling (EOS), resolving both amplitude and phase of the probe mid-IR pulses.
Download figure:
Standard image High-resolution imageAdvances in science and technology to meet challenges
The greatest challenge from an experimental point of view consists in implementing a 2D spectroscopy setup while being able to focus the optical beams onto a single plasmonic antenna that will provide the enhancement of the light matter interaction. This goal requires focal spots of around 5–10 µm, which is achievable by microscopes with all-reflective components that preserve the temporal duration of ultrashort mid-IR pulses [8]. Collinear, partially collinear, or noncollinear geometries (see insets in figure 17) are all existing possibilities for the implementation of a 2DIR spectroscopy setup. However, the coupling of the pulses to a microscope and the different robustness to background signals will require a thorough investigation. Furthermore, 2D spectroscopy in the mid infrared allows for new approaches in the detection of the probe pulse: at these frequencies, it is possible to fully resolve the electric fields of the optical pulses in amplitude and phase by means of electro-optical sampling. To this end a sampling pulse shorter than an optical half-cycle of the mid-infrared radiation is used to sample the probe in a nonlinear crystal [133]. The nonlinearity of this phase sensitive readout as well as ensuring phase stability within a measurement adds another degree of complexity to the experiment and requires considerable effort. However, the advantage of detection in amplitude and phase consists in significant gains in the sensitivity of the experimental setup. The capability and potential of such approach was demonstrated with bulk semiconductor materials [134]. For biomolecules however, the damage threshold is significantly lower than solid state materials. For this reason, and to increase the detection sensitivity, the experimental system requires high counting statistics at low peak pulse powers. To date, with laser repetition rates between 50 and 200 kHz, optical fields as low as a few tens of V cm−1 can be resolved with good sensitivity. One additional aspect of the problem is the response of the nanoantenna itself, and its coupling to the biomolecules. Weak and strong coupling regimes have profoundly different impacts on the specimen response, either enhancing the signal strength or forming mixed light-matter states, respectively [135].
Concluding remarks
Mid-IR plasmonics with heavily doped semiconductors became a very active and dynamic field in recent years, triggered by the development of excellent laser sources, nanofabrication techniques, and detection schemes, with promising results and perspectives along the way. Especially its combination with 2D mid-IR spectroscopy for sensing very diluted specimen is a promising tool for unprecedented spatio-temporal resolution and the understanding of a variety of interesting dynamics in biomolecules with the possibility to gain new insights into their structure, biological function, and interaction with their environment and other complexes.
Acknowledgments
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
11. Theory of ultrafast charge-separation phenomena in molecules and nanostructures
F E Quintela Rodriguez1, F Troiani2, C A Rozzi2 and E Molinari1,2
1 Dipartimento FIM—Università di Modena e Reggio Emilia, Italy
2 Istituto Nanoscienze—CNR, Modena, Italy
Status
In a photoinduced charge separation process, the absorption of a photon induces a redox reaction in an excited state of a system. The resulting electron transfer is at the heart of vital natural functions such as photosynthesis, vision, and DNA damage repair. Understanding and controlling these reactions is thus of paramount scientific importance, and can have a high impact in a variety of applications, ranging from solar energy conversion to photocatalysis. A fully quantum and microscopic description of these phenomena is thus in order, and represents an ongoing scientific effort [136].
A key role in the charge separation problem is played by the interplay between electron and nuclear motion (figure 18). Electron dynamics in molecules and solids can proceed on an attosecond time scale. Nuclear dynamics can involve low-frequency vibrations and slow displacements that may take hundreds of picoseconds to complete. This apparently results in a simple picture, where electron and nuclear dynamics can be 'disentangled' (Born–Oppenheimer approximation). However, on the one hand, electron transfer can occur across a wide range of time scales. On the one hand, an increasing evidence has emerged that non-Born–Oppenheimer dynamics can dramatically influence also the early stages (first few fs) of the charge separation process. The complexity of the problem is further increased by the presence of a number of energy loss and electron–hole recombination channels that compete with the charge separation process and tend to reduce its efficiency. Among them, internal conversion, inter-system crossing, and fluorescence. The understanding the dynamics of the charge separation is thus fundamental in order to increase its efficiency [137].
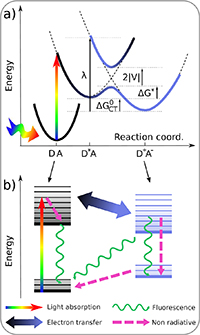
Figure 18. (a) Schematic picture of the photoinduced electron transfer energetics. ΔG0 CT is the Gibbs free energy change for the electron transfer reaction, λ the reorganization energy. Dashed black lines and thick colour lines respectively indicate diabatic and adiabatic potential energy surfaces. (b) Structure of electronic (thick horizontal lines) and vibrational states (thin horizontal lines). The channels for charge transfer, radiative and non-radiative recombination and relaxation are also shown. From [139]. Reproduced from [139]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageRecent advances in experimental techniques are achieving an unprecedented spatial and temporal resolution, and are moving towards the microscopic control of charge, energy and information flows [138]. In particular, the development of time resolved spectroscopies has allowed researchers to shed new light on the ultrafast dynamics governing charge separation processes. The development of fully quantum mechanical simulations is thus needed, both to interpret and explain the phenomena that are accessible at these space and time scales, and to optimize the relevant processes through the synthesis and engineering of suitable materials.
Current and future challenges
The final products of the excited state reaction are spatially separated and unbound charges in molecular species, or free charge carriers in bulk solids. Photoinduced charge separation proceeds through a variety of qualitatively diverse excited states and channels, having entirely different lengths and time scales, sometimes deeply interleaved. One of the current challenges is represented by the identification of the driving force that overcomes the binding energy of the photogenerated electron–hole pair. If the charge-separated state is produced by the dissociation of a highly excited ('hot') charge transfer (CT) exciton and the relaxed CT exciton represents a photocurrent loss channel, then thermalization of the hot state and its dissociation to a charge separated state compete. According to the alternative 'cold' excitons scenario, mobile carriers are generated via a multi-step process, which includes a fast CT relaxation on a 100 fs time scale, followed by a much slower CT dissociation.
Another challenge is represented by the simulation of non-adiabatic processes. In the adiabatic picture, the nuclear reconfiguration drives the reaction through transition states that gradually change their localization from the donor to the acceptor, while the electrons remain on the same nuclear potential energy surface (PES). However, when the dynamics along nuclear and electronic coordinates are strongly coupled, the adiabatic approximation fails, the time evolution from the CT to the charge-separated state occurs at a rate comparable to that of the energy dissipation, and the energy redistribution through vibronic coupling can then be faster than typical vibrations.
A further challenge is related to the role of quantum coherence [140]. The idea that quantum coherence is exploited by nature in order to optimize the efficiency of energy transfer in photosynthetic complexes is intuitively appealing [141]. Indeed, quantum coherence might play a role in enhancing charge-separation processes, both because it can produce delocalized eigenstates (spatial coherence) and because it can qualitatively modify the dynamics of the exciton transport and the interplay between electronic and nuclear degrees of freedom (coherent dynamics) [142]. The identification of the coherences (electronic, vibrational, or vibronic) that are actually responsible for this enhancement, and of those that show up in ultrafast transient spectroscopies remains an open issue.
Advances in science and technology to meet challenges
When the dynamics along nuclear and electronic coordinates is strongly coupled the inclusion of non-adiabatic couplings is mandatory. The approaches that have been developed to describe the coupled electron-nuclear dynamics can be divided into two main classes, depending on whether the nuclei are treated as classical or quantum particles [139, 143]. In the former case, the nuclear wave packet is approximated by an ensemble of particles that follow classical trajectories; quantum corrections are then added to deal with nonadiabatic effects. In the latter, the nuclear wavepacket is described including all quantum effects, such as nonlocality, tunneling and decoherence. A third class includes semiclassical methods, which add part of the missing quantum effects to the classical simulations by means of a quantum phase associated to the trajectories. Methods that incorporate nuclear quantum degrees of freedom (DoF) into a classical formulation of the dynamics are called mixed quantum–classical. Here, the 'back-reaction' of the classical DoF to the quantum DoF is to calculate the effective force on the classical trajectory through a mean potential that is averaged over the quantum DoF (Ehrenfest's theorem).
The development of methods that combine density-based electronic structure theory and the simulation of the nuclear dynamics has allowed the full simulation of molecular systems embedded in their physical environment. These methods are in general less accurate, but more computationally efficient than wave-function based methods. The main approaches are: (a) the reduction to density functionals of time-dependent Kohn–Sham propagation scheme, combined with nuclear dynamics (Ehrenfest dynamics) [144]; (b) the implementation of nonadiabatic mixed quantum–classical molecular dynamics schemes based on Tully's trajectory surface hopping (TSH); (c) the reduction to density functionals of nonadiabatic vectors (important for the detection of conical intersections on the PES and for the rescaling of the nuclear velocities after each surface hop); (d) the development of a coupling scheme for the inclusion of external time-dependent electric (laser) fields; (e) the derivation of spin–orbit couplings within time-dependent density functional theory and their combination with TSH dynamics for the investigation of inter-system crossing events.
Concluding remarks
The time resolution recently achieved in optical and electron spectroscopies has opened new windows of investigation in a wide variety of systems, ranging from photosynthetic complexes to solid-state nanostructures. Here, the need to simulate the ultrafast coupled dynamics of electrons and nuclei has stimulated a number of theoretical developments and approaches. Although a fully quantum description has only been achieved for model systems, charge separation processes in molecules and supra-molecular complexes can now be simulated from first principles by means of quantum–classical, wave-packet and trajectory-based methods. In nanostructures, the presence of low-energy scales, long-range interactions, and delocalized states makes the simulation more problematic. The large size of natural photosynthetic complexes renders fully atomistic simulations unaffordable and requires multiscale and Quantum Mechanics/Molecular Mechanics (QM/MM) approaches. Finally, substantial information about the role of quantum coherence and entanglement can be obtained by complementing the above approaches with model Hamiltonians.
Acknowledgments
This work has been partially supported by the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992 and by MIUR under the PRIN Grant No. 201795SBA3.
12. Light sources for biophotonics
Dominykas Gudavičius1,2, Eric Michele Fantuzzi3, Thomas Deckert4 and Mikas Vengris1,5
1 Light Conversion, Keramiku st. 2B, LT-10233 Vilnius, Lithuania
2 School of Physics and Astronomy, Cardiff University, The Parade, CF24 3AA Cardiff, United Kingdom
3 Aix Marseille Univ, CNRS, Centrale Marseille, Institut Fresnel, Marseille, France
4 Department of Physics and Materials Science, University of Luxembourg, 162a avenue de la Faïencerie, L-1511 Luxembourg, Luxembourg
5 Vilnius University Laser Research Center, Sauletekio av. 10, LT-10223 Vilnius, Lithuania
Status
Ever since the first demonstrations of the biological applications of optical microscopy by A. van Leeuwenhoek in the seventeenth century, this technique has been a staple of biomedical research, providing images of samples ex or in vivo. Confocal microscopy, based on one-photon-excited fluorescence, is perhaps the most widespread modern kind of microscopy, however, it has its fundamental limits in terms of resolution, selectivity, contrast and signal quality. Nonlinear imaging techniques, such as multiphoton-excited fluorescence and coherent Raman scattering (CARS and SRS), have emerged to address some of the issues. Simultaneously, femtosecond laser technology—notably the Ti:Sapphire oscillator—has matured to produce robust and reliable devices. They typically provide laser radiation as a train of 100 fs pulses tunable in the 650 nm to 1050 range at a repetition rate of 80 MHz, and produce average powers in excess of 1 W, corresponding to pulse energies exceeding 10 nJ. Despite the success of Ti:Sapphire technology, new application requirements demand higher pulse energies and a wider tuning range than it can deliver. Therefore, other laser sources are being investigated in the biophotonics labs, including widely-tunable optical parametric oscillators (OPOs), high-energy chirped pulse amplification (CPA) lasers, as well as CPA-pumped high-energy tunable optical parametric amplifiers (OPAs) [145]. Diagrams of both device architectures are shown in figure 19, while the CPA method is detailed in figure 20. An OPO is comprised of a resonator and a nonlinear crystal which converts input radiation at frequency ωp into two output beams with lower frequencies ωs and ωi . The three interacting fields are called pump, signal, and idler, respectively. The output wavelength of the OPO can be chosen by tuning the resonator and the nonlinear crystal, resulting in a wavelength tunable source whose range is not restricted by the gain medium. Wider application of OPOs is constrained by their maximum pulse energy, which is limited by the optical damage, nonlinear and thermal effects [146]. At lower repetition rates and higher pump pulse energies, tunability can be provided by OPAs, which operate similar to OPOs but do not require an external resonator, because the single- or double-pass gain is high enough to convert a substantial portion of pump light to the desired wavelength. On the other hand, fibre laser systems serve as an alternative to solid-state lasers. Their pulse energies are usually lower than those of the free-space lasers, but the fibre lasers are very compact, alignment-free and stable, making them attractive for a variety of imaging and biosensing applications [147, 148]. As imaging techniques strive for nano-scale video-rate imaging, requirements for light source wavelength tunability, pulse repetition rate, energy and stability, as well average power are growing.
Figure 19. (a) Diagram of an optical parametric oscillator (OPO). Yb: oscillator—ytterbium-doped pump oscillator. The pump is coupled into the cavity where signal and idler pulses are generated in the nonlinear crystal. Output coupler (OC) couples the signal out of the cavity, DL denotes variable resonator delay to match the pump repetition rate. (b) Diagram of an optical parametric amplifier (OPA). Yb: amplifier—Yb-doped amplified laser. The pump and white-light-generated (WLG) seed are overlapped in time (DL) in the nonlinear crystal, where signal and idler pulses are generated. Fundamental or SHG pumping allows for wide tuning in the IR (1.3–1.9 μm) and NIR (0.65–1 μm) ranges.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 20. Chirped pulse amplification (CPA) in which the optical pulse is stretched, amplified and recompressed. The maximum permissible pulse energy can typically be much higher in the compressor (e.g. with only reflective optics) compared to the amplifier, thus CPA allows for much higher peak intensities to be reached.
Download figure:
Standard image High-resolution image{kind=link}
Current and future challenges
The current laser technology matured itself in a CPA invention, which won the Nobel prize in 2018. The CPA paved the way for high-peak intensity pulse generation, which can be broken down into three parts: stretching, amplification and recompression [145]. The incoming pulse is stretched in a dispersive medium, gets amplified while it is elongated without causing optical damage, and is finally recompressed (figure 20). CPA has been historically dominated by Ti:Sapphire lasers due to their tunability and a wide gain bandwidth which is needed for short pulses. For example, a Ti:Sapphire laser emitting 30 fs 1.2 µJ pulses at 800 nm together with an OPA was used for vibrational sum frequency generation microscopy to study molecular self-assembly [149]. However, thermal management of Ti:Sapphire systems is difficult, and the short excited state lifetime of Ti:Sapphire requires high quality pulsed pump sources. Ytterbium (Yb)-based lasers have a significantly better power scaling because of their very low quantum defect. In addition, an Yb-based laser medium can be directly pumped by readily available high-power laser diodes. This makes Yb-based CPA lasers more power efficient and allows their overall footprint to be much smaller. Despite the thermal benefits, Yb-based laser systems have a narrower gain bandwidth which disallows the wavelength tunability and limits pulse duration to around 200 fs. On the other hand, the pulse duration can be reduced and wavelength tunability can be exploited in subsequent OPA stages. For example, an Yb CPA laser with an average power of 9 W emitting 220 fs pulses at 1028 nm combined with an OPA was used for super-resolution CARS microscopy, which requires a two colour laser source having synchronous pulse trains [150]. A recent study reported on the use of a 10 W Yb:KGW laser with a variable 50 kHz to 1 MHz repetition rate and three noncollinear OPAs, which allowed for broadband spectral tuning from 450 nm to 2.1 µm and pulse durations below 50 fs [151]. In another study, deep-tissue imaging of a mouse neocortex was performed using a CPA-OPA system tunable from 1.1 µm to 1.4 µm with a maximum pulse energy of 400 nJ at a 511 kHz repetition rate, with longer wavelengths and higher pulse energies leading to deeper imaging depths [152]. Where high repetition rates and low pulse energies are used, fibre lasers serve as an alternative to solid state wavelength-tunable systems. CARS microscopy applications utilizing lasers with photonic-crystal-fibre-based wavelength converters are reported in the literature [153, 154]. Their simplicity, flexibility, and the possibility for group velocity dispersion tailoring might help move CARS microscopy outside of the laboratory. However, the limited tunability of such systems combined with their high level of noise prevents high-quality video-rate SRS microscopy.
All in all, the future biophotonics imaging challenges pose a dire need for light sources with higher output power, increased pulse energies, shorter pulse durations (or longer, in the case of CARS and SRS) and increased wavelength tunability.
Advances in science and technology to meet challenges
OPOs have the benefits addressing some of the aforementioned challenges: wide wavelength tuning range and large pulse repetition rate. In the last decade, OPO performance has increased significantly, in part due to improvements in nonlinear crystals and optics. Currently, off-the-shelf devices pumped by Yb-doped oscillators offer more than 600 nm of wavelength tuning range with average powers in excess of 3 W and a pulse repetition rate of around 100 MHz with a shot-noise-limited pulse energy stability. Increased pump oscillator power output has allowed a significant part of the pump radiation (typically at 1030 nm for Yb-based systems) to be split to the output of the device. Since OPO outputs are optically derived from the pump they are synchronized in time to femtosecond precision, which is useful for multibeam applications, such as CARS and SRS, wideband fluorescence-excitation and harmonic-generation microscopy. Similarly, a single pump source can be used to pump multiple OPOs, delivering multiple synchronized wavelength-tunable beams. For example, a study used a commercially available Ti:Sapphire laser source together with an OPO to image the cerebral cortex of a mouse [155].
The start of this century has brought about a significant development in extreme light sources—laser systems producing light intensities much higher than can ever be used directly in biological applications. Their most prominent implementations first demonstrated in the early 1990s [156] combine the principles of OPA and CPA to produce pulses with durations of several femtoseconds and peak intensities of terawatts and petawatts [157]. While such light pulses are not directly applicable to biological applications, they can be used to produce secondary radiation in exotic wavelength regions—extreme UV and soft x-ray range, where coherent light is difficult to generate by other methods [158]. As these systems continue to mature, new applications with spatial resolution in single nanometres and time resolutions of attoseconds can be expected in future decades.
Meanwhile, fibre lasers have benefited from the developments in the communication industry, especially in the form of fibre amplifiers, custom fibre manufacturing and fast signal modulation. An elaborate laser system which incorporates a fast electro-optic modulator before the final power amplifier and produces 64 fs pulses at 1700 nm with an arbitrary pulse repetition rate of up to 32 MHz was used for functional brain imaging in [159]. Another study used a femtosecond fibre laser delivering 80 fs 30 nJ pulses at 1050 nm and an average power above 2 W for in vivo brain and vasculature imaging in a mouse model [160]. Furthermore, attempts to use ultra-high nonlinearity fibres which can operate with low input energies show that they can be used for CARS microscopy with sufficient long-term stability to image biological samples [161]. Such all-fibre systems are desired due to their lower price and size. Another important advancement came with hollow-core photonic-crystal fibres which can deliver high peak energy without suffering from a parasitic four-wave-mixing process. Hollow-core systems are well suited for epi-detection microscopy since the excitation and backscattered light travel in different parts of the fibre. Hollow-core photonic-crystal fibres enable coherent Raman endoscopes which can perform CARS and even SRS imaging [162].
Concluding remarks
Several predictions can be made from the current biophotonics laser applications landscape. OPOs are expected to attain higher average powers and better control of the pulse chirp. Furthermore, there is a growing need for multi-channel OPOs to replace multi-laser setups. CPA and OPA systems will surpass repetition rates beyond 10 s of MHz, closing the gap between oscillator- and amplifier-based sources, while maintaining µJ-level pulse energies and covering a wide spectral tuning range. All laser sources will have to do better in terms of beam pointing control, stability and environmental stability. Optical parametric chirped pulse amplification (OPCPA) systems are expected to shrink, paving the way for tabletop high harmonic generation sources suitable for femtosecond and attosecond imaging with sufficient flux for single-shot acquisition at kHz rates. Average power gains and noise reduction will continue to improve in fibre lasers improving their widespread and affordability, in addition to wavelength extension due to four-wave mixing amplifiers. In general, there is an exciting shift to a user-driven market, where biophotonics applications become sufficiently numerous to shape the design of future laser sources.
Acknowledgments
The authors would like to thank Dr Lukas Kontenis and Ignas Stasevičius for their helpful advice.
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 812992.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.