
Abstract
Microtia is a small, malformed external ear, which occurs at an incidence of 1–10 per 10 000 births. Autologous reconstruction using costal cartilage is the most widely accepted surgical microtia repair technique. Yet, the method involves donor-site pain and discomfort and relies on the artistic skill of the surgeon to create an aesthetic ear. This study employed novel tissue engineering techniques to overcome these limitations by developing a clinical-grade, 3D-printed biodegradable auricle scaffold that formed stable, custom-made neocartilage implants. The unique scaffold design combined strategically reinforced areas to maintain the complex topography of the outer ear and micropores to allow cell adhesion for the effective production of stable cartilage. The auricle construct was computed tomography (CT) scan-based composed of a 3D-printed clinical-grade polycaprolactone scaffold loaded with patient‐derived chondrocytes produced from either auricular cartilage or costal cartilage biopsies combined with adipose-derived mesenchymal stem cells. Cartilage formation was measured within the construct in vitro, and cartilage maturation and stabilization were observed 12 weeks after its subcutaneous implantation into a murine model. The proposed technology is simple and effective and is expected to improve aesthetic outcomes and reduce patient discomfort.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Microtia is the most frequent congenital ear anomaly and ranges in severity from mild structural abnormalities to anotia, a complete absence of the outer ear. It may occur as an isolated congenital defect or as part of a spectrum of anomalies or syndromes [1]. Microtia is often associated with hearing loss, and patients typically require treatment for hearing impairment and surgical ear reconstruction. Autologous reconstruction using costal cartilage is the current gold-standard treatment for patients with significant auricle deformation or damage. This procedure is a complex surgical technique and results in donor site morbidity and includes challenges in producing auricles with acceptable aesthetic results [2].
For over two decades, human auricle or external ear tissue engineering has been pursued as an alternative to existing auricular reconstruction methods [3–6]. Various difficulties have emerged during the fabrication process, including insufficient expansion of isolated chondrocytes, insufficient cartilage formation and mechanical instability.
In a recent study [7], patient-specific ear-shaped cartilage was engineered using expanded microtia chondrocytes and biodegradable compound scaffolds. The engineered auricle grafts were used for auricle reconstruction in five microtia patients and achieved satisfactory aesthetic outcomes, with mature cartilage formation over the 2.5 year follow-up. Despite demonstrating the typical chondrogenic potential of cells originating in aberrant microtia tissue, there were limitations to the work. The auricle design was based on a polycaprolactone (PCL) mesh with polyglycolide sponges that were pressed in between two negative molds. The process was not automated and included multiple steps, which can cause variations in the final product.
Recent attempts have been made to fabricate an auricle using direct 3D bioprinting techniques, but these are still in pre-clinical phases, with various hurdles yet to be overcome [8–13]. One of the main challenges is finding a bioink suitable for both reproducible additive manufacturing and meeting biological requirements [9]. It is essential to fabricate a stable scaffold that will preserve its shape upon implantation, sustain the forces exerted by cells in the environment, and degrade at a rate that matches the rate of cartilage formation.
Alternatively, indirect 3D printing can be applied to fabricate complex water-soluble sacrificial molds [14]. After mold printing, in a grid pattern, the desired material is cast into the mold and the entire construct is freeze-dried to achieve pores within the material; the mold is then washed with water, leaving behind both large pores from the 3D printing process and small pores from the freeze-drying step. This approach has a significant advantage compared to direct printing since the mechanical properties of the sacrificial material are highly controllable, which allows for precise printing and the fabrication of fine details. Moreover, porosity can be controlled. Areas with high porosity are essential for cell spreading and matrix secretion, whilst denser areas are crucial for shape maintenance and construct stability to withstand the surrounding stress upon implantation.
A full-sized pediatric ear requires 450 million expandable autologous auricular chondrocytes [7]. Cell quantity is a limiting factor since the differentiation potential is progressively lost with increasing numbers of cell passages [15]. Stem cells proliferate easily but should be cautiously directed to differentiate into chondrocytes [9] Recent evidence has shown that adding mesenchymal stem cells (MSCs) to the culture increases cartilage formation. MSCs can be easily isolated and expanded without losing their chondrogenic potential. Two processes drive the mechanism behind increased cartilage formation in the presence of MSCs: differentiation of MSCs into chondrocytes in the presence of chondrocytes and the influence of MSCs on chondrocytes to increase their proliferation and cartilage extracellular matrix (ECM) secretion [16].
Bone marrow-derived MSCs are extensively used to create auricle replacements. However, their use is associated with complex cell isolation and patient discomfort. Exploitation of adipose-derived MSCs can overcome these limitations. These cells can be easily isolated concurrently with the auricular chondrocytes as they are taken from the adipose tissue surrounding the auricle and hence do not require an additional procedure. These cells were previously shown to differentiate into chondrocytes in the presence of transforming growth factor beta (TGFb3) and chondrocytes [17].
In this study we aimed to fabricate auricular constructs by using 3D-printed molding that will both withstand exerted forces upon implantation and enhance cell proliferation and cartilage secretion. In addition, we examined the effect of a combination chondrocytes from different origins co-cultured with MSCs on in vitro cartilage formation within the construct and in vivo cartilage maturation and stabilization following its subcutaneous implantation in a murine model.
2. Materials and methods
2.1. Experimental design
To bioengineer patient-specific ear-shaped cartilage for microtia patients, microtia auricular chondrocytes (mACs), costal chondrocytes (CCs), microtia auricular adipose-derived mesenchymal stem cells (maMSCs), and costal adipose-derived mesenchymal stem cells (cMSCs) were isolated and expanded. The chondrogenic potential of the cells was assessed using an in vitro plug assay. Molds were 3D-printed and filled with clinical-grade PCL discs and CT scanned-based PCL auricle constructs with strategically reinforced areas. In vivo cartilage formation and stabilization post-implantation of the constructs were observed in a subcutaneous murine model (figure 1).
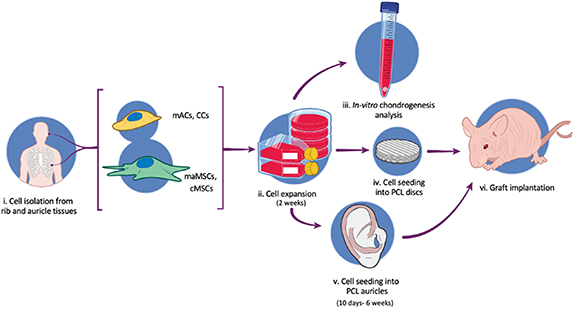
Figure 1. Experimental design. (i) mACs, CCs, maMSCs and cMSCs were isolated from the rib and auricle of patients with microtia. (ii) Cells are expanded and (iii) their chondrogenic potential was examined through in vitro plug assay. (iv) Cells were seeded into PCL discs or (v) PCL auricles created by 3D-printing molding and (vi) implanted into the subcutaneous space of athymic nude mice.
Download figure:
Standard image High-resolution image2.2. Scaffold preparation
PCL discs scaffold were molded using 3D printing and freeze-drying [14]. The discs were created by printing butenediol vinyl alcohol co-polymer (BVOH) meshed molds. Then, 600 µl medical-grade PCL solution was poured into the mold, followed by rapid construct freezing to avoid evaporation. Samples were then lyophilized to form pores within the polymer bulk, and then washed with water to remove the BVOH mold. The final scaffold was dried and sterilized (figure 2).
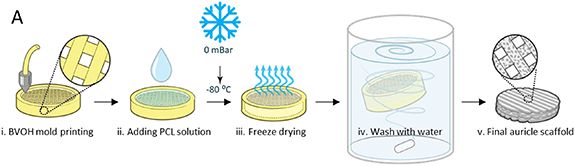
Figure 2. PCL disc fabrication and characterization. (i) BVOH mold printing. (ii) PCL solution is injected into the mold. (iii) The entire construct is then freeze-dried overnight. (iv) The mold is washed with water. (v) The PCL disc is then dried and sterilized and is ready for seeding.
Download figure:
Standard image High-resolution image2.3. Auricle scaffold preparation
The 3D files used in this work are presented in the supporting information section (available online at stacks.iop.org/BF/14/015010/mmedia).
The mold was designed as a negative auricle based on a CT scan using 3D Slicer (open-source 3D printing toolbox). The fabrication was conducted within a supporting box with inlet channels, allowing for PCL solution loading. For the molds used for the reinforced regions, the denser areas were left as a void with no infill. Solidworks and Slicer software (Prusa) were used to design the molds.
Briefly (figure 3):
- (a)A Digital Imaging and Communications in Medicine (DICOM) CT scan of an auricle was imported into the 3D slicer software to generate a 3D Stereolithography (STL) model based on the patient-specific scan.
- (b)The unrefined STL was processed in Blender (open-source 3D creation suite) to remesh and smooth the object.
- (c)The refined STL was then imported into Solidworks using the ScanTo3D add-on to generate surfaces and a solid computer aided design (CAD) object. The stiffer part was 3D sketched onto the solid auricle model, and by using the swept cut tool, the stiffer part of the auricle was subtracted from the auricle model. The auricle was then positioned in a supporting box with injection holes, and an STL file of the auricle (excluding the stiffer areas) and the supporting box (excluding the auricle) was generated and imported to the Slicer software (Prusa).
- (d)The two objects were then aligned, and slicing was applied.
- (e)The auricle was filled with 50% rectilinear infill and the supporting box was filled with 10% rectilinear infill (figure S1).To create the PCL discs, 50% infill was designed in 9 mm diameter discs. All molds were printed from water-soluble BVOH (Verbatim) using a Prusa MK2.5 printer with a 0.4 mm nozzle.
- (f)To prepare the PCL solution, medical-grade PCL (Polymed) was dissolved in dioxane (overnight, under shaking, 70 °C) to form a 10% (w/v) solution. PCL solution was then injected into the mold through the inlet channel using a 1 ml pipettor.
- (g)The filled molds were placed in a −80 °C freezer overnight, followed by lyophilization overnight.
- (h)To dissolve the BVOH molds, constructs were washed with distilled water overnight (water was replaced every few hours). The auricles were easily separated from polymer residuals that accumulated in the supporting box.
- (i)PCL auricles were soaked in 70% ethanol solution for 3 h for sterilization.
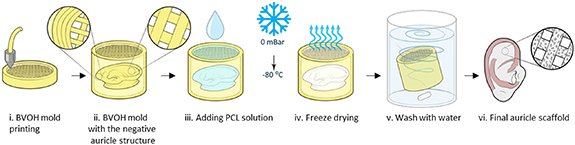
Figure 3. PCL-auricle scaffold fabrication process. (i) The BVOH mold was printed in a supporting box with the negative auricle in it. (ii) The mold was printed as a grid and the edge was completely sealed to enable separation of the auricle from the box. (iii) PCL solution was added to the mold. (iv) Constructs were then freeze-dried overnight. (v) The mold was washed with water and dissolved. (vi) The final auricle scaffold.
Download figure:
Standard image High-resolution image2.4. Cell culture
2.4.1. Cell isolation
The study protocol was approved by the Sheba Medical Center Ethics Committee (4745–17-SMC). Informed consent was obtained from parents of patients with microtia who underwent auricular reconstruction at Sheba Medical Center in Tel Hashomer. Cells used in this study were collected from four different patients (age 10–12 years, male and female). Cartilage remnants were taken from the microtic auricle, rib cartilage and adipose tissue remnants after sculpturing the new auricle. (table 1).
Table 1. A listing of all isolated cells used in this study.
| Cell type | Acronym |
|---|---|
| Microtia auricular chondrocytes | mACs |
| Costal chondrocytes | CCs |
| Microtia auricular adipose-derived mesenchymal stem cells | maMSCs |
| Costal adipose-derived mesenchymal stem cells | cMSCs |
To isolate the chondrocytes, either rib cartilage or auricular cartilage were cut into small pieces (approximately 1 mm), followed by a Phosphate-buffered saline (PBS) with 1% penicillin-streptomycin (pen-strep, Biological-Industries) wash and incubation with collagenase 2 for 12–14 h. The sample was then passed through a 100 µm strainer. Growth medium (40 ml DMEM F12 with 10% fetal bovine serum (FBS) and 1% pen-strep) were added to the filtered sample, which was then centrifuged (2000 g, 10 min). The medium was aspirated, and the cells were resuspended in a growth medium and seeded at a density of 5000 cells mm−2.
To isolate the adipose-derived MSCs, either rib fat or auricular fat were cut into small pieces and incubated with 2 mg ml−1 collagenase 1 in DMEM (10 ml collagenase for every 3 g tissue) and incubated for 1 h, 37 °C. DMEM F12 (Biological Industries) with 10% FBS was added to the solution in equal parts to the collagenase 1, and the sample was then filtrated through a 100 µm and then a 40 µm strainer. The solution was centrifuged for 7 min, 500 g. The medium was aspirated, and the cells were resuspended with the growth medium. One day later, the medium was replaced with Nutristem (Biological Industries) and exchanged every 2–3 d.
2.4.2. Flow cytometry
Flow cytometry was used to characterize mesenchymal phenotype using a MSC marker antibody panel (R&D Systems). maMSCs were fixed in 4% PFA (ChemCruz, Santa Cruz) for 10 min at 4 °C. To remove the fixation solution, the cells were centrifuged at room temperature (RT) for 2 min at 600 g and then washed in 2% FBS (Gibco) in PBS 1X (Sigma). The cells were incubated with the primary antibodies for 30 min at RT and washed in 2% FBS in PBS 1X and centrifuged at RT for 4 min at 300 g. Next, the cells were incubated with a secondary antibody, donkey anti-mouse Alx647 (Jackson, 1:400), for 30 min at RT in the dark. The cells were washed in 2% FBS in PBS 1X and centrifuged at RT for 4 min at 300 g. Cells were analyzed using the LSR-II flow cytometer (BD) instrument and the data was assessed with FlOWJO (BD) Analysis software.
2.4.3. Cell seeding
Before seeding, PCL discs and auricles were washed three times with PBS and then with a growth medium to create a hydrophilic environment for cell seeding. The construct was then stained with phenol red.
Chondrocytes and MSCs were trypsinized using 2X trypsin (Gibco) and resuspended with 7.5 U thrombin (EVICEL). BAC2 solution (15 mg ml−1; the same volume as the thrombin) was then added to the cells, which were then seeded onto the auricle or scaffold. Unloaded areas were easily detected as they remained red, whilst the phenol red color disappeared from the seeded areas. Cells were seeded at a density of 50 × 106 cells ml−1 along with fibrin to mediate cell attachment. For the auricle, 800 µl (40 × 106 cells) of the fibrin solution was used. For the PCL discs, 20 µl (875 × 103cells) of fibrin were used.
Seeded constructs were incubated (1 h, 37 °C), after which a growth medium was added. Two days after seeding, differentiation medium (DMEM F12 (pen-strep (1%), Biological Industries), TGF-β (10 ng ml−1, Prospec), ITS premix (50 mg ml−1, Corning), ascorbic acid (50 μg ml−1, Sigma), dexamethasone (100 nM, Sigma), amphotericin B (0.25 μg ml−1, Biological Industries)) was added. The volume of medium added depended on its size; 150 ml for the 100% size auricle, 100 ml for the 70% size auricle and 4 ml for the discs.
To demonstrate homogenous cell seeding and spreading, GFP expressing fibroblasts (Angioproteomie) were seeded into PCL auricle scaffolds in the same manner as described above. Constructs were imaged 5 d post-seeding using a confocal microscope (LSM700, Zeiss).
2.4.4. Chondrocyte plugs
Different combinations of chondrocytes (auricular or rib origin) and adipose-derived MSCs (auricular or rib origin) were mixed and centrifuged for 5 min at 400 g to create plugs. For the co-culture group, MSCs and chondrocytes were mixed in a 1:1 ratio. Basal medium was added to the plugs for the first 2 d, after which differentiation medium was added. The medium was changed every 2–3 d. After 6 weeks of culture, the plugs were fixed with 4% paraformaldehyde for 10 min, washed three times with PBS, incubated in 30% (wt/vol) sucrose solution, embedded in optimal cutting temperature compound (Tissue-Tek) and frozen for subsequent cryosectioning (5–7 μm). Three plugs were created per group.
2.5. Graft implantation
The animal study was approved by the Technion's inspection committee on the constitution of the animal experimentation (assigned approval number: 84–2013). Athymic nude mice (male, 7–9 weeks-old; Harlan Laboratories) were anesthetized with isofluorane. Small (∼2 cm) and large (∼4 cm) incisions were made in the skin for the discs and auricles, respectively, and grafts were transplanted in the subcutaneous space. The skin was then sutured with 5–0 absorbable sutures. Mice were sacrificed after 12 weeks, followed by excision of the grafts. The grafts were then cut into three pieces for mechanical testing, staining, and biochemical analysis.
Number of constructs used in the animal experiments:
- For the different cell combination assessment: mAC- three constructs, CCs- six constructs, CCs+ cMSCs- three constructs, mACs+ CCs- three constructs, mACs+ maMSCs- two constructs.
- For short vs long in-vitro incubation time: for the 10 d—four constructs in four mice and for the 6 weeks—two constructs in two mice.
- For the full ear experiments: three with reinforced regions and three without reinforced regions.
2.6. Immunofluorescent and biochemical staining
Constructs were dehydrated in ethanol and embedded in paraffin at 65 °C. Sections (5 µm thick) were cut, deparaffinized with xylene and rehydrated, then stained with alcian-blue, safranin-O with fast green and hematoxylin and eosin.
For immunofluorescent staining, sections were incubated in chondroitinase ABC (0.25 U ml−1, Sigma-Aldrich) in tris-acetate buffer (1X, Sigma-Aldrich) (1 h, 37 °C) and then in keratinase (0.25 U ml−1, Sigma-Aldrich) in tris-acetate buffer (1X, Sigma-Aldrich; 30 min, 37 °C). Constructs were then washed with PBS and immersed overnight in bovine serum albumin (BSA) solution (5%; Millipore).
Samples were then incubated (overnight, 4 °C) with anti-human collagen 2 (1:250; R&D), anti-human aggrecan (1:500; R&D) and anti-human elastin (1:1000, Sigma-Aldrich) antibodies. After extensive rinsing, they were then incubated (3 h, room temperature) with Cy3-labeled (1:100; Jackson Immunoresearch Laboratory), Alexa-488 (1:400; ThermoFisher Scientific), and Alexa-647 (1:400; ThermoFisher Scientific) secondary antibodies mixed with DAPI (Sigma-Aldrich).
For 2D plate immunofluorescent staining, cells were fixated in paraformaldehyde (4%) for 20 min and then permeabilized with 0.3% Triton X‐100 (Bio Lab Ltd) for 10 min. Constructs were then washed with PBS and immersed in BSA solution (5%; Millipore) overnight. Samples were then incubated with the following primary antibodies for 2 h: mouse anti‐human CD73 (1:100; Acris), mouse anti‐human 105 (1:100; BD). Cells were then treated with Alexa‐488‐labeled (1:400; ThermoFisher Scientific) secondary antibodies and DAPI (Sigma‐Aldrich), for 2 h, at room temperature.
2.7. Biochemical assays
In vitro and in vivo samples were digested with papain solution (40 µg ml−1 diluted in 20 nM ammonium acetate, 1 mM Ethylenediamine tetraacetic acid (EDTA), and 2 mM dithiothreitol) for 48 h, at 65 °C. Deoxyribonucleic acid (DNA) content was measured using the Hoechst dye-binding assay [18]. Proteoglycan concentration was quantified by measuring the amount of sulfated glycosaminoglycan (GAG) using the 1,9-dimethylmethylene blue (DMMB) dye-binding spectrophotometric assay [19]. Collagen content was quantified by adding 6 N hydrochloric acid for 18 h, 110 °C and neutralization with NaOH. Hydroxyproline level was measured using the chloramine-T/Ehrlich's spectrophotometric assay [20]. These experiments were done with at least two technical replicates for each sample.
2.8. Scanning electron microscope imaging
Scaffold morphology was examined using a scanning electron microscope (SEM). Before scanning, samples were coated with a gold-palladium mixture using a Polaron gold coater and then scanned with a Quanta 200 microscope (FEI).
2.9. microCT imaging
To further assess the macro-structure of the construct, PCL constructs were scanned using a high-resolution microCT scanner (Skyscan 1276, Bruker, Kontich, Belgium), using the following scanning parameters: source voltage 65 kV, source current 57 μA, applied 0.5 mm aluminum filter. Scanning was performed using a 0.3° rotation step with frame averaging (3), yielding a total of 1201 projections. Image acquisition was performed with a scaled pixel size of 21 μm. Back projections were reconstructed using NRecon (Skyscan, version 1.7.2.0), CTAnn Software (Skyscan, version 1.17.7.2) was used for segmentation, and CTVox (Skyscan, version 2.2.0) was used for 3D visualization.
2.10. Confocal imaging
Constructs sections with immunofluorescent staining have been imaged using a confocal microscope (LSM700; Zeiss), with 2.5X, 5X, 10X, 20X and 40X lenses. For biochemical staining, sections have been imaged using an inverted microscope (Axio Observer; Zeiss), with 5X, 10X, 20X and 40X lenses. All samples were imaged in at least three regions and representative images were presented.
2.11. Mechanical testing
To determine the stiffness of each region, two types of 6 mm disc molds were fabricated and filled with PCL solution; the first was with no infill to ensure the same structure as the reinforced region and the second was with 50% rectilinear infill to mimic the structure of the auricle bulk.
Stress–strain curves were generated using an AR-G2 rheometer (TA Instruments, New Castle, DE, USA) equipped with parallel-plate geometry. Samples were compressed to 90% of their original width, and stress was then calculated by dividing the measured force by the cross-section area. Young's modulus was determined by calculating the slope of the stress–strain curve in the linear region. For in-vivo auricle samples, samples were cut into ∼20 mm2 pieces before testing and imaged immediately after testing to find the cross-section area.
2.12. Statistical analysis
Presented data include the mean ± standard deviation. Student's t-test was performed to compare two groups. To examine differences between multiple groups, a one-way analysis of variance was performed, with posthoc Tukey's multiple comparisons. Results were considered significant for p < 0.05. Statistical analysis was performed using GraphPad Software, a computerized statistical program.
3. Results
3.1. Characterization of the chondrogenic potential of isolated cells
Imaging of mACs, CCs, and adipose-derived MSCs demonstrated viable and proliferative 2D-adherent cultures exhibiting typical morphology (figure S2(A)). The purity of the isolated MSCs was verified by immunofluorescence staining for CD105 and CD73 (figure S2(B)). Moreover, the immunophenotype of the MSCs was analyzed using flow cytometry which demonstrated positive expression of mesenchymal markers CD44, CD90, CD105 and low expression of the hematopoietic marker CD45 (figure S2(C)).
Chondrogenic potential and the effect of the addition of MSCs on cultured chondrocytes were assessed in cartilage plugs comprised of monoculture of mACs, CCs, microtia maMSCs and cMSCs. In addition, to examine additional cell combinations, three co-culture groups were seeded: mACs with maMSCs, CCs with cMSCs, and mACs with CCs. Plugs prepared with both auricular and CCs were analyzed for cartilage formation initiation; safranin-O, alcian-blue and hematoxylin and eosin staining showed typical lacunae formation and secretion of glycosaminoglycans. Safranin-O and alcian-blue staining revealed no differences between the different groups, with the exception of MSCs plugs, for which safranin-O staining was absent and alcian blue staining was pale (figures 4(A) and S3). GAGs normalized to DNA content revealed that cartilage formation from mAC plugs was higher than any other groups, CCs, mACs + maMSCs and mACs + CCs produced fewer GAGs per cells but still had more than CCs + cMSCs (figure 4(B)).
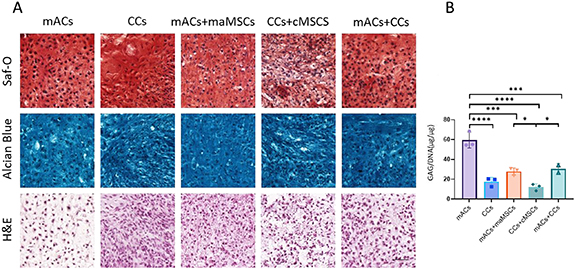
Figure 4. Characterization of the in vitro chondrogenic potential of chondrocytes and MSCs. (A) Safranin-O, alcian-blue and hematoxylin and eosin (H&E) staining of sections of scaffold-free cartilage plugs composed of different combinations of cell types that were cultured for 6 weeks in vitro. Scale bar = 50 μm. (B) The DMMB biochemical assay was performed to papain-digested plugs to measure GAG secretion. Error bars show SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Download figure:
Standard image High-resolution image3.2. Characterization of scaffold-based chondrogenic potential
Small PCL discs were designed to enable the assessment of cartilage formation under the same conditions as large auricle PCL scaffolds (figure 2). The use of small discs is simpler to produce and test and can aid in determining the optimal conditions for auricle construct fabrication. The resulting scaffold contained large pores (∼500 μm), formed from the printed mold, and small pores, formed during the freeze-drying process (∼100 μm) (figures 5(A) and (B)).

Figure 5. Cartilage formation within constructs of different cell combinations. (A) The final PCL disc. Scale = 1 mm. (B) Scanning electron microscopy (SEM) images of the PCL disc show large pores resulting from the grid mold and small pores resulting from the freeze-drying step. Scale bar = 500 µm (left image), 100 µm (right image). The large pore diameter is approximately 500 µm, while the small pore diameter range was 10–100 µm. (C) Disc extracted 12 weeks after implantation into athymic nude mice. Scale = 1 mm. (D) Constructs seeded with different cell combinations were cultured in vitro, implanted into the subcutaneous space of athymic nude mice, extracted 12 weeks post-implantation and then stained for aggrecan, elastin, coll-2, Saf-O, and alcian blue. Scale bar = 25 μm. (E) GAG levels normalized to DNA content, collagen levels normalized to DNA content, and collagen levels normalized to GAG levels. Error bars show SEM.
Download figure:
Standard image High-resolution imageDifferent cell combinations of mACs, CCs, ACs with maMSCs, CCs with cMSCs, mACs with CCs were mixed with fibrin solution and seeded into 6 mm PCL disc scaffolds, cultured in vitro for 6 weeks, and implanted into the subcutaneous space of athymic nude mice. Mice were sacrificed 12 weeks post-implantation, followed by dissection of the graft area (figure 5(C)). Staining of the constructs extracted 12 weeks post-implantation revealed lacunae formation within all groups (figure 5(D)). No significant differences in GAGs and collagen secretion were observed between the different cell combinations (figure 5(E)). Therefore, other cell combinations can be integrated into the construct when needed.
3.3. In vitro culture time effect on cartilage formation
Due to the logistic and technical benefits of shortening the in vitro culture time, including reduced time to implantation, reduced culturing costs and reduced possibility of graft contamination, the impact of in vitro culture time on cartilage formation was assessed for two different mACs grafts. Grafts cultured for 10 d or for 6 weeks prior to implantation exhibited similar morphology, with typical lacunae formation (figure 6(A)). In addition, the two sets of grafts showed similar Young's modulus (figure 6(B)). No significant differences in GAGs and collagen levels of the two implant groups (figures 6(C)–(E)). Taken together, a short in vitro incubation time is sufficient to achieve cartilage production.
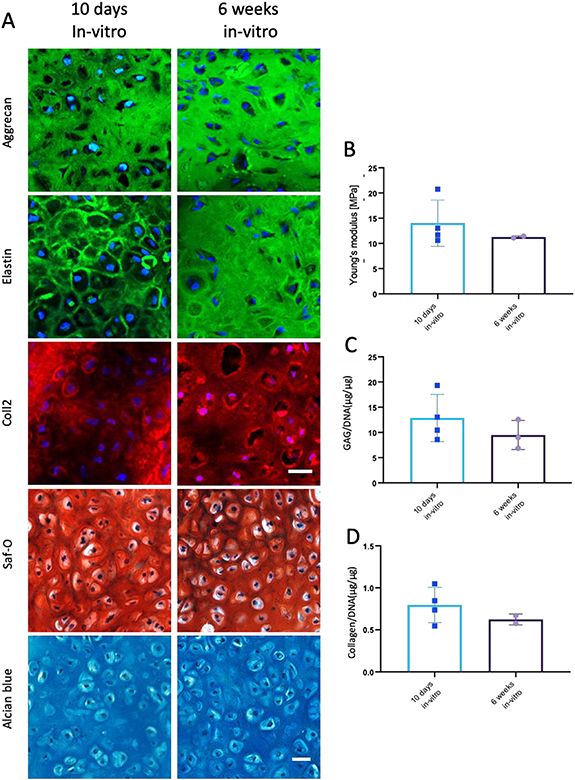
Figure 6. Cartilage formation within constructs cultured for 10 d versus 6 weeks in vitro. (A) mAC constructs were cultured in vitro for 10 d (left) or 6 weeks (right) and implanted into the subcutaneous space of athymic nude mice for 12 weeks. Extracted constructs were stained with aggrecan, elastin, and coll-2 antibodies and Saf-O and alcian blue. Scale bar = 25 μm. (B) Young's modulus of mAC constructs cultured in vitro for 10 d or 6 and 12 weeks in vivo. (C) GAG and (D) collagen secretion quantification in papain-digested constructs of mAC constructs cultured in vitro for 10 d or 6 and 12 weeks in vivo. GAG and collagen levels were normalized to DNA content.
Download figure:
Standard image High-resolution image3.4. Ear-shaped scaffold characterization
When designing a scaffold for auricle reconstruction, scaffold porosity must be sufficient to enable cell attachment and matrix production (figure 7(A)) while being sufficiently mechanically stable to withstand the surrounding stress upon implantation. To ensure these traits, reinforced regions were introduced into the auricle design (figure 7(B), left) by printing the BVOH molds at lower densities in specific areas, which would enable increased polymer accumulation within them (figure 3). The resulting auricle was subsequently composed of two areas—the supporting frame with a highly dense polymer with small pores ∼100 µm, formed during the freeze-drying process (figure 5(B)) and the bulk area with pores formed during both the freeze-drying process and by the BVOH mold. In this manner, the auricle scaffold shape was close to the native auricle, with reinforced regions to withstand pressure upon implantation and varying interconnected pores to allow cell spreading, migration and ECM secretion (figure 7(B), right).

Figure 7. Ear-shaped scaffold characterization. (A) The auricle design, PCL-auricle scaffold and a microCT-scan of the PCL-auricle scaffold. (B) The auricle design and the PCL-auricle scaffold with reinforced regions (shown in blue). (C) Stress–strain curve of the reinforced (with the printed grid) versus the nonreinforced (without the printed grid) area. (D) The measured slope of the graph (Young's modulus). Error bars show SEM, ****p < 0.0001. (E) Confocal microscopy images of a full-sized auricle seeded with green fluorescent protein (GFP)-labeled cells. Scale bar = 5 mm.
Download figure:
Standard image High-resolution imageRheology testing was performed to demonstrate the difference in stiffness between the two regions (figures 7(C) and (D)). A stress–strain analysis identified a different mechanical response of the two areas to compression. Slope measurement of the first linear range revealed a significant difference in Young's modulus between the areas of the construct with higher (∼2 MPa) versus lower PCL density (∼0.2 MPa) (figure 7(D)).
To ensure the seeding process resulted in an even distribution of the cells, GFP-expressing cells were seeded on the PCL auricle constructs; confocal images revealed that the cells formed homogenous layers in all dimensions of the constructs (figure 7(E)).
3.5. In vivo implantation of human auricle grafts in a murine model
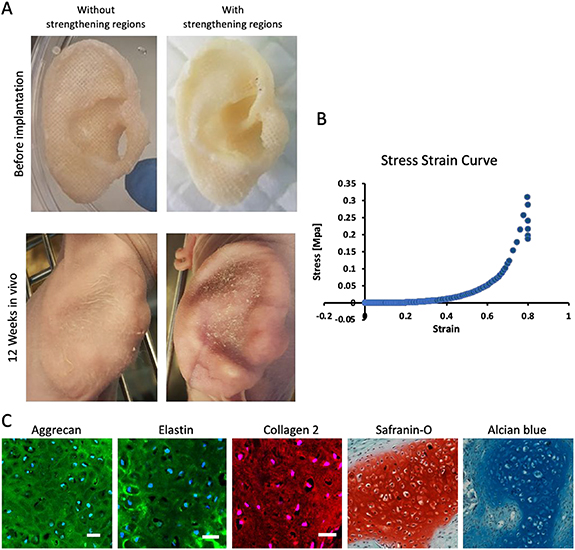
After validating the chondrogenic potential of embedded cells and designing the appropriate auricle scaffold, the mechanical and morphological stability of mAC-embedded auricle grafts were assessed 12 weeks after their subcutaneous implantation into athymic nude mice. Images of the implanted auricle demonstrated that the reinforced regions played a crucial role in maintaining graft shape; all curvatures and details were maintained, whereas without the reinforced regions, the auricle deformed and did not retain its original shape (figure 8(A), lower panel). The constructs' Young's modulus value extracted from the stress strain curve (figure 8(B)), 2.55  0.793 MPa, was similar to that of a native auricle, 1.73 MPa [21]. The samples contained lacunae and typical cartilage ECM secretion (figure 8(C)).
0.793 MPa, was similar to that of a native auricle, 1.73 MPa [21]. The samples contained lacunae and typical cartilage ECM secretion (figure 8(C)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Cartilage formation within human auricle grafts. (A) Images of grafts without and with reinforced regions. (B) Representative stress–strain curve of a 12 week implant. (C) Aggrecan, elastin, coll-2, Saf-O and alcian blue section staining of the implants with reinforced regions. Scale bar = 50 μm.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
Auricular defects pose one of the most significant challenges in reconstructive surgery of the head and neck. This is due to the unique 3D anatomical architecture of the auricle, with its multiple concavities of the cartilage and the thin, delicate skin cover. The current gold-standard treatment for such defects is a surgical reconstruction from autologous harvested rib cartilage sculpted into an auricle structure. However, this technique causes donor site morbidity, increases infection risk, and relies on the surgeon's artistic skills.
This work developed an efficient method to bioengineer a full-scale human autologous auricle. The CT scan-based auricle construct was comprised of a 3D-printed clinical-grade biodegradable PCL scaffold loaded with patient‐derived chondrocytes produced from either auricular cartilage or costal cartilage biopsies combined with adipose-derived MSCs and auricular perichondrium cells. In addition, it included strategically reinforced regions to ensure the mechanical stability necessary to withstand the surrounding stress upon implantation. Cartilage formation within the construct and cartilage maturation and stability were demonstrated in vivo using a subcutaneous immune-deficient murine model.
Previous attempts were performed to construct anatomically shaped cell-seeded auricular constructs, including scaffold‐free auricular tissues [22], bioprinting multiple cell-laden hydrogels with supporting PCL polymer [11], incorporating decalcified bone matrix with cartilage sheets [23], creating cartilage shells that cover alloplastic implants [24].
Choosing a suitable material for the auricle construct plays a crucial role in cartilage formation and graft integration. To increase cartilage formation, decellularized extracellular matrix bioinks were used. Bhamare et al digested goat auricular cartilage polymerized with polyvinyl alcohol and gelatine and used it as a bioink for extrusion printing by computer-aided design [25]. In another study, porcine auricular cartilage was methacrylated to form a photo-cross-linkable hydrogel and suspended with chondrocytes to form a bioink [26]. While these constructs are optimal for cell growth and cartilage formation, their mechanical stability upon implantation might not be sufficient. To overcome this, a combined strategy should be applied; Chang et al used laser sintering-3D printing of PCL with porcine cartilage tissue inserts to create auricle constructs [27]. Otto et al used fuse-deposition printing to combine PCL with a photocrosslinked human auricular cartilage progenitor cell-laden gelatin methacryloyl hydrogel; this approach was shown to preserve the construct's shape over 30 d of culturing in-vitro [28]. In another study, direct PCL printing was used to fabricate an auricle construct seeded with porcine chondrocytes with collagen gel [29]. In another study, 3D-printed PCL mesh was combined with two non-woven polyglycolic acid (PGA) meshes and seeded with human chondrocytes [30]. However, these approaches are limited in creating fine structures and pore size might be too large to be filled with cells. In the approach presented in this study, one medical-grade material was used at varying densities, which was achieved by 3D-printing molding. This resulted in a construct with predetermined reinforced regions that were shown to maintain its delicate structure upon implantation. The complexity of the production process was minimal and enabled rapid production of a large mass of constructs with a fine structure and pores appropriate for cell attachment. Moreover, the use of fibrin gel was previously shown to support neocartilage formation [31]. Another study showed that fibrin enhanced cell distribution and viability in the chondral composites [32].
As previously mentioned, one of the main obstacles to creating cartilage replacements is the limited expansion capacity of chondrocytes. Cell passaging results in cells dedifferentiation into fibroblast phenotypes, which secrete a fibrous ECM, resulting in impaired mechanical properties [16]. In addition, mACs were shown to have impaired cartilage formation when compared to healthy auricular chondrocytes [33, 34]. These conditions render it impossible to engineer an auricle from mAC cultures alone and require integrating other cells in the construct.
Previous studies have investigated the differences of cartilage production by auricular versus articular chondrocytes; Osch et al reported on an advantage for auricular chondrocytes, in that cartilage formation was double for the same number of cells [35].
Studies have shown that chondrocytes can induce chondrogenesis from MSCs in vitro and in vivo by secreting soluble chondrogenic factors [30].
While our in vitro assessments demonstrated a significant advantage for the mACs monoculture, which correlated with an earlier report [36], neither GAG nor collagen secretion, nor typical lacuna formation was significantly different between the constructs of various cell types upon their implantation into the subcutaneous space of athymic nude mice. This result is in correlation with Morrison et al who co-cultured porcine adipose-derived stem cells and chondrocytes (in different ratios, at 1:1, 2:1, 5:1, 10:1, and 0:1) within hyaluronic acid/collagen hydrogel seeded into 3D-printed PCL scaffolds. The group found no difference in sulfated-glycosaminoglycan production between the different groups [37]. These findings also correlated with those reported by Zhang et al who found no biomechanical or biochemical differences between the monoculture of microtia chondrocytes and the co-culture of microtia chondrocytes and bone marrow stromal cells [30].
Although mACs formed cartilage-forming constructs, auricular chondrocytes are often unavailable due to severe trauma or lack of auricle tissue under severe microtia conditions. In such cases, the use of CCs and costal adipose-derived MSCs (cMSCs) could be beneficial, as these alternative cell sources decrease the need for cell expansion and improve cartilage formation. Previous studies have documented contradicting data about GAG formation from chondrocytes of different origins. In one study, GAG formation by costal and auricular cartilage sheets was similar in vitro; however, there was an advantage to the auricular grafts in vivo [38]. In another study, constructs composed of CCs had significantly higher secretion of GAGs than auricular chondrocyte constructs [39]. These differences may be due to the different biomaterials used in the studies. The synthetic polymer used in our study could have caused variations in the in vivo results. However, this should be further studied to reach definite conclusions.
In vitro culturing time can be a crucial factor in the clinical translation of the proposed approach. Hence, we were interested in examining whether culturing the engineered auricle constructs for a longer time period would result in increased secretion of cartilage components, as previously reported [32]. To this end, mACs were cultured within PCL-disc constructs for either 10 d or 6 weeks and then implanted into the subcutaneous space of athymic nude mice for 12 weeks. Surprisingly, no significant differences in lacuna development, or collagen-2, elastin, GAG and collagen expression were observed between the two constructs. In fact, a slight advantage was measured for the 10 d in vitro constructs, which aligned with the observations of Zhang et al who implanted engineered auricles after a 1 week in vitro incubation period [30].
This work presented a fast and simple method for engineering a medical-grade auricle. Use of 3D-printing molding with tissue-specific reinforced regions, along with a short in vitro culture period of scaffolds with auricular cartilage cells, yielded patient-specific ear-shaped cartilage, suitable for transplants for microtia patients. In cases of a complete lack of auricular cartilage, such as anotia and trauma, the method can be modified to integrate costal cartilage cells. Moreover, this technique could be expanded into more products in the clinic, such as nasal reconstruction or other orthopedic implants.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 818808). We thank Abigail Newman for proofreading the manuscript.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Author contributions
S L A, A A S B K, M M, Y S, I R, O H S, S D W, S I D, S L E designed research; S L A, A A S Y S, I R, M M, M B, J Z, O K K, S I D performed research; S L A, O K K, S L analyzed data; and S L A, O H S, S L E wrote the paper.
Conflict of interest
The authors declare no conflicts of interest and do not have any financial disclosures.