
Abstract
Microvasculature is essential for the exchange of gas and nutrient for most tissues in our body. Some tissue structures such as the meniscus presents spatially confined blood vessels adjacent to non-vascularized regions. In biofabrication, mimicking the spatial distribution of such vascular components is paramount, as capillary ingrowth into non-vascularized tissues can lead to tissue matrix alterations and subsequent pathology. Multi-material three-dimensional (3D) bioprinting strategies have the potential to resolve anisotropic tissue features, although building complex constructs comprising stable vascularized and non-vascularized regions remains a major challenge to date. In this study, we developed endothelial cell-laden pro- and anti-angiogenic bioinks, supplemented with bioactive matrix-derived microfibers (MFs) that were created from type I collagen sponges (col-1) and cartilage decellularized extracellular matrix (CdECM), respectively. Human umbilical vein endothelial cell (HUVEC)-driven capillary networks started to form 2 d after bioprinting. Supplementing cartilage-derived MFs to endothelial-cell laden bioinks reduced the total length of neo-microvessels by 29%, and the number of microvessel junctions by 37% after 14 d, compared to bioinks with pro-angiogenic col-1 MFs. As a proof of concept, the bioinks were bioprinted into an anatomical meniscus shape with a biomimetic vascularized outer and non-vascularized inner region, using a gellan gum microgel suspension bath. These 3D meniscus-like constructs were cultured up to 14 d, with in the outer zone the HUVEC-, mural cell-, and col-1 MF-laden pro-angiogenic bioink, and in the inner zone a meniscus progenitor cell (MPC)- and CdECM MF-laden anti-angiogenic bioink, revealing successful spatial confinement of the nascent vascular network only in the outer zone. Further, to co-facilitate both microvessel formation and MPC-derived matrix formation, we formulated cell culture medium conditions with a temporal switch. Overall, this study provides a new strategy that could be applied to develop zonal biomimetic meniscal constructs. Moreover, the use of ECM-derived MFs to promote or inhibit capillary networks opens new possibilities for the biofabrication of tissues with anisotropic microvascular distribution. These have potential for many applications including in vitro models of vascular-to-avascular tissue interfaces, cancer progression, and for testing anti-angiogenic therapies.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Biofabrication has great potential to produce living three-dimensional (3D) constructs that mimic native tissues, and that can be applied for tissue replacement, repair, or disease modeling [1]. Complex structures can be recreated by the precise deposition of selected biomaterials, cells, and bioactive molecules. However, the generation of vascular components remains a major challenge towards the generation of full-thickness tissue equivalents [2]. Networks of open channels that mimic vasculature have been produced and enable perfusion, for instance via bioprinting of sacrificial materials [3, 4]. Yet, a major challenge remains in steering the formation of pervasive capillaries in a region-specific fashion, since functional microvessels are needed to provide nutrients throughout most, but not all, large living tissue constructs.
The recreation of capillaries in vitro has been widely studied. Endothelial cells can self-assemble into a capillary-like network, when cultured in a 3D environment with specific growth factors and mural cells [5–7]. Previous studies have shown how such microvessels can be guided and matured, by culturing the abovementioned cells in presence of type I collagen (col-1) microfibers (MFs). With these MFs, capillaries showed more open lumen and larger capillary diameters, contributing to the ability to recreate a more biomimetic in vitro engineered capillary network [6]. Such collagen MFs are promising candidates as bioink components to carry instructive signals for endothelial cells, as these can be easily resuspended in hydrogels or aqueous formulations without the need for chemical modifications.
Where the majority of native tissues is dependent on a vascular capillary network, in partly vascularized tissues the spatial distribution of microvessels is tightly defined. The coexistence of precisely confined vascular and avascular regions is a defining element of many tissues and organs, including the eye, the intervertebral disc (IVD), the osteochondral boundary, and the menisci [8–10]. Replacement strategies of such tissue structures upon damage with (bioprinted) biomimetic living constructs, requires the recapitulation of their microvascular distribution. Specifically, the knee joint meniscus, a specialized tissue that provides knee joint stability and transfers mechanical forces from the femur to the tibia, consists of a non-vascularized inner, transitional, and vascularized outer zone [11]. Menisci are the most frequently damaged tissue structure of the knee, and while their outer zone has the capacity to repair (small) injuries, this ability is lacking in the inner zone, a difference which is in part explained by the local presence or absence of vasculature [12]. Thus, meniscus injuries in the inner zone, or of a critical size, that are unable to heal by intrinsic repair mechanisms or otherwise surgical interventions, can be eligible for replacement therapies. Importantly, when microvasculature is not properly confined in its intended region, microvessel ingrowth accompanied with nerve infiltration can take place into non-vascular regions [13–15]. This can lead to changed local tissue characteristics, such as cellular composition and altered (extracellular) matrix deposition by resident cells, as typically observed in osteoarthritic (OA) menisci, in the degenerating IVD, and OA cartilage [10, 14–17]. Therefore, it is essential to prevent infiltration of microvasculature into non-vascularized zones when generating an engineered tissue equivalent in which adjacent vascularized and non-vascularized zones natively co-exist.
Strategies to spatially control microvasculature patterning are currently limited [18]. As such, it is paramount to develop precise (bio)fabrication strategies that guide the spatial formation of microvasculature, and where necessary, prevent the formation or infiltration of capillaries in avascular biofabricated structures during growth and maturation. Previously identified anti-angiogenic molecules often reside in tissues like articular cartilage or the IVD, indicating a role in the preservation of their avascular nature [19–21]. Endostatin, a type XVIII collagen fragment present in (fibro)cartilage, inhibits vascular network formation in vitro and in vivo [22, 23]. Also, chondroitin sulphate (CS), an important glycosaminoglycan (GAG) in cartilage tissue, inhibits endothelial-cell network formation in vitro [24]. Next, chondrocyte-derived extracellular matrix (ECM) reduces endothelial cell-derived network formation in vitro, and prevents microvessel ingrowth in vivo [25]. Such tissue fractions or molecules can thus be promising supplements for printable bioinks to prevent the formation of endothelial-cell mediated network formation.
In this study, we present a versatile strategy to steer endothelial cells to spatially reorganize into capillaries within a hydrogel, by the supplementation of MFs with pro- and anti-angiogenic potential. To generate constructs with a zonal vascular distribution and complex anatomical geometries, we used extrusion-based bioprinting to bioprint multiple bioinks in a microgel suspension bath. To demonstrate the application of such bioinks, 3D bioprinted meniscus-shaped constructs were produced with a distinct outer vascular and inner avascular fibrocartiligeous region. The outer meniscal zone was comprised of a fibrin-based pro-angiogenic bioink with human umbilical vein endothelial cells (HUVECs), mesenchymal stem cells (MSCs), and col-1 MFs to support capillary formation. The inner meniscal zone was comprised of a fibrin-based anti-angiogenic bioink with meniscus progenitor cells (MPCs), and cartilage decellularized ECM (CdECM) MFs to inhibit formation of capillaries.
The printable bioinks with pro- and anti-angiogenic potential presented in this study allow for the spatial distribution of distinct vascularized and non-vascularized tissue components, in complex-shaped architectures using our presented 3D bioprinting setup. This platform can be used for various applications, including the recreation of tissues with zonal vascularity, and testing models for mechanisms of angiogenesis, or new therapeutic strategies, including drug testing and screening.
2. Materials and methods
2.1. HUVEC and MSC co-cultures with MFs
2.1.1. Collagen MF preparation
Type I (col-1) and type II collagen (col-2) MFs were prepared as described previously [26]. In short, porcine type I collagen sponges (Nippon ham, Osaka, Japan) were dehydrated and crosslinked at 200 °C for 24 h. Next, samples were homogenized in deionized water for 6 min at 30.000 rpm (VH-10 homogenizer, Violamo, S10N-10G: 10 mm diameter and 115 mm probe length). Then, samples were subjected to ultrasonication (Ultrasonic processor VC50, 50 W, 20 kHz) for 100 rounds of 20 s, with 10 s of rest in between, while the tube was maintained in ice water. Lastly, the fibers were filtered through a 42 μm pore size nylon mesh (PA-42u, AS ONE, Osaka, Japan) and lyophilized. Chicken-derived type II collagen (Nippon ham, Osaka, Japan) was crosslinked at 200 °C for 6 h, and similarly to type I collagen samples homogenized, sonicated, filtered and lyophilized.
CdECM MFs were derived from decellularized equine stifle articular cartilage powder, prepared as previously described [27]. In short, articular cartilage was removed from healthy equine stifle joints, with permission from the owners and in line with the ethical regulations of the institution. Cartilage slices were cut into chips of 1 cm2, lyophilized, ground in liquid nitrogen, and sieved through 0.71 mm pores. Decellularization was initiated with six cycles of 4 h 0.25% trypsin (25200072, Thermo Fischer Scientific), followed by a 4 h incubation with 50 U ml−1 DNase (DN-25, Sigma-Aldrich) and 1 U ml−1 RNase (R6513, Sigma-Aldrich) in 10 mM Tris/HCl pH 7.5, and finalized with a 24 h exposure of 1% Triton X-100 (X100, Sigma-Aldrich). The decellularized powder was washed in deionized water for six cycles of 8 h, and freeze-dried. After, the powder was further processed into MFs, by homogenization, sonication, filtering and freeze-drying, similarly as the col-1 and col-2 MFs. Before the use in cell-culture, all fibers were ultraviolet light -sterilized for at least 20 min, and then resuspended in medium without fetal bovine serum (FBS).
2.1.2. Raman spectroscopy and atomic force microscopy (AFM)
For AFM measurements, col-1, col-2, and CdECM MFs were resuspended in a vial with distilled water, and deposited on a SiO2 substrate by spin coating. The SiO2 substrate was treated by oxygen plasma before the spin coating. For Raman spectroscopy lyophilized MFs were used.
AFM measurements were performed using a Bruker Icon system. A ScanAsyst AFM tip with nominal stiffness of ∼0.7 N m−1 and tip radius of ∼2 nm was used for the measurements. The topography and stiffness obtained with the quantitative nanomechanical mapping mode available in the Bruker's software which directly converts the indentation data (i.e. force-displacements curves) to values of modulus. The theoretical approach for the conversion of the f–d curves to modulus has been described in detail elsewhere [28]. The maximum applied indentation force was kept in the range of ∼1–4 nN, depending on the sample. For this force regime the mechanical response is linear elastic, thus the applied forces did not cause plastic deformation on the sample and did not affect the topography of the samples. The scan rate varied in the range of 0.3–0.6 Hz. A relatively slow scan rate was required to obtain the modulus maps with sufficient clarity.
Raman spectra were collected using a 785 nm laserline with a 100× objective. The exposure time and laser power were adjusted to collect noise free spectra depending on the sample. Constituents were revealed by assignment to specific frequencies found in literature for the Raman bands, including amide I and III, GAGs, amino acids and proteins.
2.1.3. Isolation and culture of green fluorescent (GFP)-HUVECs, hMSCs, hMCs, and hMPCs
Green fluorescent (GFP-HUVECs (cAP-001GFP, Angioproteomie)) were expanded in type I collagen pre-coated culture flasks at a density of 3 × 103 cells cm−2 up to 80% confluency in endothelial cell growth medium-2 (EGM-2) BulletKit Medium (CC-3162, Lonza). Culture flasks were pre-coated with 50 μg ml−1 collagen I rat tail (354236, Corning) in 0.01 M HCl for 1 h at 37 °C, followed by two washes of phosphate-buffered saline (PBS). GFP-HUVECS in experiments were used in passage 7, and cultured in EGM-2 medium for expansion and differentiation.
Human iliac crest bone marrow-derived MSCs were harvested from a patient (age 66, male) receiving total hip arthroplasty or spondylodesis surgery as described before. Informed consent was given, and a protocol was used approved by the local medical ethical committee. First, the mononuclear bone marrow fraction was separated by centrifugation with a Ficoll-Paque density gradient (GE Healthcare, The Netherlands). Then, they were cultured up to passage 4 in T175 culture flasks at a density of 3 × 103 cells cm−2 up to 80% confluency, in Minimum Essential Medium (MEM) alpha (22561, Life Technologies) supplemented with 10% heat-inactivated FBS (Biowest), 0.20 mM L-ascorbic acid 2-phosphate (A8960, Sigma-Aldrich), 1% penicillin/streptomycin (100 U ml−1, 100 μg ml−1, 15140122, Gibco), and 1 ng ml−1 recombinant human basic bFGF (233-FB, R&D Systems). The hbMSCs were assessed for their ability to form HUVEC-supporting pericytes by the expression of α-smooth muscle actin (SMA), and a pericyte-like morphology [5]. In experiments human bone marrow-derived MSCs (hbMSCs) were used at passage 5, and culture medium was refreshed twice per week.
Human meniscus cells (MCs) and human MPCs were isolated from OA menisci (MCs: male, 62 years; MPCs: female, 54 years) as described before [29]. In short, meniscus tissue was digested with 0.2% pronase for 2 h at 37 °C, and then 0.075% collagenase overnight. MCs were expanded at 3 × 103 cells cm−2 up to passage 2 in MEM alpha, 10% heat-inactivated FBS, and 1% penicillin/streptomycin. MPCs were further selected by their differential adhesion to fibronectin, by culturing them on fibronectin coated culture flasks for 20 min, after which unadhered cells were washed away. Polyclonal expansion was performed at 2 × 103 cells cm−2 up to passage 4 in Dulbecco's Modified Eagle Medium (DMEM), high glucose, GlutaMAX (319566, Gibco), 10% fetal calf serum (HZ), 1% penicillin/streptomycin, non-essential amino acids (11140050, Gibco), 0.20 mM L-ascorbic acid 2-phosphate, and 5 ng bFGF per ml medium. Chondrogenic differentiation was induced by culturing in DMEM, high glucose, GlutaMAX, 1% ITS Premix (354352, Fisher Scientific), 0.039 μg ml−1 dexamethasone (D8893, Sigma-Aldrich), 0.20 mM L-ascorbic acid 2-phosphate, 1% penicillin-streptomycin (15140122, Gibco), and 10 ng ml−1 transforming growth factor (TGF)-β1 (100-21, Peprotech).
2.1.4. GFP-HUVECs and MSCs co-culture in fibrin gels
GFP-HUVECs and hbMSCs were co-cultured in fibrin bioinks, consisting of 6.6 mg ml−1 bovine fibrinogen (F8630, Sigma-Aldrich) and 2 U ml−1 bovine thrombin (T4648, Sigma-Aldrich). At this stage, gelatin was not added to the formulation yet, as its main role was enabling bioink printability. Importantly, gelatin is rapidly and spontaneously dissolved as soon as the hydrogels are placed at optimal cell culture conditions (37 °C) and thus diffuses out of the constructs [30], therefore it is not meant to contribute to the cell behavior during the 14 d of duration of our experiments.
First, cells were collected in a pellet at a concentration of 2.5 million GFP-HUVECs and 5 million hbMSCs ml−1 fibrin gel. Optionally, 0.25%, 0.5%, or 1% w/v MFs were collected in the same pellet. Secondly, the cell (and MF) pellet was resuspended in fibrinogen dissolved in saline with 1% sterile filtered bovine serum albumin (BSA) (Roche) and 10 mM N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) (15630080, Gibco). Lastly, thrombin dissolved in medium without FBS was added to the mixture, and 20 μl droplets were pipetted quickly at the bottom of a culture plate. After 30 min of gelation and adhesion, EGM-2 medium was added. Each group was performed in triplicate or quadruple. The co-cultures were kept for 14 d, refreshing the medium twice per week.
2.1.5. Effect of medium formulations on the co-culture of meniscal (progenitor) cells with HUVECs and MSCs
In order to identify suitable co-culture conditions needed for the maturation of a heterocellular meniscus-like construct, comprising a vascularized outer and fibrocartilage-like inner zone, the effect of 13 different cell culture medium compositions was tested on the performance of HUVEC-driven network formation, and meniscus (progenitor) cell differentiation. For this, a 20 μl fibrin hydrogel (6.6 mg ml−1 fibrinogen crosslinked with 2 U ml−1 thrombin), not supplemented with MFs, containing GFP-HUVECs and MSCs was cultured in the same well as a fibrin gel with either MCs, MPCs, or a combination of MCs and MPCs. The MCs and MPCs were cultured at 2 million cells ml−1 fibrin gel, and the MC and MPC (1:1) at 1 million cells ml−1 of each cell type. MCs were used at passage 3, and the MPCs at passage 5 in experiments. In this experiment, no col-1 or CdECM MFs were incorporated into the fibrin gels, to make sure that the experimental outcomes were initiated by the medium conditions.
The fibrin gels were cultured in cell culture medium compositions: 100% EGM-2 medium, 100% chondrogenic differentiation medium with TGF-β1, 75% EMG-2 medium with 25% chondrogenic differentiation medium with TGF-β1, 50% EGM-2 medium with 50% chondrogenic differentiation medium with TGF-β1, 25% EMG-2 medium with 75% chondrogenic differentiation medium with TGF-β1, and 100% EGM-2 medium with growth factors from chondrogenic differentiation medium (1% ITS Premix, 1.95 μl (1 mg ml−1) dexamethasone per 50 ml medium, and 10 ng ml−1 TGF-β1). A medium switch was applied in several formulations: control groups with 14 d EGM-2, 14 d chondrogenic differentiation medium (with or without TGF-β1), 7 d EGM-2 and 7 d 75% EGM-2 with 25% chondrogenic differentiation medium (with or without TGF-β1), 7 d EGM-2 and 7 d 90% EGM-2 with 10% chondrogenic differentiation medium (with or without TGF-β1), 7 d EGM-2 and 7 d EGM-2 with either 25% ITS-Premix, dexamethasone, or TGF-β1.
All samples were stained for proteoglycans using safranin-O/fast green, and immunohistochemistry was performed for type I, and type II collagen. The extent of GFP-HUVEC network formation was measured using Angiotool [31]. Each group was performed in quadruplicate.
2.2. 3D bioprinting of MF-laden bioinks into multi-material constructs
2.2.1. Bioink preparation
The bioink for extrusion-based bioprinting was composed of 6.7 mg ml−1 fibrinogen, 2% w/v filter sterilized gelatin (type A, bloom 300, G1890, Sigma) all dissolved in a saline solution supplemented with 1% BSA and 10 mM HEPES, and 2.5 million GFP-HUVECs ml−1 with 5 million hbMSCs ml−1. After transferring the bioink into the printing cartridge, the mixture was placed at 4 °C for 10 min, and then in a water bath at 22 °C for 20 min (room temperature).
2.2.2. Gellan gum suspension bath
A microgel suspension bath was prepared from 1% w/v gellan gum solution. First, 2% gellan gum (G1910, Sigma) was dissolved in sterile deionized water at 90 °C for approximately 1 h, to ensure sterilization. Then sterile 2× PBS was added in a 1:1 ratio, and heated to allow homogeneous mixing. After cooling down to room temperature, the solidified gel was transferred into a sterilized mixer jar. To incorporate a final thrombin concentration of 1 U ml−1 into the gellan gum, an equal volume of 1× PBS with 2 U ml−1 thrombin was added to the jar. The mixture was milled for one minute, and transferred into 50 ml tubes with a maximum volume of 25 ml. After 30 min of incubation, the tubes were spun down at 2576 relative centrifugal force for 10 min, and the supernatant was removed. After vortexing, this step was repeated until a volume of 12.5 ml gellan gum microgel remained.
2.2.3. Bioprinting
Three-dimensional bioprinting experiments were performed with the 3D discovery bioprinter (regenHu, Switzerland), using two pneumatic-driven extrusion printheads. The nozzles used for both bioinks were conical with a diameter of 0.25 mm. G-codes were generated using BioCAD and BioCAM software (regenHu, Switzerland). During optimization, immortalized MSCs (iMSCs) were encapsulated at 7.5 × 106 in the bioinks, representing the sum of HUVECs and MSCs, and stained with brilliant blue. The filament fusion test represents distances from 0.300 to 1.050 mm with steps of 0.050 mm, from which also a filament diameter was extracted (n = 52). The filament collapse test was performed as before [32]. Briefly, pillars were 3D printed with digital light processing (ATUM 3D), with gap sizes of 16, 8, 4, 2, and 1 mm between pillars, over which the bioink was suspended. The 9 × 9 mm squared 3D bioprinted structures have five horizontal and five vertical layers, with a z-step distance of 0.25 mm, a printing speed of 10 mm s−1, and an inter-line distance of 1.5 mm. A layer stacking analysis was performed on four constructs after printing and removal of the gellan gum microgel suspension bath. A meniscus model was obtained from an equine meniscus-derived μ-computed tomography (CT) scan, and converted into a dual zone digital 3D model using ImageJ. Meniscus constructs were bioprinted with the pro-angiogenic bioink in the outer zone, containing 2.5 × 106 HUVECs ml−1 and 5.0 × 106 MSCs ml−1 with 0.5% w/v col-1 MFs, and an anti-angiogenic bioink in the inner zone, containing 2.0 × 106 MPCs ml−1 with 0.5% w/v CdECM MFs. Bioink filaments were programmed to have a distance of 0.600 mm between fiber centers, and a z-step distance of 0.25 mm. Large, rectangular-shaped blocks with a pro-angiogenic bioink and anti-angiogenic bioink compartment were designed using BioCAD, to assess matrix deposition after 14 d of culture (L: 3.6 mm for the pro-angiogenic compartment +7.2 mm for the anti-angiogenic compartment; D: 7.2 mm; H: 6 mm (24 layers)).
2.3. (Immuno)histochemistry and metabolic activity
2.3.1. Histology
After 14 d, fibrin gels were fixated in 4% buffered formalin, and further processed by dehydration through graded ethanol series and xylene washes, and paraffin embedding. Samples were cut in slices of 5 µm, and rehydrated before staining. Proteoglycans were stained with 0.125% safranin-O (Merck), and counterstained with 0.4% fast green (Sigma-Aldrich) and Weigert's hematoxylin (Clin-Tech). Presence of collagens was evaluated by immunohistochemistry using antibodies for type I collagen (EPR7785, Abcam; 1:400 in PBS/BSA 5%) and type II collagen (II-II6B3; DHSB; 1:100 in PBS/BSA 5%). First, sections were blocked with 0.3% H2O2, followed by antigen retrieval with 1 mg ml−1 pronase (Sigma-Aldrich) for 30 min at 37 °C and 10 mg ml−1 hyaluronidase (Sigma-Aldrich) for 30 min at 37 °C. Then, sections were blocked using 5% BSA in PBS w/v for 30 min, and incubated overnight with the primary antibody at 4 °C. The next day, type II collagen sections were incubated with a 1:100 goat-anti-mouse IgG horseradish peroxidase (HRP)-conjugated in 5% BSA in PBS w/v (P0447, DAKO) for 1 h, and type I collagen sections with Envision + System-HRP anti-rabbit (K4003, DAKO), for 30 min at room temperature. Both stainings were visualized using 3,3'-diaminobenzidine (Sigma-Aldrich), counterstained with Mayer's hematoxylin (Klinipath) and mounted in DPX mounting medium (Merck).
2.3.2. CD31/α-SMA immunohistochemistry
The presence of endothelial cell marker CD31 and pericytes was shown with a CD31 immunohistochemistry staining. Samples were fixated in 4% buffered formalin, permeabilized with 0.2% triton for 30 min, and blocked with 5% BSA for 30 min. Then, samples were incubated with the primary antibody CD31 (1:20, mouse anti-human, M0823, Dako) for 1.5 h, washed with PBS-Tween, and incubated with a biotinylated secondary antibody (1:200, sheep anti-mouse, GE Healthcare, RPN1001v1) for 1 h. After, samples were incubated with a tertiary antibody (steptavidin Alexa fluor 488, Thermofischer S32354) and α-SMA (1:300, clone 1A4, Cy3, C6198, Sigma Aldrich) for 1 h, and finally incubated with 4',6‐diamidino‐2‐phenylindole dihydrochloride staining (100 ng ml−1, Sigma). Antibodies were all diluted in 5% BSA in PBS w/v.
2.3.3. Imaging
Imaging of fluorescent samples was performed using the SP8 confocal microscope (Leica), Thunder microscope (Thunder, Leica), or fluorescent microscope (Olympus IX 53). Samples (immuno)histologically stained for type I and II collagen, and safranin-O/fast green, were imaged using the Olympus BX51 microscope (Olympus DP70 camera, Hamburg, Germany).
2.3.4. Metabolic activity assay
Metabolic activity was assessed with an alamar blue assay on live samples. First, 5.54 mg Resazurin sodium salt (B21187, Alfa Aesar) dissolved in 50 ml PBS was sterile filtered. Samples were washed with PBS twice, and then incubated with culture medium supplemented with 10% resazurin sodium salt solution for 4 h at 37 °C. Fluorescence was measured with an excitation of 544 nm and emission of 620 nm using a fluorescence microplate reader (Fluoroskan Ascent FL, Thermo Labsystems). After, samples were washed with PBS and proceeded in culture.
2.4. Statistical analyses
The vessel length and number of junctions were measured using AngioTool v. 0.6a (02.18.14) [31]. All statistical analyses were performed with GraphPad Prism 8.3.0 (538). Results were presented as mean ± standard deviation, or median (inter quartile range (IQR)). The MF length and diameter measurements needed transformation following ynew = log(y). Statistical differences were tested using a Kruskal-Wallis test and Dunn's multiple comparison post-hoc test, separately comparing between concentrations within one MF type, different MF types with the same concentration, and lastly comparing all groups to the group without MFs. The effect of MFs on total vessel length and number of junctions was tested using a mixed model, and Sidak's multiple comparison test with a single pooled variance. A mixed-model with Geisser–Greenhouse correction, and post-hoc Tukey's multiple comparison test was performed for measurements on the metabolic activity of HUVECs and MSCs when cultured with 0.25%, 0.5% and 1% MFs, and as well for the measurement on metabolic activity of HUVECs and MSCs that were bioprinted into squared 3D constructs, in four different concentrations. The effect of cell concentrations on construct shrinkage was tested using a mixed model with Geisser–Greenhouse correction, and Dunnett's multiple comparison test. Generally, statistical significance was considered when p ⩽ 0.05.
3. Results
3.1. Characterization of the MFs
Although all the source materials were processed under the similar conditions, MFs from different ECM components displayed different geometrical features (figure 1). Namely, the median (IQR) length of CdECM MFs was significantly higher (109.5 (63.0–167.5) μm) compared to the median lengths of col-1 (11.5 (7.0–24.3) μm) and col-2 MFs (28.0 (10.0–50.0) μm) (figure 1(a)). Where the median diameter of CdECM MFs (4.0 (3.0–6.0) μm) was similar to col-1 MF (4.0 (3.0–6.0) μm), it was lower than the diameter of col-2 MFs (5.0 (4.0–8.0) μm) (figure 1(b)).
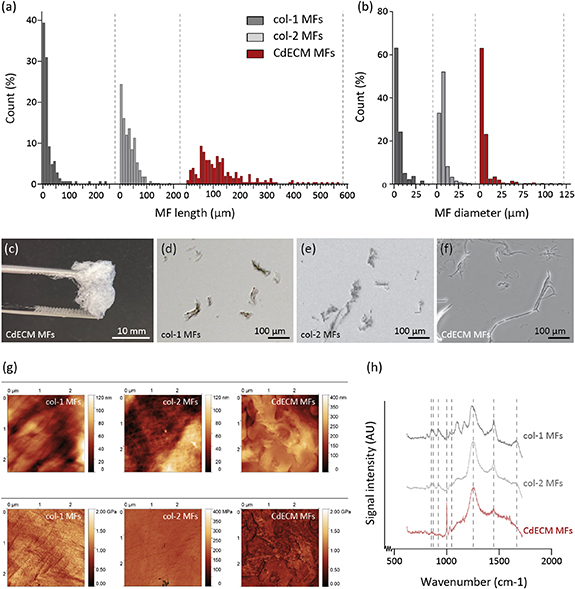
Figure 1. Characterization of col-1, col-2 and CdECM MFs. (a), (b) The length and diameter distribution of col-1 (n = 142 and n = 136), col-2 (n = 435 and n = 336), and CdECM MFs (n = 204 and n = 293) is presented. (c), (f) Macroscopic and microscopic photographs capture the col-1, col-2 and CdECM MF morphology. (g) AFM displays surface roughness (top) and elastic modulus (bottom) of the MFs on a squared surface of approximately 9 μm2. (h) Raman spectroscopy revealed content differences between MFs, indicated by the presence of peaks at 853 (typically associated with proline), 873 (hydroxyproline), 920 (proline), 1001 (phenylalanine), 1044 (GAGs), 1250 (amide III), 1450 (CH2 and CH3 bending for collagen species, or lipids), and 1670 (amide I) cm−1.
Download figure:
Standard image High-resolution imageThe AFM topography measurements revealed that both the col-1 and col-2 MFs were composed of assemblies of smaller nanoscale fibers (figure 1(g)). The smaller fibers within the col-1 MFs were aligned, while within the col-2 MFs they formed a randomly oriented mesh-like structure. The CdECM MFs did not consist of individual nanofibers, but presented round, edge-shaped structures. The local elastic modulus of the MF, mapped by nanoindentation with the AFM device, was found to be in the range of ∼0.58–1.1 GPa for the col-1 MFs, ∼0.13–0.17 GPa for the col-2 MFs, and between ∼0.22 and 0.52 GPa for CdECM MFs. These values agree very well with experimental and theoretical findings presented in other studies concerning collagen fibrils [33].
Raman spectroscopy demonstrated that all experimental groups share their most prominent peak in the frequency ∼1250 cm−1, which corresponds to amide III (figure 1(h), supplementary table 1 available online at stacks.iop.org/BF/14/034104/mmedia). Comparing the peak ratios with other bands for the same material, it follows that amide-III is more abundant in col-2 and CdECM MFs compared to col-1 MFs. Common spectral bands are identified also in the frequency ∼1000 and ∼1670 cm−1, corresponding to vs (C–C) phenylalanine and amide I, respectively and the CH2 and CH3 bending bands at ∼1450 cm−1. The latter are ascribed primarily to collagen chemical species, however respective bands of lipids may also contribute in the same spectral range. Two additional weaker bands at ∼1044 cm−1 have been used in order to predict the amount of GAGs in cartilage samples [34] and in this study this peak was only observed for the col-2 MFs while it was not detected in the col-1 and CdECM MF samples (supplementary table 1). The latter was expected, as it was previously shown that the trypsin treatments, part of the decellularization process, deprives cartilage tissue from GAGs [27]. In order to compare across samples, ratio intensities of common spectra across samples were used (table 1). When comparing the ratios of other peak intensities with interest to the application, we determined that intensity ratios of I920/I1675 reflected a difference in proline residues with respect to the amide I peaks. Where col-2 and CdECM MF ratios were similar, the ratio of col-1 MFs showed a large difference in respect to the other two conditions. In addition, the same correlation was seen for I873/I1675 and I1001/I1675 reflecting the ratios of peak intensities that are characteristic of hydroxyproline and amide I, and phenylalanine and amide I respectively [35]. These findings suggest that col-2 and CdECM MFs possess a lower presence of the aminoacids proline, hydroxyproline and phenylalanine in their collagen fibers than col-1 MFs.
Table 1. Intensity ratios of selected Raman peaks characteristic of proline, hydroxyproline and amide I.
| Proline (I853)/amide I (I1675) | Hydroxyproline (I873)/amide I (I1675) | Phenylalanine(I1001)/amide I (I1675) | |
|---|---|---|---|
| Col-1 MFs | 0.94 ± 0.12 | 0.65 ± 0.13 | 0.98 ± 0.15 |
| Col-2 MFs | 0.34 ± 0.22 | 0.23 ± 0.13 | 0.31 ± 0.18 |
| CdECM MFs | 0.18 ± 0.08 | 0.22 ± 0.09 | 0.26 ± 0.11 |
3.2. CdECM MFs reduce HUVEC-driven network formation
We assessed the effect of col-1, col-2 and CdECM MFs on HUVEC-driven network formation at 0.25%, 0.5%, and 1% w/v, by encapsulating HUVECs and MSCs with MFs in a 3D fibrin gel (6.6 mg ml−1) (figure 2(a), n = 9 for all groups). The endothelial cells that formed capillary-like structures were surrounded by pericyte-like, α-SMA positive MSCs (figure 2(b)). By quantitative measurements of GFP-HUVEC networks, it was found that CdECM MFs significantly reduced the total vessel length and number of junctions in a dose-dependent manner, as compared to the same concentrations of col-1 and col-2 MFs (figures 2(c) and (d)). In fact, the supplementation of 1% w/v CdECM MFs resulted in the strongest reduction of HUVEC-driven network formation compared to col-1 MFs, more specifically, reducing the total vessel length by 34% (after 7 d) and 29% (after 14 d). Additionally, the total number of junctions decreased with 41% (after 7 d) and 37% (after 14 d) (figure 2(d)). Based on both the measured total vessel length and total number of junctions, beside 1%, also 0.25% (on day 7) and 0.5% (day 7 and 14) CdECM MFs significantly reduced HUVEC-driven network formation compared to concentration-matched col-1 and col-2 MFs.

Figure 2. Effect of CdECM MFs on HUVEC-driven capillary network formation. (a) Fluorescent microscopy photographs of GFP-HUVECs, co-cultured with MSCs in a fibrin gel with 0.5% w/v col-1, col-2, or CdECM MFs after 14 d. From each sample photographs were taken from the edge of the gel. (b) Confocal micrographs showed that GFP-HUVECs formed capillary-like structures in presence of col-2 MFs, surrounded by MSCs-derived pericytes. (c), (d) The total vessel length and number of junctions were presented for the co-cultures in a fibrin gel, supplemented with col-1, col-2, and CdECM MFs at 0.25%, 0.5% and 1% w/v, and a group without MFs. Each group is represented by a symbol. Statistical significance was only tested between groups with the same concentration of MFs, and between concentrations within one MF type. Significant differences between groups were indicated by the presence of the group specific symbols above the graph bar of the other group. In general, symbols for col-1 MFs were presented in dark gray, for col-2 MFs in light gray, and for CdECM MFs in red. Circles indicate a group with 0.25% w/v MFs, squares 0.5% v/w MFs, and triangles 1% w/v MFs. (e) The metabolic activity was measured on day 1, 7, and 14 of culture (n = 9), following the same indication for significant differences between groups as in figures (c) and (d). In general, statistical significance was considered p ⩽ 0.05.
Download figure:
Standard image High-resolution imageIn fact, except for 0.25% w/v CdECM on day 14, the total vessel length on day 7 and 14 was significantly lower in presence of CdECM (day 7: 0.25%: 399.7 ± 55.1 mm; 0.5%: 385.6 ± 64.8; 1%: 288.9 ± 62.0) of the corresponding concentration (figure 2(c)). Also, the total vessel length was reduced significantly in presence of 1% w/v CdECM compared to CdECM 0.25% and 0.5% w/v on day 7 and 14. Also, the group with encapsulated col-1 MFs showed a significantly increased total vessel length on day 14, compared to the 0.25% and 1% w/v col-1 groups, as well as the no-MF group. Similar significant differences were found for the total number of junctions, with a significant reduction when cultured with of 0.5% and 1% w/v of CdECM MFs. The MFs did not show any auto-fluorescence when exposed to the 470 nm light source used to visualize the GFP-positive cells, and did thus not interfere with the vessel length measurements (supplementary figure 1). In general, regardless of the material of origin of the fibers, the addition of MFs reduced metabolic activity of the embedded cells significantly on day 1 and 14, compared to no addition of MFs (figure 2(e)). Only on day 1, a concentration-dependent reduction in metabolic activity was found related to col-2 and CdECM MF concentration. In fact, the HUVEC and MSC co-cultures supplemented with 1% w/v col-2 MF was decreased with 12% compared to both col-2 0.25% and 0.5% w/v. For CdECM MFs, the group with 1% MFs w/v presented a 9% and 11% drop compared to 0.25%, and 0.5% CdECM MFs respectively. On day 7, no significant differences in metabolic activity were found that were related to the type of MF or increase in MF concentration. On day 14, only within the col-2 MF group a concentration dependent significant reduction was observed (0.25% and 1% w/v), but not for the other MFs. The 0.25% CdECM MF group showed a significantly lower metabolic activity than the 0.25% w/v col-2 MF group. Lastly, in line with the results on total vessel length and number of junctions, on day 14, the 0.5% w/v col-1 group showed a significantly higher metabolic activity than the other col-2 MF groups, as well as the 0.5% w/v col-2 and CdECM MFs.
3.3. 3D printing of the bioinks
An optimized bioink consisting of 2% w/v gelatin, and 6.6 mg ml−1 fibrin was successfully extrusion based-bioprinted into a gellan gum microgel-based supporting environment, serving as a base for an angiogenic and anti-angiogenic bioink (figure 3(a)). To increase the viscosity of the ink, the 2% w/v gelatin concentration was selected over 2.5%, 3%, 3.5%, 4%, and 5% w/v, since it allowed to extrude the ink as a continuous filament in the support bath at room temperature (data not shown). A filament collapse test was performed to assess the deformation that the printed filaments undergo when spanning across distanced support structure and the tendency of the material to sag under its own weight, as an indicator of its ability to preserve shape fidelity in the z-direction [32]. This assay was performed using the fibrinogen-gelatin bioink-laden with iMSCs to mimic the final HUVEC and MSC cell concentration (figure 3(b)). We observed that, without the gellan gum support bath, filaments could not span the smallest distance of 1 mm at 10 mm s−1, whereas within the support bath filaments could span over a distance of 16 mm without any gravity-induced deformation. Then, using the same iMSC-laden bioink, a filament fusion test showed that the average diameter of a printed filament (ft) was 0.44 + 0.07 mm. This specific test estimates the tendency of the ink to undergo deformation in the xy plane due to surface tension and viscoelasticity driven flow of the material prior to stabilization by crosslinking [32]. This information can be used to estimate the capacity of the ink to resolve sharp angles and to identify the smallest distance at which to adjacent filaments can be placed [32].
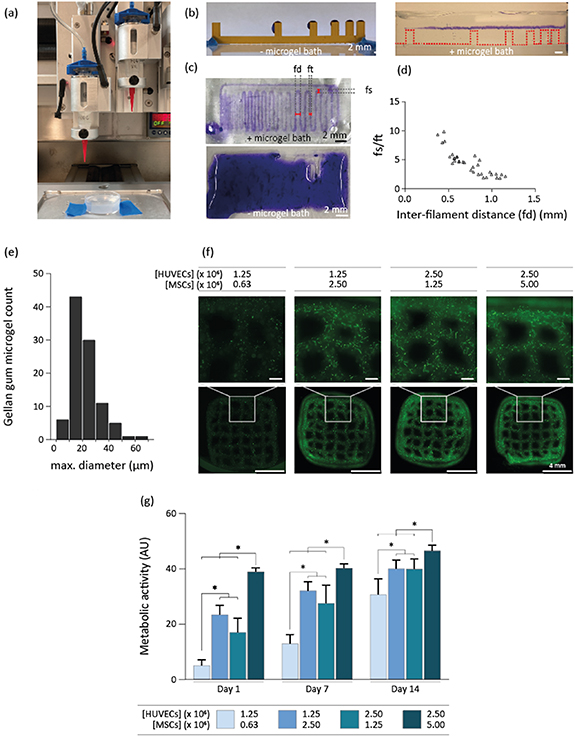
Figure 3. Extrusion-based bioprinting of fibrinogen and gelatin cell-laden bioink. (a) Macroscopic photograph of the extrusion-based bioprinting setup with a microgel support bath. (b) A filament collapse test with an iMSCs-laden fibrin and gelatin bioink revealed that filaments collapsed without gellan gum support bath, but remained intact when printed in the support bath (n = 2). (c) A filament fusion test showed that this bioink could only be bioprinted into a gellan gum support bath (top), as filaments did not retain shape fidelity without it (bottom) (n = 2). (d) The ratio between the fused filament segment (fs) and the diameter of an individual filament (ft), was plotted as a function of the inter-filament distance (fd) (distance from center to center) (n = 33 fs/ft measurements). In a printed meandering pattern, fs measures the length of the portion of the pattern in which two adjacent filaments fuse together when connected by a corner of the meander, and therefore in an ideal print, fs/ft = 1 for all fd > ft. This plot reveals the minimum inter-filament distance at which adjacent filaments do not fuse at all, and, at the same time, indicates the extent of the deviation of the print from a sharp 90° turn, as a function of the designed inter-filament spacing. (e) The distribution of gellan gum microgel maximum diameters. (f) GFP-HUVECs network formation was visualized after 14 d of bioprinting four different HUVEC and MSC concentrations in the fibrinogen-gelatin formulation (n = 8 samples for all groups except for the 2.5/5 million cells ml−1, where n = 6 samples). (g) Metabolic activity of the groups were measured on day 1, 7, and 14.
Download figure:
Standard image High-resolution imageIt also revealed this minimum possible distance between two parallel printed filaments (measured from filament center to center) (fd), was 0.6 mm (figure 3(c)). When also taking into account that the filament fusion length in the corner should not exceed 2× the filament diameter, the optimal inter-filament distance was approximately 1 mm (figure 3(d)). In this way, an inter-filament distance can be defined, while maintaining shape fidelity within 3D printed structures.
It was expected that the addition of MFs would not influence the bioink printability in a relevant way, since gelatin primarily defines the bioink's viscosity, as the sole fibrinogen solution, with and without MFs, behaves as a non-viscous fluid, and is therefore not printable. Also, it was shown before that the addition of 0.5% and 1% w/v col-1 MFs to a fibrin-based bioink resulted in similar rheological properties as compared to the bioink without MFs [6].
As the support bath contributes to the printed filament resolution also by 'templating' the filament with a surface roughness, it's profile being dependent on the size and shape of the microgel particles [36], the geometrical features of the gellan gum microgel suspension bath particles were characterized. The particles displayed an average (maximum) diameter of 20.7 ± 8.6 μm, and showed an irregular, polygonal morphology (figure 3(e)). For 9 × 9 mm squared constructs of ten layers high with a programmed z-stacking distance of 0.25 mm, which theoretically leads to a construct height of 2.5 mm, a layer stacking characterization showed that the construct height was 2.42 ± 0.22 mm. Additionally, no delamination has taken place between layers. The optimal ratio of HUVECs and MSCs for network formation after bioprinting with the fibrin-gelatin bioink was found to be 1:2, more specifically embedding 2.5 × 106 HUVECs ml−1, and 5 × 106 MSCs ml−1. This was compared to a 2:1 ratio with the same HUVEC concentration, and a 1:2 and 2:1 ratio using 1.25 × 106 HUVECs ml−1, by subjective qualitative fluorescent microscopy observations (figure 3(f)). The metabolic activity of 3D printed constructs increased over time for all the experimental groups, confirming cell viability over a 14 d period in all groups (figure 3(g)). The higher metabolic activity levels were detected in the groups homing the highest cell density, and the highest increase in metabolic activity over time in the group homing the lowest cell density. This indicates a higher rate of cell proliferation when seeded at a lower cell density, which is consistent with what was previously reported for MSCs [37]. This implies that in all groups individual cell metabolic activity increased and/or cell proliferation took place, and that, most likely, in the lowest cell density group mainly MSCs proliferation led to the increase in metabolic activity.
Altogether, a fibrinogen-based bioink was optimized for bioprinting in a thrombin containing microgel support bath, enabling the formation of fibrin-based filaments with an average diameter of 0.44 ± 0.07 mm.
3.4. Co-culture of HUVECs with MSCs and meniscus (progenitor) cells, and their 3D bioprinting into a zonal meniscus-shaped construct
We demonstrate that both the successful formation of HUVEC-driven networks, and type I collagen deposition by MPCs was achieved by culturing the cells in EGM-2 medium first, and then in a combination of EGM-2 and chondrogenic differentiation medium (figure 4). In total, 13 medium conditions have been assessed to investigate the differentiation capacity of HUVECs with MSCs to form capillary networks, and of meniscus (progenitor) cells to deposit matrix proteins. For this screening, the HUVECs and MSCs were encapsulated in a fibrin gel (6.6 mg ml−1), and cultured in the same well as a fibrin gel in which meniscus (progenitor) cells were encapsulated. More specifically, HUVECs with MSCs were encapsulated in a 20 μl fibrin-based hydrogel (6.6 mg ml−1 fibrinogen and 2 U ml−1 thrombin), and cultured in the same well as MPCs encapsulated in a similar fibrin-based hydrogel. MFs were not added to the fibrin gels yet, to make sure the observed matrix formation was initiated by the medium conditions alone. First, EGM-2 medium was applied for 7 d, followed by either a 75/25 or 90/10 v/v ratio of EGM-2/chondrogenic differentiation medium containing TGF-β1. In general, medium groups that contained TGF-β1, facilitated the expression of type I collagen by the MPCs, but not type II collagen (figure 4(b)). Interestingly, the same medium formulations without TGF-β1 did not stimulate MPCs to deposit type I collagen. Also, in presence of a higher concentration chondrogenic differentiation medium, the MPCs expressed more type I collagen (75/25 compared to 90/10) (figure 4(b)). Beside the use of multiple medium formulations during the 14 d culture time, we also assessed the use of single culture medium formulations during 14 d. The application of five ratios of EGM-2/chondrogenic differentiation medium (100/0, 75/25, 50/50, 25/75, and 0/100 v/v), revealed that MCs, MPCs, and MCs with MPCs produce type I collagen in presence of 25% or more chondrogenic differentiation medium that included TGF-β1 (supplementary figure 2(a)). The MCs with MPCs in EGM-2 medium (100/0) expressed very little type I collagen. Type II collagen and proteoglycan deposition were not or hardly detected in any of the medium conditions (supplementary figures 2(b) and (c)).
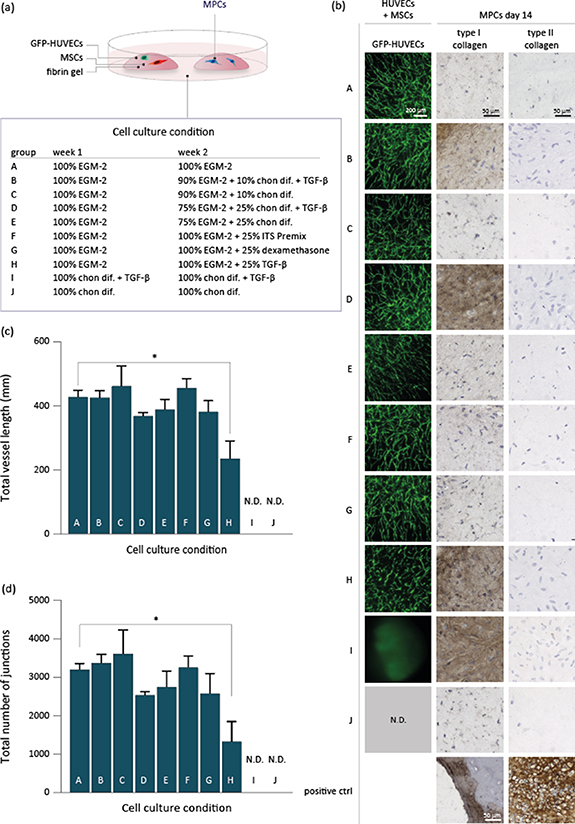
Figure 4. The effect of ten culture medium conditions on the co-culture of HUVECs with MSCs and MPCs. (a) One fibrin hydrogel with HUVECs and MSCs and a second hydrogel with MPCs were cultured in one well in medium compositions A–J for 14 d. Medium was composed of EGM-2 medium in combination with chondrogenic differentiation medium (chon. dif.), with or without TGF-β1. (b) Fluorescent micrographs of GFP-HUVECs, as well as micrographs of MPC type I and II collagen immunohistochemistry were presented for each cell culture condition. (c), (d) Total vessel length and number of junctions of HUVEC-driven network formation was measured on day 14, and significantly lower in group H compared to control group A. GFP-HUVECs were not detectable (N.D.) in groups I and J. For group A–H n = 5, for groups J and I n = 4.
Download figure:
Standard image High-resolution imageHUVEC-driven network formation was stable in almost all culture medium conditions that initiated with 7 d of EGM-2 medium (figures 4(b)–(d)). Only the supplementation of 25% TGF-β1 to EGM-2 medium on day 8–14 (group H) resulted in a significant reduction of the quantified total vessel length (day 1–14 EGM-2 medium: 427 ± 21 mm, group H: 234 ± 55 mm) and total number of junctions (day 1–14 EGM-2 medium: 3186 ± 157 mm, group H: 1319 ± 521 mm) (figures 4(c) and (d)). During day 8–14, the addition of 10% or 25% v/v chondrogenic differentiation medium with or without TGF-β1, or the supplementation of 25% ITS-Premix or dexamethasone to EGM-2 medium did not significantly reduce the abundance of microvessels. When HUVECs and MSCs were cultured in five different ratios of EGM-2/chondrogenic differentiation medium (75/25, 50/50, 25/75, 100/0 v/v) for 14 d, the microvessel network either underwent a collapse before day 14 (75/25, 50/50, 25/75 v/v), or did not initiate network formation (0/100 v/v) (supplementary figure 3, fluorescent micrographs not shown). The presence of chondrogenic differentiation medium induced HUVEC and MSC-laden hydrogel edges to shrink and eventually curl up. This effect was concentration dependent, resulting in spherical shaped hydrogels when the samples were cultured in chondrogenic differentiation medium (0/100 v/v) (data not shown). Since microvessel network formation was not supported in this condition, we visualized deposition of type I, II collagen and proteoglycan deposition to determine whether MSC may have entered the chondrogenic lineage. Type I collagen was observed in all five medium conditions, yet, in presence of EGM-2 medium, it was mainly aligned along the capillary-like structures. In general, type II collagen was hardly detectable, and proteoglycans seemed absent in all groups (supplementary figure 3).
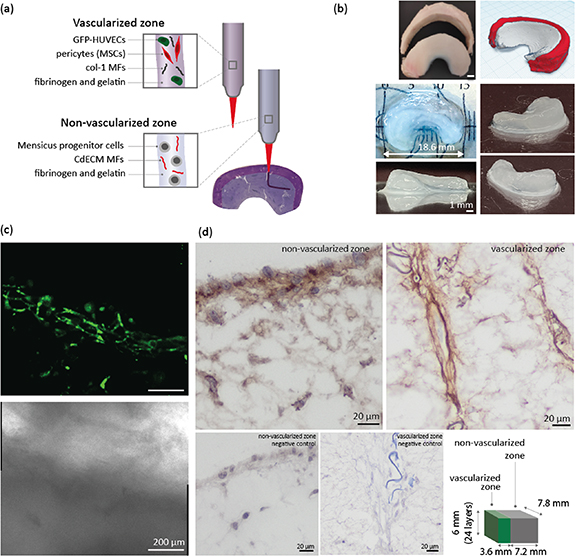
In order to prove that the pro- and anti-angiogenic bioink can be 3D bioprinted into a complex 3D structure, an anatomical meniscus construct was bioprinted, using a μCT-image of a native equine meniscus as a blueprint. Col-1 MFs have previously been shown to enhance the formation of endothelial cell-derived microcapillary-like structures [6], and was therefore selected as a suitable MF for the vascularized zone, as compared to col-2 MFs. In our MFs screening study, col-2 MFs served as a control to exclude that this specific collagen component could be responsible for the inhibition of vascularization observed in the CdECM MFs. Regarding the choice of cells, MPCs were preferred over MCs, due to their superior proliferative ability and stability over extensive passaging when compared to MCs [29]. Moreover, this ability is particularly beneficial for bioprinting applications, for which high cell numbers are typically required. The bioprinted model consisted of a vascularized outer and non-vascularized inner zone (figures 5(a) and (b)). The pro-angiogenic bioink, consisting of 6.6 mg ml−1 fibrinogen and 2% w/v gelatin, used for the outer zone was supplemented with 0.5% w/v col-1 MFs, and contained HUVECs and MSCs, while the inner zone anti-angiogenic bioink, also consisting of 6.6 mg ml−1 fibrinogen and 2% w/v gelatin, was supplemented with 0.5% w/v CdECM MFs, and further consisted of MPCs (figure 5(a)). The bioinks were bioprinted into a gellan gum support bath supplemented with thrombin. With 2.5 ml of pro- and anti-angiogenic bioink, approximately 5 larger meniscus constructs can be bioprinted. The repeatability of bioprinting the constructs was good, with the different constructs displaying comparable shape and size. Moreover, no noticeable difference in terms of cell behavior (i.e. formation of vascular networks by the encapsulated HUVECs) could be observed between the first and the last construct to be printed, despite of the extra time out of the optimal culture conditions experienced by the cells in the printing cartridge. The 3D bioprinted meniscus models were cultured for up to 14 d, and HUVEC-driven networks formed in the outer region, but not in the inner non-vascularized region (figure 5(c) and supplementary figure 4). In addition to dual zone meniscus constructs, large rectangle-shaped constructs were bioprinted with an inner and outer zone composed of the same bioinks as the meniscus construct. The sizes of the vascularized and non-vascularized block-shaped zones was designed in a way that the maximum distance of cells to the cell culture medium was similar to that of the large meniscus construct (18.6 mm in length). An advantage of this block-shaped construct over a more organic-shaped meniscus construct, is that during histological assessments, the vascularized and non-vascularized zone were more easily distinguishable from a macroscopic assessment. The larger printed constructs were used to determine type I collagen matrix deposition after 14 d of culture (figure 5(d)). Constructs were cultured in medium formulation D, including EGM-2 medium from day 1–7 and 75% EGM-2 with 25% v/v chondrogenic differentiation medium from day 8–14. This formulation was successful in enabling MPCs to deposit type I collagen in a small fibrin gel (figure 4(b)). We observed that in the middle of the construct, the vascularized zone presented open lumen from capillary-like structures, around which type I collagen was deposited (figure 5(d)). In the non-vascularized zone, MPC-derived type I collagen deposition was found in very small amounts in the middle of the construct, but more abundantly on the edges of the construct. The type I collagen deposition was expected, due the large construct size, less abundant compared to the 20 μl casted fibrin gels (figure 4(b)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Bioprinting a meniscus-shaped construct with a pro- and anti-angiogenic bioink. (a) A meniscus-shaped construct was bioprinted with a pro-angiogenic outer zone consisting of HUVECs, MSCs, and col-1 MFs, and an anti-angiogenic inner zone, consisting of MPCs with CdECM MFs. (b) A meniscus stl file was obtained from an equine meniscus μCT scan, and used as a model to bioprint a meniscus construct of 24 layers with the developed bioinks. (c) The interface of the outer vascularized, and inner non-vascularized zone is visualized with fluorescent (top), and brightfield (bottom), micrographs, obtained with confocal microscopy on day 14, where the vascularized zone is located in the lower part of the micrograph, and the non-vascularized in the upper part, directly above the GFP-HUVECs. The construct was cultured in EGM-2 medium (day 0–7) and 75% EGM-2 with 25% (v/v) chondrogenic differentiation medium (day 8–14). (d) Type I collagen immunohistochemistry was performed for the vascularized and non-vascularized zone. The staining for the non-vascularized zone was performed on an 18 mm long meniscus construct cultured up to day 14, where type I collagen deposition was present on the outer sides of the construct. The vascularized zone type I collagen staining was performed on a large bioprinted block-like structure, that resembles the thickness of the 18 mm meniscus construct. Microvessels with open lumen were observed in both the edge and the middle of the construct, and stained positive for type I collagen. Construct dimensions were 6.0 mm high, 7.2 mm in depth, and 3.6 mm in length for the vascularized zone, and 7.2 mm for the non-vascularized zone.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
The possibility to precisely position vascular features, and subsequent engineered vessel networks, holds great potential for bioprinting constructs that capture the anisotropic composition of native tissues. While larger vessel structures can be included through the printing of sacrificial materials, patterning microcapillaries remains elusive [4]. Addressing this unsolved challenge has direct implications for the biofabrication of structures with a boundary between vascularized and avascular regions, for example as found in the different zones within menisci, or at the osteochondral interface.
In this study, we investigated a versatile, bioink design-based strategy to enable spatial control over capillary microvasculature formation within 3D bioprinted constructs. Two classes of bioprintable bioinks were introduced that either facilitate (pro-angiogenic) or inhibit (anti-angiogenic) microvessel formation in vitro, by the incorporation of extrudable MFs derived from different ECM components. Using extrusion-based 3D bioprinting, the two bioinks were deposited in a complex 3D organization, and able to locally steer the behavior of encapsulated co-cultures of HUVECs and MSCs, guiding their ability to self-assemble into microvascular networks.
First, MFs from different origins and chemistry were investigated in combination with fibrin, a biomaterial extensively characterized for its ability to sustain endothelial cells in culture, and a major component for several bioink formulations [35, 36]. The fibrin-based pro-angiogenic bioink, consisting of HUVECs and MSCs, was supplemented with col-1 MFs, derived from porcine type I collagen sponges. Previously, it was shown that the addition of these col-1 MFs resulted in the formation of capillaries with larger diameters and open lumen in vitro [6]. The col-1 MFs are suggested to act as a bioactive adhesion side for endothelial cells. Increased expression levels of integrin-β1 by endothelial cells in the presence of col-1 MFs suggest a role of the receptor in this connection [6].
The anti-angiogenic fibrin-based bioink was obtain via the supplementation of CdECM MFs, derived from equine CdECM. These MFs effectively reduced HUVEC-driven network formation in a dose-dependent manner, compared to col-1 MFs. Noteworthy, this inhibiting effect did not correlate with a cytotoxic effect of the MFs. In fact, for all the bioink formulations, the cultured cells showed comparable levels of metabolic activity, regardless of the concentration or nature of embedded MFs. This indicates that the inhibitory effect is likely due to the CdECM MFs biochemical composition and/or physical properties. In addition, bioinks supplemented with col-2 MFs, did not lead to an impaired HUVEC-derived growth of capillary networks, suggesting that type II collagen, the most prominent protein in cartilage tissue and in the CdECM, does not seem to have anti-angiogenic properties.
The reason for the different effects of col-1, col-2 and CdECM MFs on angiogenesis may be sought in the biochemical compositions of the MFs. Raman spectroscopy has previously been used to assess quality characteristics of tissue engineered as well as native tissues such as cartilage tissues known to contain type II collagen [38, 39]. Data obtained from Raman spectroscopy analyses confirmed that col-2 and CdECM MFs were more similar to each other than to col-1 MFs, considering that the relative peak intensities that correspond to proline, hydroxyproline, and phenylalanine were similar, but also lower in col-2 and CdECM samples, as compared to col-1 MFs. The resemblance between the two MFs is consistent with the abundance of type II collagen in native cartilage, even though col-2 and CdECM MFs were harvested from avian and equine sources respectively. The CdECM MFs were deprived from most GAGs and proteoglycans as an effect of the decellularization process [27], whereas residual GAGs could be identified in the Raman spectrum of the col-2 MFs. Yet these MFs did not successfully inhibit capillary formation in the fibrin bioinks, even though, some GAGs, including CS have been reported to exert anti-angiogenic activity [40]. Beside GAGs, other molecules with inhibiting potency may still be present in the CdECM MFs. Previously, microvascular inhibiting molecules have been identified in tissues with an avascular nature, such as the inner meniscal zone, cartilage tissue, and the nucleus pulposus [22–24, 40, 41]. These include (shark) cartilage fractions, chondrocyte-derived ECM, or the soluble secretome from cells residing in avascular tissues [15, 16, 20, 21, 41]. Specific molecules identified in cartilaginous tissue include endostatin or collagen XVIII fragments, and the type IIB collagen NH2-propeptide [22, 40, 42]. Beside its biochemical content, the generation of MFs by homogenization and sonication resulted in CdECM MFs with a significantly higher and less homogeneous length distribution compared to col-1 MFs. The CdECM MF median diameter was not significantly different from col-1 MFs, yet the presence of a few CdECM MFs with larger diameters may influence the physical bioink structure at micrometer level within the fibrin gels. These larger MFs can result in denser and a less homogeneous distribution in the hydrogels, and thereby potentially influencing the ability for HUVECs to self-assemble [6].
The extrusion-based bioprinting of complex cell-laden constructs poses the challenge of formulating a cell-friendly hydrogel with low stiffness, that, at the same time, displays high shape fidelity after printing [43–45]. Because the filament collapse test showed that the bioinks were unable to form a stable filament upon conventional extrusion, and thus failed to bridge gaps of ⩾1 mm, the bioinks were printed into a gellan gum microgel support bath. The microgel slurry provided a sufficient support to enable free-form fabrication of suspended filaments of any length that experience no collapse or gravity-induced deformation. Previous works in the field of suspended bath printing have demonstrated that higher printing resolutions and low surface roughness of the filament can be achieved by using smooth, spherical-shaped microgels, and by decreasing microgel size and polydispersity [36, 46]. Even so, printing resolution and shape fidelity are also dependent on the rapid gelation of the bioink upon contact with the suspension bath, limiting the percolation of the ink into the space in between microgels. As a consequence, constructs with filament size as low as 20 μm have been obtained with limited classes of materials, such as ionically crosslinked alginates or collagen inks prepared at collagen concentrations significantly higher (24 mg ml−1) compared to what conventionally used for the culture of encapsulated cells [36]. Conversely, the printing settings and microgel bath compositions identified in this study enabled to effectively print fibers with diameters in the range of 440 ± 70 μm, even with these fibrin inks with low mechanical properties. Such low-polymer content structure was proven suitable to permit cell proliferation and migration, which are needed to sustain angiogenesis from HUVEC and MSC co-cultures, leading to rapid neo-capillary formation within 2 d after printing.
The two formulated bioinks were used to 3D-print a complex meniscus-shaped construct, with an outer vascularized and inner non-vascularized zone. The outer zone was created using the pro-angiogenic bioink, and the inner zone was composed of the anti-angiogenic bioink with encapsulated MPCs, representing fibrochondrocytes and fibroblast-like cells that are normally present in the inner zone [47]. After 14 d, successful formation of HUVEC-driven networks was visible, including a separated inner and outer zone, and the presence of open lumen-like structures in the middle of a six mm high 3D bioprinted construct.
HUVEC-driven network formation is highly dependent on the presence of selected growth factors, that allow single endothelial cells to self-assemble in to capillary-like structures, and supporting cells to become pericytes. Yet, inducing MCs and MPCs to commit into a (fibro)chondrogenic lineage, requires a different set of media supplements [29], which can potentially interfere with the commitment of MSCs to differentiate into pericytes, as well as with endothelial cells to function. To the best of our knowledge, an optimized co-culture of endothelial cells with MSCs, and meniscus (progenitor) cells has not been reported before. In previous studies, co-cultures of HUVECs with MCs were only performed in endothelial cell culture medium, to assess MC migration, with no provided insight on the impact of this medium formulation on MC and MPC matrix deposition [46]. Co-cultures of pre-vascularized osteogenic constructs, instead, have been setup with success, allowing the formation of endothelial cell-based networks and differentiation of MSCs into the osteogenic lineage, even with osteogenic medium alone [5, 48].
In this study, HUVEC-driven networks formed successfully when cultured in EGM-2 medium, but failed in presence of ⩾25% v/v chondrogenic differentiation medium with TGF-β1, whereas the meniscus (progenitor) cells expressed type I collagen only in presence of ⩾25% v/v. This suggests that other cell culture medium conditions are required to promote microvessel formation and meniscus (progenitor) cell matrix deposition in the same construct. Since the collapse of HUVEC-driven networks in presence of chondrogenic differentiation medium growth factors may be explained by the failure of MSCs to differentiate into pericytes, we investigated the use of a temporal switch of cell culture medium. We found that an initial 7 d culture period in EGM-2, followed by 7 d in 10% or 25% v/v chondrogenic differentiation medium facilitated both a stable HUVEC-driven network, as well as the expression of type I collagen by MPCs. Thus, while the initial presence of chondrogenic differentiation medium prevents the building of a stable capillary network, the introduction of the same formulation at a later stage during culture, results in the preservation of the stability of the vessels. This indicates that the HUVEC-MSCs are sensitive to the timing of presented media supplements, which can be a useful starting point for setting up co-culture conditions for other tissue-specific cells with microvessels. In addition, although our data suggests that TGF-β1 supplementation in the second week of culture can impair vessel formation and stability, this same negative effect was not observed when this growth factor was added to EGM-2 together with 25% of the complete chondrogenic media. This underlines how different components in the media, besides the main growth factors, can significantly influence cell behavior.
In terms of fibrocartilage neo-synthesis, while the inner meniscal zone consists of both type I and II collagen, we found little to no type II collagen deposition. Similarly, no proteoglycan deposition was seen by any of the meniscus (progenitor) cells, in none of the medium conditions. It was, however, shown before that the MPCs used in this study can transcribe COL2A1 and aggrecan (ACAN) messenger riboncleic acid (mRNA), and express small amounts of type II collagen and proteoglycans, observed by respectively immunohistochemistry and histochemistry [29]. The absence or limited deposition of type II collagen, and GAGs in this study could be explained by the shorter culture time of 14 d compared to 28 d, or the use of a fibrin gel instead of a pellet culture, influencing the MPC cell density, and thereby potentially their growth and differentiation potential. It should be noted that, proteoglycan levels in the human inner meniscal zone are generally much lower than articular cartilage and comprise less than 1% of a meniscus wet weight [49]. Moreover, proteoglycan levels increase with age and (initially) with progress of degenerative pathologies [49, 50]. The MPCs in this study are derived from adult donors with OA menisci, which may be the explanation for the potential of the MPCs to produce GAGs [29].
We also observed type I collagen deposition in the large 3D bioprinted structures (6 mm high), composed of the pro-angiogenic and anti-angiogenic bioink. The culture medium composition included a temporal switch to a partial chondrogenic differentiation medium formulation after 7 d, which we demonstrated to be effective for MPC-mediated type I collagen deposition in fibrin gels (figure 4(b)). Within the non-vascularized MPC-laden zone, type I collagen staining was especially marked in regions closer to the edge of the construct, and with noticeably lower intensity at locations in the middle of the constructs after 14 d of culture. The intensity of the staining, indicative of type I collagen abundance was, expectedly, lower in the 6 mm high 3D bioprinted construct compared the 20 μl fibrin gels loaded with MPCs. Due to the diffusion limit of nutrients and gasses across longer distances in cell-laden constructs and tissues, cells located further away from a capillary network, or, in vitro settings from the direct contact with the culture media, may receive a lower number of metabolites, which can lead to reduced matrix biosynthesis. Consistently, this behavior was also observed in our large bioprinted block-shaped constructs with dimensions of 10.8 × 7.2 × 6.0 mm (L × D × H). Considering the size of the construct and culturing in a static condition, it is likely that cells too far away from the medium were cultured under limited nutritional conditions. Dynamic culture strategies (i.e. involving automated exchange of media or the use of bioreactors) together with enhanced porosity introduced by 3D printing strategies could address such challenges [50]. In the vascularized zone of the large 3D bioprinted construct, type I collagen deposition was abundantly visible around stretched areas containing open lumen-like structures most likely part of an immature capillary network. We already observed that the small fibrin hydrogels, laden with endothelial and mural cells, allowed the growth of capillary structures with open lumen after 14 d of culture. We here show that also in the formulated bioink, consisting of fibrin gel and col-1 MFs, such structures form, even in the middle of a larger bioprinted construct. It is possible that the porosity of the bioprinted construct introduced by the distance between printed filaments, allows the diffusion of media components, and thus contributes to the formation of HUVEC-mediated capillary-like structures.
While providing an important proof of concept and generalizable strategies to modulate angiogenesis in printed constructs with the help of ECM-derived MFs, this study has several limitations. First, the col-1, col-2, and CdECM MFs are derived from different species: bovine, chick, and equine. While the isolated col-1 and col-2 sponges and the CdECM should be deprived from cellular materials, the MFs may harbor species-specific moieties, that could not be identified in this study, and that could influence the behavior of the human-derived HUVECs, MSCs, and MPCs, compared to if the MFs were derived from the same species or individual. Still, the availability of post-mortem healthy human tissues is limited compared to animal tissues, posing extra challenges when choosing to use allogeneic or autologous, instead of xenogeneic tissue products. Second, in order to bring cell-laden tissue engineered constructs into the clinic, it is of importance to consider which cell types and sources should ideally be used. The MPCs used in this study are derived from menisci of OA knee joints, meaning that they have resided in an inflamed environment prior to their isolation. While such cells were shown to express differential gene expression patterns and surface markers, such as CD318, compared to cells derived from healthy menisci, it has also been shown that the expression patterns comparable to those of MPCs derived from healthy menisci can be recovered upon in vitro expansion in culture media supplied with TGFβ1 [51]. Furthermore, rejection of implanted tissue equivalents must be avoided, hence autologous primary, or iPSC-derived cells are preferred over allogeneic cell sources. This, however, brings up the challenge of donor-variability, meaning that cells that are isolated from different individuals do not perform in a similar way [5]. Within the scope of this study, one donor per cell type was used for the experiments, where both the MSCs and MPCs are primary cells derived from human patients. In future studies, also patient-derived endothelial cells could be implemented to improve the study's translational perspectives. Additionally, fibrinogen and thrombin are bovine-derived, and may therefore be less clinically translatable, compared to human fibrinogen and thrombin. Since fibrinogen is highly conserved between species, except for the α-carboxy region, the origin may have an influence on material (mechanical) properties after crosslinking [52–54]. Nevertheless, the implementation of fibrin from different sources should always be optimized within future studies. Another challenge is the co-culture of different cell types in one tissue construct. We demonstrated that the co-culture of HUVECs, MSCs, and MPCs by simple mixture of cell culture media does not provide a suitable environment to stimulate HUVEC-driven capillary formation, the differentiation of MSCs into pericytes, and at the same time the differentiation of MPCs. For this setup we found a solution in the temporal switching of medium composition. Still, to further facilitate optimal functionality of different cell types in a co-culture, additional solutions need to be sought, for example, the local incorporation of bioactive molecules strong enough to steer cell differentiation locally. Finally, in light of future applications for meniscus replacement strategies, the constructs produced in this study would need to be reinforced with adequate supporting materials, to match the mechanical properties of native menisci. Elegant strategies for mechanical reinforcement of bioprinted hydrogels have been thoroughly described in the literature by co-printing or (melt) electrowriting of thermoplastic polymers [55–62]. While these may be complex to perform in presence of suspended baths, alternative and effective strategies may involve the co-printing with supporting strong hydrogels [63, 64], which would not require major engineering efforts to be printed in particular media.
Moreover, the versatile strategy proposed in this study introduces new opportunities to exploit the biological response to ECM-derived MFs, in order to steer the fate and bioactivity of the bioprinted cells. MFs derived from a broader range of tissues or matrix components could be used as bioink supplement, and their geometrical and topographical features could be exploited as well, for instance to induce cell alignment in response to MF orientation, which could be modulated by the shear stresses produced by the printing process, or via the application of external stimuli i.e. magnetic field for fibers functionalized with magnetoresponsive compounds [65, 66].
In summary, we present pro- and anti-angiogenic fibrin-based biomaterials that can be combined with cells and printed into complex shapes, as was presented here by bioprinting a meniscus construct with a vascularized and non-vascularized zone as a proof of concept. These bioinks can be utilized for the spatial control of capillary formation in a 3D construct, and the recreation of anisotropic tissue constructs with embedded prevascular capillary networks. With the use of col-1 and CdECM MF, the angiogenic capacity of the bioinks can be easily adjusted, without the need for chemical modification of the ink. These versatile bioinks can further be used for in vitro models for drug testing or studying mechanisms of angiogenesis.
Acknowledgments
The authors acknowledge Liu Hao, Leanne de Silva, Margot Rikkers, and Jasmijn Korpershoek for their help with the HUVEC co-culture and for providing MSCs, MCs and MPCs. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 949806, No. 647426, and No. 814444). The authors acknowledge funding from ReumaNederland (LLP-12 and 22), and R L acknowledges support from to the 2019 Hofvijverkring Fellowship. The authors also acknowledge financial support by Mirai-Program (21-201031456) from JST and Grant-in-Aid for Scientific Research (A) (20H00665) from JSPS. The antibody against collagen type II, developed by T F Linsenmayer, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Ethical statement
Meniscus tissue was collected according to the Medical Ethics regulations of the University Medical Center Utrecht and the guideline 'Human Tissue and Medical Research: Code of Conduct for responsible use' of the Dutch Federation of Medical Research Societies. Human bone marrow was isolated from the iliac crest of patients after their informed consent, when receiving spondylodesis or hip replacement surgery. Isolation and distribution were performed in accordance with protocols approved by the Biobank Research Ethics Committee (isolation 08-001, distribution protocol 18-739, University Medical Center Utrecht). Protocols used are in line with the principles embodied in the Declaration of Helsinki.