

Abstract
Human pluripotent stem cells (hPSCs) are required in large numbers for various biomedical applications. However, the scalable and cost-effective culturing of high quality hPSCs and their derivatives remains very challenging. Here, we report a novel and physiologically relevant 3D culture system (called the AlgTube cell culture system) for hPSC expansion and differentiation. With this system, cells are processed into and cultured in microscale alginate hydrogel tubes that are suspended in the cell culture medium in a culture vessel. The hydrogel tubes protect cells from hydrodynamic stresses in the culture vessel and limit the cell mass smaller than 400 μm in diameter to ensure efficient mass transport, creating cell-friendly microenvironments for growing cells. This system is simple, scalable, highly efficient, defined and compatible with the current good manufacturing practices. Under optimized culture conditions, the AlgTubes enabled long-term culture of hPSCs (>10 passages, >50 days) with high cell viability, high growth rate (1000-fold expansion over 10 days per passage), high purity (>95% Oct4+) and high yield (5.0 × 108 cells ml−1), all of which offer considerable advantages compared to current approaches. Moreover, the AlgTubes enabled directed differentiation of hPSCs into various tissue cells. This system can be readily scaled to support research from basic biological study to clinical development and the future industry-scale production.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs), are attractive cell sources for various biomedical applications including cell therapies [1], tissue biofabrication [2–4], drug screening and toxicity tests [5, 6]. These applications require large numbers of high quality cells [7]. For instance, it is estimated that ∼109 cardiomyocytes, 109 β cells, 1012 red blood cells, and 1011 platelets are needed for treating one myocardial infarction or diabetic patient, or one blood or platelet transfusion, respectively [1]. Approximately 1010 cells are required for engineering a human-size heart or liver [5], or for screening a library with one million compounds once [6]. Considering the large patient population and the large numbers of chemical libraries that can be screened, massive numbers of cells are therefore needed [7].
However, the scalable and cost-effective culturing of high quality hPSCs and their derivatives, especially for clinical applications, remains a challenge [1, 7, 8]. In vivo, majority of human cells including the hESCs reside in 3D microenvironments that have plenty of cell–cell and cell–ECM (extracellular matrix) interactions, sufficient supply of nutrients, oxygen and growth factors, and no or minimal hydrodynamic stresses [9–13]. The current hPSC culturing methods, however, provide culturing conditions that are very different from these physiological microenvironments, leading to low culture efficiency and difficulty to culture cells at large scales. Two-dimensional (2D) cell culturing (e.g. with cell culture flasks) is the most widely used method for culturing hPSCs. However, a 2D environment is very different from an in vivo environment. Additionally, 2D culturing is labor-, space- and reagent-consuming, and considered only suitable for preparing small scale cells [1, 7, 8]. Three-dimensional (3D) suspension culturing (e.g. with stirred-tank bioreactors), in which cells are suspended in the culture medium, has been widely studied to scale up the production and has achieved some attractive results [1, 7, 8]. However, a critical problem with 3D suspension culturing is the uncontrolled cell agglomeration (figures S1(A) and (B) are available online at stacks.iop.org/BF/10/025006/mmedia) [1]. hPSCs have strong cell–cell interactions that make them aggregate [12, 13]. Suspended single hPSCs first associate to form small cell clusters (i.e., initial cell clustering), which is important for the survival and growth of dissociated hPSCs [12, 13]. These small cell clusters grow as spherical aggregates (i.e., spheroids) when cells divide. Spheroids, however, frequently fuse to each other to form large cell agglomerates (i.e., agglomeration) (figures S1(A) and (B)). Agglomeration leads to inhomogeneity in cell aggregate size and is detrimental to cell culture [1]. For instance, the transport of nutrients, oxygen, and growth factors to, and the metabolic waste from cells located at the center of large cell agglomerates (e.g., >400 μm diameter) become insufficient, leading to slow cell growth, apoptosis, and uncontrolled differentiation [1, 14]. While agitating the culture can enhance mass transport and reduce cell agglomeration, agitation also generates critical hydrodynamic stresses (e.g. shear stresses) that negatively impacts cells [1, 15, 16]. With the uncontrolled cell aggregation and hydrodynamic stresses, significant cell death, low cell growth and low volumetric yield are common in 3D suspension culturing [7]. For instance, we and others showed hPSCs typically expanded 4-fold in 4 days to yield around 2.0 × 106 cells ml−1 (table S1) [17–19]. These cells occupy ∼0.4% of the bioreactor volume [20].
Furthermore, the hydrodynamic conditions (e.g. the medium flow direction, velocity, shear force and chemical environment) generated by the agitation in 3D suspension culturing are very complicate [1, 7, 8, 15–19, 21]. They are spatially and temporally varied, and sensitive to many factors including the bioreactor design (e.g. impeller geometry, size and position, vessel geometry and size, positions of probes for pH, temperature, oxygen), medium viscosity and agitation rate [1, 21]. They are currently not well understood and hard to control [1, 15, 16, 21]. Additionally, how cells respond to these complicate hydrodynamic conditions is not well known and is hard to study [1, 15, 16]. These complicate conditions and knowledge gap lead to culture inconsistency and difficulty in scaling up, as shown by recent studies on producing cardiomyocytes from hPSCs in stirred-tank bioreactors [22, 23]. For three batches (∼100 ml suspension cultures) using a hESC line, the final yield ranged from 40 million to 100 million cells, and final cardiomyocyte purity ranged from 54% to 84%. Using the same bioreactor and a different hPSC line, the final yield ranged from 89 million to 125 million cells, and final cardiomyocyte purity ranged from 28% to 88% [22, 23]. When the culture volume was scaled from ∼100 ml to ∼1000 ml, the yield and differentiation efficiency were altered. Re-optimizing bioreactor design, agitation rate and culture protocol was required [22, 23]. This indicates the challenge of further scaling up the culture volume (e.g. to hundreds liters) since optimizing multiple factors in large culture volume is costly. To our best knowledge, the largest 3D suspension culture volume demonstrated to date for hPSCs is less than 10 l [1, 24].
Researchers have tried to encapsulate and culture hPSCs in various hydrogel scaffolds with the goal of using scaffolds as physical barriers to isolate cells from agglomeration and hydrodynamic stresses [7, 17, 25–29]. We recently found hPSCs could be effectively cultured in a very soft and thermoreversible hydrogel matrix (Mebiol® Gel) [7, 17, 29]. Within this hydrogel matrix, single hPSCs clonally grew into uniform spheroids (diameter ∼100–150 μm) (figures S1(C) and (D)). The hydrogel scaffold enabled serial expansion of hPSCs with good cell viability, growth rate (e.g. 20-fold/5 days), yield (e.g. 2.0 × 107 cells ml−1) and purity, all of which offered improvements over 3D suspension cultures. However, this system has some limitations that make it inappropriate for large-scale cell production. First, it is not universally applicable. For some hPSC lines and human cell types, the dissociated cells cannot survive and clonally grow in the hydrogel. Though the exact reason is still unknown, we speculate that the scaffold prevents the initial cell–cell interactions that are important for cell's survival [7, 17, 29]. Second, the volumetric yield and expansion per passage are moderate, probably due to the fact that cells encapsulated in hydrogels have no free space for expansion and they have to deform scaffolds to create spaces for the new cells [7, 17, 29]. Third, the hydrogel is mechanically fragile, making it hard to be processed into formats (e.g., beads) suitable for large-scale cell cultures. And lastly, the hydrogel remains stable for about two weeks, which prohibits its use for long-term cell culture, such as differentiating hPSCs into neurons, which requires more than one month. The results of this study showed that eliminating the cell agglomeration and hydrodynamic stresses could significantly improve the culture efficiency, however, free spaces that allow the initial cellular interactions and subsequent cell expansion were also important to achieve high culture outcomes.
We here report a novel technology that can overcome the limitations of current hPSC culturing methods and provide physiologically relevant cell culture microenvironments. With this technology, hPSCs are processed into and cultured in microscale alginate hydrogel tubes (or AlgTubes) that are suspended in the cell culture medium in a culture vessel (figures 1(A) and (B)). The hydrogel tubes create cell-friendly microspaces that allow cells to interact with each other and expand. Meanwhile, they protect cells from hydrodynamic stresses in the culture vessel and confine the cell mass less than 400 μm (in radial diameter) to ensure efficient mass transport during the entire culture (figures 1(A) and (B)). Additionally, this technology is simple, scalable, defined and compatible with the current good manufacturing practices that make it commercially viable. We showed that, under optimized culture conditions, the AlgTubes offered paradigm-shifting improvements in cell viability, growth, yield, culture consistency and scalability over current hPSC culturing technologies.
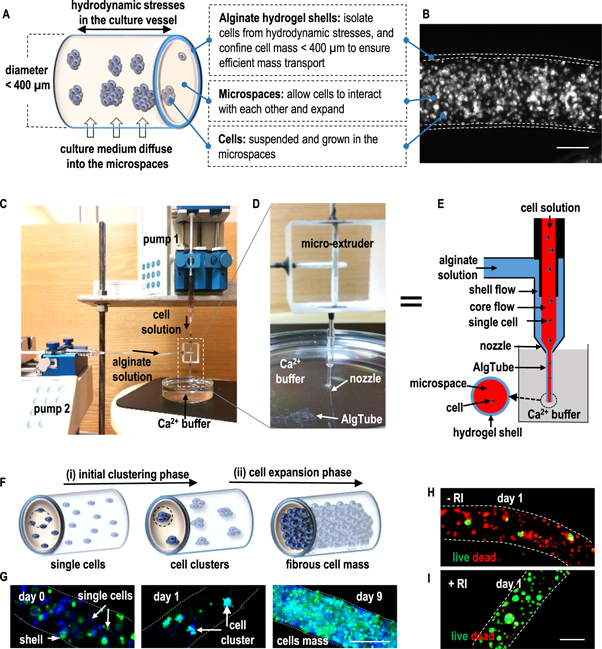
Figure 1. Overview of the AlgTube cell culture system. (A), (B) Design principles of AlgTubes. Cells are processed into and cultured in microscale alginate hydrogel tubes that are suspended in the cell culture medium in a culture vessel. The hydrogel tubes protect cells from hydrodynamic stresses (e.g. shear stresses) in the culture vessel and confine the cell mass less than 400 μm (in radial diameter) to ensure efficient mass transport. They also create physiologically relevant microspaces for cells to interact with each other and expand. Cell culture medium can efficiently diffuse through the alginate hydrogel shell. An illustration (A) and microscope picture (B) of an AlgTube are shown. (C)–(E) Extruding AlgTubes. The setup for extruding AlgTubes consists of two syringe pumps, a custom-made micro-extruder and a CaCl2 buffer. To make AlgTubes, a cell solution (e.g. single cells suspended in hyaluronic acid solution) and an alginate solution is pumped into the central channel and side channel of the micro-extruder, respectively, to form coaxial core–shell flows that are extruded through the nozzle of the micro-extruder into the CaCl2 buffer. The shell alginate flow is instantly crosslinked by Ca2+ ions to form an alginate hydrogel tube. (F) Proposed cell growth mechanism in AlgTubes. After extruding the AlgTubes, individual cells first associate to form small cell clusters (i.e. the initial clustering phase). Subsequently, cells proliferate and the small cell clusters expand to form fibrous cell mass (i.e. the cell expansion phase). (G) This mechanism was confirmed with experiment. Two vials of H9-hESCs, stained with blue and green fluorescent dyes, respectively, were mixed at 1:1 and cultured in AlgTubes. Single cells (day 0), small cell clusters (day 1), and fibrous cell mass (day 9) were seen. (H), (I) ROCK inhibitors (RIs) are required for the survival of single H9-hESCs. Live/dead staining showed majority of cells went apoptosis after 24 h without RIs (−RI) (H). Cells survived and grew well with RIs (+RI) (I). Scale bar: 200 μm.
Download figure:
Standard image High-resolution imageMethods
Materials
Fib-iPSCs and MSC-iPSCs were obtained from Human Embryonic Stem Cell Core, Harvard Medical School. MSC-iPSCs and Fib-iPSCs were reprogrammed from mesenchymal stem cells and fibroblasts, respectively, by George Q. Daley Lab (Children's Hospital Boston, MA) and have been well characterized and described in the literature [56]. H9-hESCs (Cat #: WA09) were purchased from WiCell Research Institute. Authentication and test for the free of mycoplasma were performed by WiCell Research Institute. A cell bank was established after we received the cells for each line. For experiments, each aliquot from the cell bank was used for less than 10 additional passages. For hPSCs, the cell growth kinetics and expression of pluripotency markers (e.g. Oct4, Nanog), karyotype, and differentiation capability were tested to ensure these cells had no major genetic mutation or loss of their differentiation capabilities. L Wnt3A cells (ATCC® CRL-2647™) were acquired from ATCC.
Reagents and their supplies: PNIPAAAm-PEG (cat # MBG-PMW20-5005, Cosmobio USA); E8 medium (cat # A1517001, life technologies); Accutase: (cat # A1110501, life technologies); Y-27632 (cat # 129830-38-2, Sigma); Matrigel (cat # 08-774-552, Corning); Sodium Hyaluronate: (cat # HA 700K-5, Lifecore Biomedical); Methylcellulose stock solution (cat # HSC001, R&D system); Sodium alginate (cat # 194-13321, 80–120cp, Wako Chemicals). Vybrant multicolor cell-labeling kit (cat # V22889, Molecular Probes); DMEM (cat # SH30003.03, GE Healthcare); FBS (cat # s11150, Atlanta biologicals); G418 (cat # BP6731, Fisher); Luciferase report assay kit (cat # K801-200, Biovision); Calcein AM viability dye (cat # 50-169-52, eBiosicence); Ethidium homodimer I (cat # 40010, Biotium); DAPI (cat # D9542, Sigma). RNeasy mini kit (cat # 74104 QIAGEN). Propidium Iodide (cat # 195458, MP Biomedicals, LLC). Antibodies used in this study were summarized in table S2.
Culturing hPSCs in 3D PNIPAAm-PEG hydrogel
hPSCs cells were maintained in PNIPAAm-PEG hydrogel before culturing in AlgTubes as described in our previous publications [7]. Briefly, single hPSCs cells were mixed with 10% PNIPAAm-PEG solution at 4 °C and cast on a 12-well plate, then incubated at 37 °C for 15 min to form a stable hydrogel before adding warm E8 medium plus 10 μM Y-27632 (Rock inhibitor). To passage cells, 2 ml of ice cold PBS was added to each well for 2 min to dissolve the hydrogel. Spheroids were collected by centrifuging at 100 g for 3 min, treated with Accutase at 37 °C for 12 min and dissociated into single cells with pipettes.
Processing AlgTubes
A custom-made micro-extruder was used to process AlgTubes. A hyaluronic acid (HA) or methylcellulose (MC) solution containing single cells and an alginate solution was pumped into the central and side channel of the home-made micro-extruder, respectively, and extruded into a CaCl2 buffer (100 mM) to make AlgTubes. Subsequently, the CaCl2 buffer was replaced by cell culture medium.
Culturing hPSCs in the AlgTubes
For a typical cell culture, 20 μl of cell solution in AlgTubes were suspended in 2 ml E8 medium in a 6-well plate and cultured in an incubator with 5% CO2, 21% O2 at 37 °C. Medium was changed daily. To passage cells, medium was removed and alginate hydrogels were dissolved with 0.5 mM EDTA for 5 min. Cell mass was collected by centrifuging at 100 g for 5 min, treated with Accutase at 37 °C for 12 min and dissociated into single cells for following culture.
Culturing L-Wnt3A-cells in AlgTubes
For a typical cell culture, 20 μl of cell solution in AlgTubes were suspended in 2 ml DMEM medium plus 10% FBS in a 6-well plate and cultured in an incubator with 5% CO2, 21% O2 at 37 °C. Medium was changed daily and collected for quantifying Wnt3A proteins. To quantify Wnt3A proteins, MDA-468 cells (ATCC® HTB-132™) were stably transfected with a luciferase reporter for the canonical Wnt signaling (Addgene, # 24308). These MDA-468-TFP cells were plated in a 96 well plate (5000 cells/well/200 μl medium). 24 h later, 150 μl of fresh DMEM plus 10% FBS and 50 μl of L-Wnt3A-cells conditioned medium was added and incubated for another 18 h. Medium was then removed and cells were washed with PBS once before 200 μl of cell lysis buffer was added and incubated for 10 min at room temperature. 50 μl of cell lysates, 50 μl of substrate A and 50 μl of substrate B from the luciferase assay kit were mixed and the light signals were immediately read with a luminometer. The quantity of Wnt3A protein was calculated with a standard curve.
Culturing hPSCs in the AlgTubes with bioreactor
2 ml of cell solution in AlgTubes were suspended in a home-made bioreactor. Cells were cultured in an incubator with 5% CO2, 21% O2 at 37 °C for 10 days. Medium was stored in a bellow bottle that was periodically pressed to flow the medium into, or released to withdraw the medium from, the container.
Staining, flow cytometry and imaging
Cells cultured on 2D surfaces were fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, and permeabilized with 0.25% Triton X-100 for 15 min, and blocked with 5% donkey serum for 1 h. Cells were then incubated with primary antibodies at 4 °C overnight. After extensive washing, secondary antibodies and 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI) were added and incubated for another 1 h at room temperature. Cells were washed with PBS three times before imaging with a Zeiss Axio Observer Fluorescent Microscopy. The percentage of Oct4+ nuclei was quantified with Image J software. At least 1000 nuclei were analyzed. To stain 3D fibrous cell mass, the cell mass was harvested and fixed with 4% PFA at room temperature for 30 min, and then incubated with PBS + 0.25% Triton X-100 + 5% goat serum + primary antibodies at 4 °C for 48 h. After extensive washing, secondary antibodies in 2% BSA were added and incubated at 4 °C for 24 h. Cells were washed with PBS three times before imaging with Nikon A1 Confocal Microscopy. For flow cytometry, single cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, permeabilized with 0.25% Triton X-100 for 15 min, and blocked with 5% donkey serum for 1 h. Cells were then incubated with primary antibodies at 4 °C overnight. After extensive washing, secondary antibodies were added and incubated for another 2 h at room temperature. Cells were washed with PBS two times before assessment using Cytek DxP 10 flow cytometer (BD Biosciences). Analysis was performed using FlowJo. LIVE/DEAD® Cell Viability staining was used to assess live and dead cells, according to the product manual.
EB differentiation
hPSCs released from the AlgTubes were suspended in DMEM + 20% FBS + 10 μM β-mercaptoethanol in a low adhesion plate for 6 days. The cell masses were then transferred into plates coated with 0.1% gelatin and cultured in the same medium for another 6 days, followed by fixation and staining as above.
Teratoma formation in vivo
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Nebraska–Lincoln. All experimental procedures involving animals were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Nebraska–Lincoln. 2 × 106 hPSCs were suspended in 25 μl of PBS plus 25 μl of Matrigel and injected subcutaneously at the back of the neck of the NOD-SCID mice (female, age 7 weeks, Charles River Laboratory). Tumors were harvested after 6–12 weeks. The tumors were fixed with 4% PFA for 48 h and sequentially dehydrated with 70%, 95%, and 100% ethanol, and deffated with xylene for 2 h before embedding in paraffin. Then, 10 μm thick sections were cut and stained with hematoxylin and eosin.
Karyotype
Karyotyping was performed by WiCell Research Institute.
Mesodermal induction
hPSCs in AlgTubes were cultured in E8 medium for 7 days, and then in DMEM/F12 medium with 1% B27 minus insulin and 12 μM CHIR99021 for 24 h before fixation and staining.
Endodermal induction
hPSCs cells in AlgTubes were cultured in E8 medium for 7 days; then in RPMI 1640 medium with 1% GlutaMAX, 1% B27 minus insulin and 4 μM CHIR99021 for 24 h; then in RPMI 1640 medium with 1% GlutaMAX and 1% B27 minus insulin for an additional 24 h before fixation and staining.
Cardiomyocyte differentiation
hPSCs cells in AlgTubes were cultured in E8 medium for 7 days, then in DMEM/F12 with 1% B27-insulin for 6 days, then DMEM/F12 with 1% B27 for 9 days. The following small molecules were added during the differentiation: 12 μM CHIR99021 for days 0–1; 5 μM IWR1 for days 3–4. Cell mass were released on day 11 to a 6-well plate. Beating cardiomyocytes were filmed on day 15.
Direct comparison of culturing hPSCs in 2D, static 3D and dynamic 3D suspension and the AlgTubes
For 2D culturing, 2 × 105 hPSCs were seeded in six-well plate coated with Matrigel and cultured for 7 days. For static 3D suspension, 2 × 105 cells ml−1 hPSCs were suspended in low attachment 6-well plates and cultured for 7 days. For dynamic 3D suspension, 2 × 105 cells ml−1 hPSCs were suspended in a spinner flasks (∼75 rpm) and cultured for 7 days. For AlgTubes, 1 × 106 cells ml−1 hPSCs were seeded and cultured for 9 days. The culture medium was changed daily and collected for measuring dead cells. Adenylate kinases (AKs) are ubiquitous proteins present in all eukaryotic and prokaryotic cells. They are rapidly released into the culture medium upon damage of the plasma membrane of cells. AKs in the cell culture medium were quantified with the bioluminescence cytotoxicity assay kit (cat # JM-K312-500, MBL medical & biological laboratories) according to the product instruction, and normalized with a standard curve to calculate the dead cells in the culture medium every 24 h. Samples were harvested and live cells were counted with trypan blue. Single cells were fixed with 70% ethanol for cell cycle analysis with propidium iodide staining and flow cytometry.
RNA sequencing and data analysis
Total RNAs of day 3 hPSCs cultured in 2D, static 3D, dynamic 3D and AlgTubes were prepared with RNeasy mini kit (cat # 74104 QIAGEN) according to the manufacturer's instruction. Libraries were prepared with TruSeq Stranded mRNA Library Prep Kit and sequenced with Illumina NextSeq 500. 20 million 75 bp paired-end reads were generated for each sample. The reads were trimmed to make sure the average quality score was larger than 30 and the minimum length was 40 bp. All trimmed short reads were mapped to the human Genome (version hg19) using TopHat, allowing up to two base mismatches per read. Reads mapped to multiple locations were discarded. Numbers of reads in genes were counted by the HTSeq-count tool using corresponding human gene annotations and the 'union' resolution mode. For normalization and pair-wise comparisons, differentially expressed genes were identified by using DEseq to analyze the numbers of reads aligned to genes. The thresholds for differential expression were set at fold-change >2 and adjusted P-values <0.001 for the null hypothesis.
Principle component analysis (PCA) analysis
PCA was implemented with R function prcomp and the 3D PCA plot was produced with R package rgl. For PCA analysis, genes with zero reads were discarded and a natural log transformation was applied to the number of reads for each gene. The proportion of total variance accounted for by the first three PCs was 64.46% (PC1 29.75%, PC2 18.06%, PC3 16.64%), which suggested that those three PCs are sufficient to describe the major changing pattern of all samples. Heatmap for gene expression. For each pathway, the expression values of genes in the given pathway were extracted and, to further stabilize the variance, a natural log transformation was applied to normalized read numbers for genes. The heatmap of expression profiles for genes in a given pathway was plotted with R package heatmap. In the heatmap, red indicates high expression while green represents low expression.
Statistical analysis
The data are presented as the mean ± S.D.. We used an unpaired t-test to compare two groups and one-way ANOVA to compare more than two groups. P < 0.05 was considered statistically significant. Homogeneity of variances among groups were confirmed using Bartlett's test. Conformity to normal distribution was confirmed using the Kolmogorov–Smirnov test. For in vitro experiments, a sample size of 3 or 5 was selected so that at a significance level of 0.05 there at is least a 90% chance of detecting a two standard deviation difference in cell numbers between the groups.
Data availability
The final processed data and raw fastq files were submitted to Gene Expression Omnibus (GEO) with the accession number GSE99776. All other data supporting the findings of this study are available within the paper and its Supplementary Information.
Results
A micro-extruder was designed and made for processing AlgTubes (figure 1). To make AlgTubes, a cell solution and an alginate solution is pumped into the central channel and side channel of the micro-extruder, respectively, to form coaxial core–shell flows that are extruded into a CaCl2 buffer. The shell alginate flow is instantly crosslinked by Ca2+ ions to form an alginate hydrogel tube (figures 1(C)–(E)). Subsequently, the CaCl2 buffer is replaced by the cell culture medium and cells are grown in the tubes. The micro-extruder can have multiple nozzles for simultaneously processing multiple AlgTubes to make large-scale AlgTubes (figure S2, and supplementary video 1). It was found that the cell solution and alginate solution should have close viscosity to process defect-free AlgTubes. Both hyaluronic acid (HA) and methylcellulose (MC) solutions could be used to suspend cells for this purpose. Similar to 3D suspension culturing (figures S1(A) and (B)), single hPSCs in AlgTubes quickly (e.g. within 24 h) associated to form small cell clusters that subsequently grew and filled the tubes (figures 1(F) and (G)). We termed these two phases as the initial clustering phase and subsequent cell expansion phase, respectively. In 2D and 3D suspension culturing of hPSCs, ROCK inhibitors (e.g. Y-27632) are added to the culture medium for the first 24 h after passaging to inhibit the dissociation-induced cell death [30]. We found ROCK inhibitors (Y-27632) were also required for the survival and growth of the single hPSCs in AlgTubes. Without ROCK inhibitors, majority of cells died within 24 h (figures 1(H) and (I)). To passage cells, the AlgTubes could be dissolved with the cell-compatible ethylenediaminetetraacetic acid (EDTA) solution (0.5 mM, 5 min at room temperature) to release the micro cell mass, which could be further treated with Accutase (10 min at 37 °C) and dissociated into single cells for the following passage.
We found AlgTubes made with a wide range of alginate concentrations, tube diameters and hydrogel shell thicknesses all supported efficient culturing of hPSCs (figures S3–S5). For instance, hPSCs grew efficiently and similarly in AlgTubes made with 1.0%, 1.5% and 2.0% alginate solutions (figure S3). When seeded at 1.0 × 107 cells ml−1, cells expanded ∼14-, 23- and 50-fold to yield ∼1.4×, 2.3 × and 5.0 × 108 cells ml−1 on day 5, 7 and 9 of the culturing with above 94% cells expressed the pluripotency marker Oct4 at all three alginate concentrations. The AlgTube's outer diameter is roughly equal to the inner diameter of the nozzle of the micro-extruder and can be controlled through adjusting the nozzle's diameter (figure S2). At a given AlgTube outer diameter, the hydrogel shell thickness could be controlled by varying the ratio of the cell solution flow rate and alginate solution flow rate and predicted by using the equation described in figure S4(A). When seeded at 1.0 × 107 cells ml−1 and cultured in AlgTubes (with ∼400 μm diameter) with 30, 40, and 70 μm shells or AlgTubes (with 30–50 μm shells) with diameter of 400 μm, 250 μm and 120 μm for 9 days, hPSCs all expanded around 50-fold to yield ∼5.0 × 108 cells ml−1 with above 90% cells retained the pluripotency marker Oct4 (figures S4 and S5). Since both HAs and MCs could be used to suspend cells, we studied whether they differentially influenced cell culture. Our research showed HAs and MCs resulted in similar cell viability, expansion and pluripotency (figure S6). The capability to support consistent hPSC culturing under a wide range of AlgTube conditions gives users large flexibility in processing the tubes.
AlgTubes supported efficient hPSC culturing with a wide range of seeding densities (figures 2 and S7). When seeded at 1.0×, 2.0×, 5.0×, and 10.0 × 106 cells ml−1, hPSCs expanded 499-, 247-, 104- and 51-fold by day 9, respectively, yielding around 5.0 × 108 cells ml−1 (figure 2). For all conditions, cells grew through the initial clustering phase and the subsequent cell expansion phase. At 24 h after processing the AlgTubes, the cell cluster size was larger for higher seeding density but the numbers of cell clusters per volume were similar for different seeding densities (figure 2(A)). These results showed hPSCs grew faster at lower seeding density (figures 2(B) and (C)). However, the seeding density did not influence pluripotency. >95% cells expressed the pluripotency marker Oct4 for all seeding densities (figures 2(D) and (E)). It is extremely exciting that hPSCs seeded at ultralow densities could grow as well without sacrificing cell viability and pluripotency (figure S7). When seeded at 1.0×, 3.0 × and 5.0 × 105 cells ml−1, hPSCs expanded 4200-, 1700- and 1000-fold to yield ∼4.2×, 5.1×, and 5.0 × 108 cells ml−1 by days 14, 12 and 10, respectively.
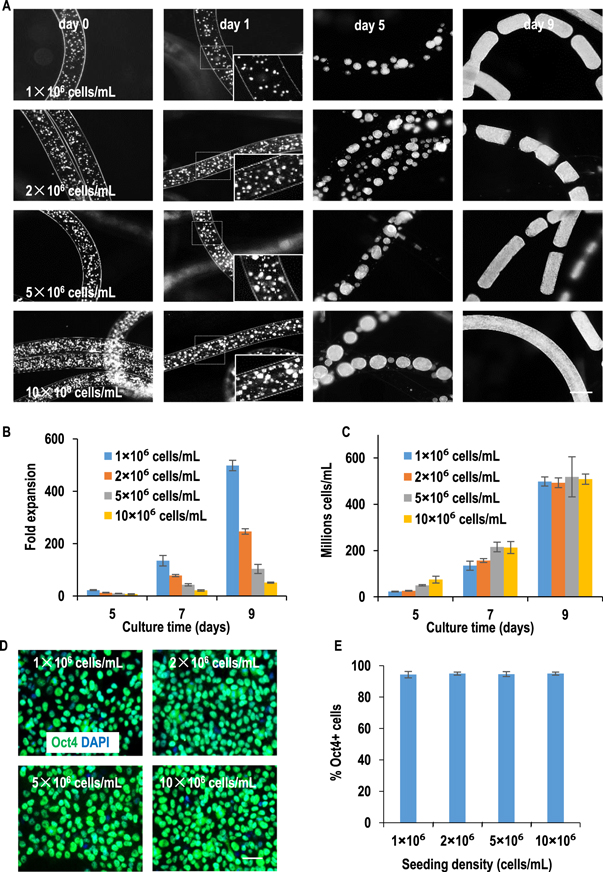
Figure 2. The growth kinetics of H9-hESCs seeded at various seeding densities in AlgTubes. H9-hESCs were seeded at 1×, 2×, 5× or 10 × 106 cells ml−1 in AlgTubes and cultured for 9 days. (A) Phase images of cells on day 0, 1, 5 and 9 in AlgTubes. After 24 h, single cells formed small clusters. The cell clusters were bigger at higher seeding density, but the number of clusters were similar (insert, day 1). Clusters grew into spheroids (day 5) and then fibrous cell mass (day 9). (B) The expansion fold on day 5, 7 and 9 showed hPSCs grew faster at lower seeding density, (C) while the final volumetric yields on day 9 were very close. (D) Immunostaining showed ∼95% cells expressed the pluripotency marker Oct4 after the 9 day culture. H9-hESCs were released from AlgTubes on day 9 and plated on a Matrigel-coated plate overnight before fixing and staining. (E) % of Oct4+ cells were quantified with Image J Data are presented as mean ± SD of three independent replicates (n = 3). Scale bar: (A) 400 μm; (D) 50 μm.
Download figure:
Standard image High-resolution imageAfter performing these optimizations, the AlgTubes were evaluated for culturing multiple hPSC lines for long-term (e.g. 10 passages, figures 3, S8 and S9). All hPSCs grew well in AlgTubes, and cell morphology, viability, growth rate, and pluripotency among multiple hPSC lines were similar. In the 10-passage culture, when seeded at 1.0 × 107 cells ml−1, hPSCs consistently expanded ∼15-fold/passage/5 days and >95% of cells expressed the pluripotency marker Oct4 (figures 3(A) and (B)). The long-term culture did not alter cell phenotype as shown by the similar cell morphology, viability, growth kinetics and pluripotency of hPSCs at passage 1 and 10 (figures 3, S8 and S9). When seeded at 1.0 × 107 cells ml−1, hPSCs expanded ∼50-fold to yield 5.0 × 108 cells ml−1 on day 9 at both passage 1 and 10. In vitro embryoid body (EB) differentiation and in vivo teratoma formation confirmed their pluripotency after the long-term culture. All hPSCs were successfully differentiated into endodermal, mesodermal and ectodermal cells in the EB assay (figures 4(A), S10(A) and (B)). All hPSCs formed teratomas containing the three germ layer tissues in immune-deficient mice in the teratoma assay (figures 4(B), S10(C) and (D)). In addition, after long-term culture, all hPSCs retained normal karyotypes (figures 4(C), S10(E) and (F)). These results show that AlgTubes can support long-term culturing of hPSCs.
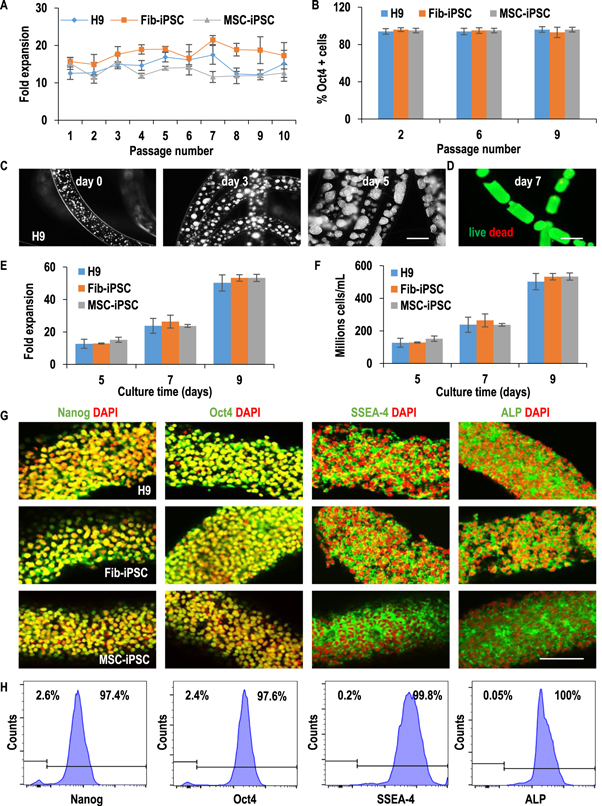
Figure 3. AlgTubes supported long-term culturing of hPSCs. H9-hESCs, Fib-iPSCs and MSC-iPSCs were cultured in the AlgTubes for 10 passages. Fib-iPSCs and MSC-iPSCs are iPSCs made from human fibroblasts and mesenchymal stem cells, respectively. (A) When seeded at 1.0 × 107 cells ml−1, all three hPSCs consistently expanded ∼15-fold per passage per 5 days and (B) >95% of the cells expressed Oct4 during the 10-passage culturing. Data are presented as mean ± SD of three independent replicates (n = 3). (C)–(H) The growth kinetics of hPSCs at Passage 10. (C) Phase images of day 0, 3 and 5 H9-hESCs and (D) live/dead staining of day 7 H9-hESCs in AlgTubes. (E), (F) Seeded at 1.0 × 107 cells ml−1, all three hPSCs expanded ∼15-, 24-, 53-fold to yield ∼150×, 240×, 530 × 106 cells ml−1 on day 5, 7 and 9, respectively. Data are presented as mean ± SD of five independent replicates (n = 5). (G) Confocal microscope images and (H) flow cytometry analysis showed majority cells on day 9 at passage 10 expressed pluripotency markers: Nanog, Oct4, SSEA-4 and alkaline phosphatase (ALP). Scale bar: (C), (D) 400 μm; (G) 100 μm.
Download figure:
Standard image High-resolution image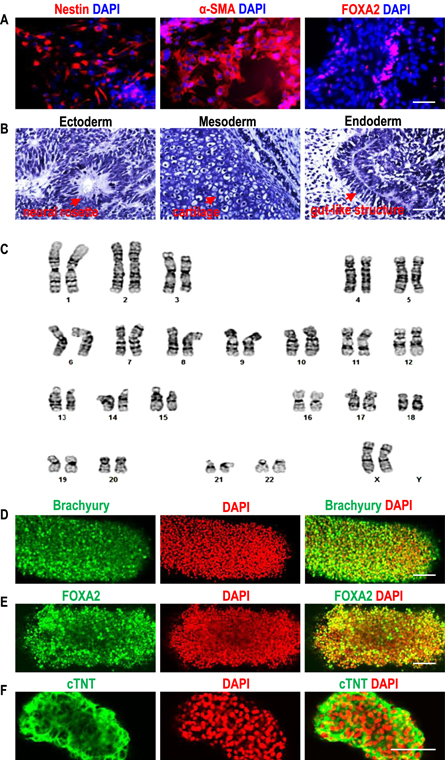
Figure 4. hPSCs retained pluripotency after long-term culturing in AlgTubes. After culturing in AlgTubes for 10 passages, H9-hESCs could be differentiated into the Nestin + ectodermal, α-SMA + mesodermal and FOXA2+ endodermal cells in the EB assay (A); form teratomas containing the three germ layer tissues in teratoma assay (B); and had normal karyotype (C). By further culturing in a mesodermal or endodermal or cardiomyocyte differentiation medium, H9-hESCs in AlgTubes could be differentiated into the corresponding brachyury + mesodermal cells (D) or FOXA2+ endodermal cells (E) or cTNT + cardiomyocytes (F). Fib-iPSCs and MSC-iPSCs showed similar results (supplementary figure 10). Scale bar: (A), (B) 50 μm; (D)–(F) 100 μm.
Download figure:
Standard image High-resolution imageAfter expansion and further culturing in a mesodermal [31, 32] or endodermal [33] or cardiomyocyte [31, 32] differentiation medium, hPSCs in the AlgTubes could be differentiated to corresponding mesodermal or endodermal cells or cardiomyocytes at high efficiency, indicating the AlgTubes support hPSC differentiation (figures 4(D)–(F), and supplementary video 2). Additionally, cells other than hPSCs can be cultured in the AlgTubes. For instance, murine L cells engineered to express Wnt3A proteins were efficiently cultured without notable cell death [34], yielding around 6.0 × 108 cells ml−1 (figure S11). Wnt3A proteins were consistently expressed during a 16 day culture. These results demonstrate AlgTube' potential as a generally applicable culture system.
A prototype bioreactor was built to show AlgTubes' scalability (figure 5). Two milliliters of AlgTubes with hPSCs were processed and contained in a bioreactor. The cell culture medium was stored in a plastic bellow bottle that could be pressed to flow the medium into the bioreactor or released to withdraw the medium from the bioreactor, respectively (figure 5(A)). The pressing and releasing speed, as well as the duration of the interval between the pressing and releasing can be programmed and controlled by the controller (figure 5(B)). Since AlgTubes have similar density with the cell culture medium, they were uniformly suspended and dispersed in the medium when the medium was pumped into the bioreactor. They became collected and contact with each other when the medium was withdrawn from the bioreactor. This alternating dispersion and collection is designed to enhance the medium mixing. Within the bioreactor, hPSCs grew well and yielded ∼5.0 × 108 cell ml−1 by day 10 (figures 5(C) and (D)). >95% of cells expressed the pluripotency markers (figures 5(E) and (F)). This prototype bioreactor could be further scaled up in the future.

Figure 5. An AlgTube-based prototype bioreactor. (A) AlgTubes with cells were suspended in a bioreactor. Medium was stored in a plastic bellow bottle that could be pressed to flow the medium into, or released to withdraw the medium from the bioreactor. (B) Images of the mechanic stage for pressing and releasing the bellow bottle; the controller that can be programmed for the pressing and releasing speed, as well as the duration of the interval between the pressing and releasing; and the bioreactor and bellow bottle. (C) Image of the white fibrous cell mass in the bioreactor on day 10. (D) ∼1.0 × 109 H9-hESCs were produced with 2.0 ml AlgTubes on day 10. (E) Confocal microscope images and (F) flow cytometry analysis showed majority the cells on day 10 expressed pluripotency markers: Nanog, Oct4, SSEA-4 and alkaline phosphatase (ALP). Scale bar: (C) 1 cm; (E) 100 μm.
Download figure:
Standard image High-resolution imageTo assess the superiority of the AlgTubes over current hPSC culturing technologies, we directly compared culturing hPSCs in 2D, 3D suspension without agitation (static 3D), 3D suspension with agitation (dynamic 3D) and the AlgTubes (figure 6). Cells were cultured for 9 days in AlgTubes to allow cells to fill the tubes. Cells were cultured for 7 days in 2D, static 3D and dynamic 3D culturing since the cell numbers had no further increase after 5 days in these cultures. Medium was changed daily. Live cells in the cultures were counted daily. Cells died in each day were quantified through measuring the adenylate kinases (AKs) in the exhausted cell culture medium. AKs, which are ubiquitous proteins presented in cells, are rapidly released into the culture medium upon damage of the plasma membrane. The ratio of dead cells over the dead cells plus the live cells was calculated (e.g. the % dead cells) and used to assess the cell viability each day. Cell proliferation was also evaluated through quantifying the percentage of cells in S and G2/M phases. Seeded at 2.0 × 105 cells ml−1, hPSCs expanded only 5-fold to yield a maximum of ∼1.0 × 106 cells ml−1 in static 3D culturing. At the same seeding density, hPSCs expanded ∼9-fold to yield a maximum of ∼1.8 × 106 cells ml−1 in dynamic 3D culturing. In 2D, cells could expand ∼51-fold to generate ∼1.0 × 107 cells per well of the 6-well plate. In AlgTubes, hPSCs expanded ∼500-fold to yield ∼5.0 × 108 cells ml−1 (figures 6(A) and (B)). Large cell death was observed in static 3D, dynamic 3D and 2D culturing, especially after day 3 or when the cell density reached ∼1.0 × 106 cells ml−1 (figures 6(D) and (E)). In static 3D culturing, the % of dead cells reached ∼43%, 53%, 59% and 62% on day 4, 5, 6 and 7, respectively. In dynamic 3D culturing, the % of dead cells reached ∼28%, 29%, 35% and 40% on day 4, 5, 6 and 7, respectively. In AlgTubes, cell death was low and the % of dead cells was <10% during the entire 9 day culture. Cell cycle analysis showed static 3D, dynamic 3D and 2D culturing did not decrease the % of cells in proliferation (figure 6(C)). To study whether the global gene expression are different, mRNAs of the day 3 cells from the 4 culturing methods were sequenced. On day 3, cell death was not significant and majority of cells were proliferating in the 4 cultures (figures 6(C)–(E)). Hierarchical clustering analysis showed the gene expression profiles were very similar between the 4 cultures (figures 6(F) and S12). These results show that large cell death in 3D suspension culturing leads to low culture efficiency, and the AlgTubes improve the culture outcome through reducing the cell death without significantly altering the global gene expression and cell phenotype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. A direct comparison of culturing H9-hESCs in 2D, 3D suspension without agitation (static 3D), 3D suspension with agitation (dynamic 3D) and AlgTubes. Cells were cultured for 7 days in 2D, static 3D and dynamic 3D culturing, and 9 days in AlgTubes. (A) Expansion fold, (B) live cell density, (C) percentage of cells in S + G2/M phases, and (D) the ratio of dead cells over (dead cells + live cells) or % dead cells in the cultures were quantified. (E) % dead cells was also plotted versus live cell densities. Data are presented as mean ± SD of 3 (n = 3 for (A)–(C)) or 5 (n = 5 for (D), (E)) replicates. Seeded at 2.0 × 105 cells ml−1, H9-hESCs expanded ∼5- or 9-fold to yield a maximum of ∼1.0 or 1.8 × 106 cells ml−1 in static or dynamic 3D suspension, respectively. Large cell death was detected. Starting on day 4 or at 1.0 × 106 cells ml−1 live cell density, the % dead cells reached 40% to 60% for static 3D suspension and 20%–40% for dynamic 3D suspension. Starting on day 4, cell death became significant in 2D culturing. In AlgTubes, cells grew to ∼5 × 108 cells ml−1 with low cell death. The % dead cells was <10% during the entire culture in AlgTubes. The AlgTubes did not enhance cells' proliferation. (F) mRNAs of day 3 cells (3 biological replicates for each culture method) were sequenced. Hierarchical clustering analysis of transcriptomes showed cells from the 4 culture methods had similar gene expression profiles. Note for 2D culturing, the unit for cell density in (B), (E) is cells/well (of 6-well plate).
Download figure:
Standard image High-resolution image{kind=link}
We also directly compared encapsulating and culturing hPSCs in solid agarose, HA, and alginate hydrogels with suspending and culturing hPSCs in AlgTubes (figure S13). When seeded at 1 × 106 cells ml−1, little cell expansions were observed in HA and agarose hydrogels. Small numbers of spheroids were formed and hPSCs expanded ∼3.5-fold in solid alginate hydrogel fibers. In AlgTubes, hPSCs expanded ∼500-fold to yield ∼5.0 × 108 cells ml−1. These results show the importance of the free microspaces for achieving high cell expansion and yield.
Discussion
Based on the results of culturing hPSCs in 2D, 3D suspension and hydrogels [7, 17, 25–29], we proposed that 3D culture microenvironments having free space for the initial cell–cell interactions and subsequent cell growth, but no cell agglomeration and hydrodynamic stresses could significantly improve the culture efficiency. The AlgTubes are designed to provide these physiologically relevant microenvironments (figures 1(A) and (B)). First, cells in AlgTubes are protected from hydrodynamic stresses/conditions by the hydrogel shells. This reduces the hydrodynamic-conditions-induced negative effects [1, 15, 16, 21–23]. Second, cell masses in AlgTubes are controlled to be less than 400 μm to ensure efficient mass transport during the entire culture. Additionally, the cell masses are monodisperse (in radial diameter), which can improve the homogeneity of hPSC expansion and differentiation. Other methods, such as culturing cells with AggreWell™, can precisely control cell aggregate size, however, are not scalable. Third, unlike encapsulating and culturing cells in hydrogels (figure S13), the AlgTubes provides free space for hPSC expansion and does not hinder the initial cellular interaction, leading to high volumetric yield and general applicability.
The use of alginates makes this new technology scalable, cost-effective and GMP-compatible. Alginates are affordable; available in large quantity; non-toxic to cells; have been used in clinics; and can be instantly crosslinked with Ca2+ ions to process large-scale AlgTubes with the scalable extrusion technology (figure S2) [35]. The resulting AlgTubes are mechanically and chemically stable for months and suitable for large-scale and long-term cell cultures; can be dissolved easily with cell-compatible EDTA solution to release the product; and are transparent to allow observation of cell growth [36].
The conceptual and technical innovations of AlgTubes lead to high culture efficiency. We demonstrated long-term culturing (>10 passages) of multiple hPSC lines without uncontrolled differentiation and chromosomal abnormalities (figures 3, S8 and S9). Cultures between batches and cell lines were very consistent (figure 3(A)). hPSCs in AlgTubes had high viability, growth rate (1000-fold/10 days/passage in general) and yield (∼5 × 108 cells ml−1 microspace). The expansion per passage (e.g. up to 4200-fold/passage was achieved in figure S7) and volumetric yield are much higher than current 3D suspension culturing (table S1) [7, 8, 17, 37, 38]. For comparison, in 3D suspension culturing, we typically achieve 4-fold expansion per 4 days to yield around 2.0 × 106 cells ml−1. As discussed later, the high yield and high expansion fold have high impact on large-scale cell production since they significantly reduce the culture volume and time, numbers of passaging operations, and the production cost. hPSCs could be efficiently differentiated into various tissues cells (figure 4 and supplementary video 2). In addition, AlgTubes-based scalable bioreactors could be readily built (figure 5). Our comparative study showed the AlgTubes did not significantly alter hPSCs' gene expression profiles, but significantly reduced cell death, resulting in high cell expansion and yield (figures 6 and S12).
It should be noted that pluripotent stem cells have been cultured in hollow fibers made from dry polymer membranes [39–41]. However, their performance offers no advancements over stirred-tank bioreactors. In addition, harvesting cells from these fibers were very challenging. For instance, it required treating the cells with trypsin for 30–60 min. Moreover, cells could not be observed with microscopy and sampling was hard during the culture. Solid hydrogel fibers (e.g. not hollow) have been studied for processing tissues [42–44]. However, the AlgTubes are hollow and conceptually and technically different from these systems.
The AlgTubes will be of broad interest to individual laboratories, institutions, and biotechnology companies working on regenerative medicine and cancer therapies. First, AlgTubes provide laboratories an easy-to-use tool for studying biology in 3D, which can better replicate the in vivo biology than 2D cultures [45]. For instance, AlgTubes may be used to culture organoids [46, 47], study cellular assembly into tissues [48] and reprogram cells [49], etc. Second, the AlgTubes can greatly benefit translational research. Using hPSC-derived cardiomyocytes for treating myocardial infarction as an example [50], 2D culturing was first used for developing the differentiation protocol and the initial demonstration of efficacy and safety in rodents [51]. Subsequently, large effort was spent to develop 3D suspension culturing for preparing sufficient cardiomyocytes for tests on large animals [52, 53]. If successful, substantial amounts of effort will be required to develop a new and large bioprocess for making sufficient cells for clinical trials and eventually clinics [50]. Since cell's phenotype, safety and efficacy are bioprocess-related, extensive and expensive in vitro and in vivo characterization of cell phenotype, safety, and efficacy are required after each change of the culture method [54]. These efforts make developing cellular therapeutics slow and expensive. AlgTubes is scalable and can be used for culturing cells for all these development stages. For instance, ∼200 μl AlgTubes can be used for developing the differentiation protocol. ∼2 ml AlgTubes are sufficient to produce ∼109 cells for preclinical small animal studies. ∼200 ml AlgTubes can yield ∼1011 cells for tests on large animals and clinical trials. The AlgTubes can save significant time and investment for developing cellular therapeutics. Third, the AlgTubes are very attractive for industry-scale cell production. A simple comparative calculation of producing ∼1.5 × 1014 hPSCs (from ∼108 hPSC seeds) with stirred-tank bioreactors and AlgTubes shows the significant impact of AlgTubes' high cell expansion per passage and volumetric yield (figure S14). For the calculation, we assumed 4-fold expansion per 4 days per passage for stirred-tank bioreactors and 1000-fold expansion per 10 days per passage for AlgTubes as well as a seeding density of 5 × 105 cells ml−1 and a passaging efficiency (i.e., % of cells remaining viable after one passaging) of 80% for both. These assumptions are based on data from my lab and others (table S1) [7, 17, 24, 55]. The production requires ∼104, 811 liters of total culture volume, 11 passaging operations, and 48 days with stirred-tank bioreactors, which is technically and economically challenging (figures S14(A) and (C)). As mentioned, building large stirred-tank bioreactors will be very challenging. Passaging is highly unwanted for large-scale cell production since it requires large amounts of time, labor, and reagent; increases the risk of contamination and culture failure; loses large numbers of cells; and causes difficulties during automation. The production can be done with 320 l of AlgTubes in 20 days and 1 passaging (figures S14(B) and (C)). The reductions in culture volume, time, and passaging not only make the production technically feasible, but also lead to enormous cuts in labor, reagents, equipment, cGMP facility size, and overall production cost.
In summary, the AlgTube technology combines physiologically relevant culture microenvironments, high performance, high scalability, cGMP compliance and commercial viability, and has potential to address the hPSC manufacturing challenge. Advances in biology have developed protocols for efficiently differentiating hPSCs into many human cell types [7, 31]. Future research can explore integrating these protocols into AlgTubes to produce various human cell types. It will be valuable to systematically study culturing other cell types, such as human adult stem cells and T cells in AlgTubes.
Acknowledgments
Aaron Howell at the Department of Chemical and Biomolecular Engineering at University of Nebraska, Lincoln drew the 3D graphics. Leonard Akert at the Department of Chemical and Biomolecular Engineering at University of Nebraska, Lincoln assisted QL with building the extruders and prototype bioreactors. Drs Stephen D Kachman and Zheng Xu at the Department of Statistics, University of Nebraska, Lincoln assisted the statistical analysis. Confocal microscope imaging was done in the Morrison Microscopy Core Research Facility at University of Nebraska, Lincoln. Drs You Zhou and Christian Elowsky assisted the confocal imaging. Flow cytometry was done in the Morrison center, the Flow Cytometry core, University of Nebraska, Lincoln with the assistance of Dirk Anderson. RNA sequencing was done at the UNMC deep sequencing core. Drs Hendrik J Viljoen and Anuradha Subramanian at the Department of Chemical and Biomolecular Engineering at University of Nebraska, Lincoln offered discussion and advice on the manuscript.
Author contributions
YL conceived the idea. YL and CZ directed the work. YL and QL designed the experiments and wrote the manuscript. QL, HL, OW, CE and HC performed experiments. QL analyzed the data. QD, KL and CZ analyzed the sequencing data.
Competing financial interests
The University of Nebraska has filed a patent application related to the technology described in this work to the United States Patent and Trademark Office.