

Abstract
Natural biopolymers have found success in tissue engineering and regenerative medicine applications. Their intrinsic biocompatibility and biological activity make them well suited for biomaterials development. Specifically, keratin-based biomaterials have demonstrated utility in regenerative medicine applications including bone regeneration, wound healing, and nerve regeneration. However, studies of structure-function relationships in keratin biomaterials have been hindered by the lack of homogeneous preparations of materials extracted and isolated from natural sources such as wool and hair fibers. Here we present a side-by-side comparison of natural and recombinant human hair keratin proteins K31 and K81. When combined, the recombinant proteins (i.e. rhK31 and rhK81) assemble into characteristic intermediate filament-like fibers. Coatings made from natural and recombinant dimers were compared side-by-side and investigated for coating characteristics and cell adhesion. In comparison to control substrates, the recombinant keratin materials show a higher propensity for inducing involucrin and hence, maturation in terms of potential skin cell differentiation.
Export citation and abstract BibTeX RIS
| rhK31 | recombinant human hair keratin 31 |
| rhK81 | recombinant human hair keratin 81 |
| IF | intermediate filament |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel |
1. Introduction
Biopolymeric scaffolds for tissue engineering and regenerative medicine have garnered much interest over the past few decades [1–11]. Natural biopolymers offer several advantages over traditional implant materials and synthetic polymer-based scaffolds. Natural biopolymeric scaffolds are often constructed from proteins found in the extracellular matrix (ECM) and other native tissues, which contributes to their biocompatibility and minimizes undesirable immune responses. Furthermore, many natural biopolymers have inherent self-assembly properties, functional cell binding motifs, and other important regulatory functions, such as control over cell migration and proliferation that provide them with intrinsic biological activity [6, 10, 12–16]. Conversely, synthetic polymers provide excellent structural support, but lack the intrinsic biological activity [17] and often elicit an unwanted foreign body response [18]. Additionally, synthetic polymers often require chemical modifications in order to improve their biocompatibility and impart biological functions for subsequent use in regenerative medicine applications. Functionalization of scaffolds has proven difficult [19, 20] as it is challenging to control the degree of functionalization and the spatial organization of functional groups [21, 22]. Therefore, natural biopolymers are promising alternatives as biomaterials for regenerative medicine due to their inherent characteristics (e.g. self-assembly, cell binding) [23].
When designing biomaterials for regenerative medicine and tissue engineering, certain criteria should be considered [24]. First, biomaterials need to provide adequate mechanical and structural support. Second, the ability to facilitate and control cellular adhesion, migration, proliferation, and differentiation is essential [25]. Lastly, materials that degrade over time and resorb into the body allow for temporary implants. The ability to control the rate of degradation further enhances the utility of the material as the degradation rate can be tailored to the body's healing process [26]. Biopolymers such as elastin [16, 27], collagen [12], keratin [28], and silk [29] have all been used for preparation of biomaterials. These biopolymers are characterized by their hierarchical structures and exquisite and tunable mechanical properties [15, 27, 30, 31]. Furthermore, important biological functions, including the ability to promote cellular attachment through specific cell-binding motifs in their primary amino acid sequences, induction of cell proliferation, as well as regulation of cellular differentiation and protein synthesis can be imparted in the process of biomaterial design [27, 32–34].
In the past decade keratin biomaterials have demonstrated utility as a suitable scaffold for tissue engineering and regenerative medicine [35–46]. The inherent self-assembly of keratin biopolymers into fibrous nanostructures allows for processing keratins into materials with excellent mechanical properties. Furthermore, keratins' biological and regulatory functions enhance its biocompatibility and provide useful bioactivity. For example, it has been proposed that keratin biopolymers contain cell binding motifs, specifically the leucine-aspartic acid-valine motif [31], and participate in regulation of protein synthesis, cell growth, and proliferation [47, 48]. Consequently, keratin has been employed in bone regeneration [49], wound healing [50], and nerve regeneration [51] applications. A notable example of keratin-based materials demonstrated its utility as a scaffold for bone regeneration through the delivery of recombinant human bone morphogenetic protein 2 (rhBMP-2) via a keratin hydrogel system [49]. Another recent application of keratin-based scaffolds is in drug delivery. Ham et al [52] used human hair keratins to fabricate hydrogels with controlled degradation profiles to allow for delivery of recombinant human insulin-like growth factor 1. However, despite the apparent utility of keratin biomaterials, structure-property relationship investigations have been mired by the lack of homogeneous biopolymer preparations. During extraction from sources such as hair and wool fibers, natural keratins are subject to extensive processing conditions that can lead to biopolymer damage. Additionally, the quality of the materials is highly source-dependent. The harsh processing methods required for keratin extraction may result in protein damage leading to alterations in network assembly and undesirable immune response despite the biocompatibility of the biopolymer. Furthermore, major biopolymer components, K31 and K81, co-purify with low molecular weight constituents such as melanin and keratin associated proteins.
We have recently reported cloning, expression, and purification of recombinant human hair keratin 31 (K31) and keratin 81 (K81) [53]. K31 and K81 have been identified as the major components of extracted hair keratin materials [54], which fall into the category of 'hard' keratins. Hard keratins are found in epidermal appendages, such as hair, skin, and nails [55]. The defining feature of hair keratins comes from their high cysteine content, which contain 5% or more cysteine residues [56]. This is in stark contrast to epithelial or 'soft' keratins, which contain less than 1% of cysteines [56]. Consequently, hard keratins form more rigid structures, compared to the loose bundles formed by soft keratins, which result from extensive intermolecular disulfide bonds formed during assembly. Keratins extracted from natural sources are obtained either through oxidative extraction (so-called keratose or KOS) or through reductive extraction (so-called kerateine or KTN). In KOS samples, disulfide bonds are not formed due to the conversion of the cysteine thiol groups to sulfonic acid. KTN cysteines do contain thiol groups and readily form disulfide crosslinks. As a result of this chemical difference, KOS materials are generally less stable than KTN materials as there are no covalent bonds formed in the material [52]. Herein, we present the side-by-side comparison of the solution characterization of recombinant human hair keratins K31 and K81 (rhK31 and rhK81) to KTN. In addition, coatings made from KTN nanomaterials and dimers of recombinant rhK31 and rhK81 were characterized and tested for their ability to adhere epithelial cells. To that end, we used the epithelial cell line (HaCaT, immortalized keratinocytes) and connective tissue cells (fibroblasts) to follow the focal adhesion formation. Furthermore, we measured involucrin, an early marker of terminal differentiation for keratinocytes.
2. Materials and methods
2.1. Gene design and cloning of recombinant K31 and K81
Amino acid sequences for K31 and K81 were reverse translated to the resultant DNA sequence and optimized for E. coli codon usage. Synthetic genes corresponding to each protein sequence were synthesized by GeneWiz Inc. The gene sequence contained restriction sites for subsequent cloning into the expression vector pProExHtam. BamHI and HindIII, at the 5' and 3' ends, were employed to ligate the gene into the plasmid, following digestion and isolation of the gene from the commercial plasmid puc57. Enzymes were purchased from New England Biolabs (Ipswich, MA). In order to confirm successful cloning of each gene, sequencing was completed by the Virginia Tech Bioinformatics Institute, which confirmed the correct gene sequences were contained in the plasmids. The same procedure was followed for cloning of the K31 and K81 genes. Plasmid pProExHtam contains an N-terminal histidine affinity tag to be used for protein purification.
2.2. Protein expression and purification of recombinant proteins
Recombinant K31 and K81 were expressed using an E. coli expression system. The same procedures were followed for both proteins. First, the proteins were expressed in BL21 (DE3) E. coli cells. Luria Broth (LB) was used for cell cultures. Cells were grown overnight for 16 h in 50 ml of media at 37 °C with shaking at 250 rpm. LB media was then used to dilute the cells in a 1:100 ratio and cells were grown to an optical density (OD) of 0.6–0.8. Once OD had been reached, 1 mM isopropyl β-D-1-thiogalactopyranoside was used to induce protein expression, which was completed at 37 °C for 4 h. Cells were then harvested by centrifugation at 5000 rpm for 15 min and subsequently resuspended in lysis buffer pH 8 containing 50 mM Tris HCl, 300 mM sodium chloride, and 1% Tween 20 and then stored at −80 °C until purification. An inclusion body purification procedure adapted from Honda et al [57]. was used to extract and purify rhK31 and rhK81. Cell pellets were thawed in a 37 °C water bath followed by a 30 min incubation with 10 mg ml−1 of lysozyme. Following this step, 10 mM MgCl2, 1 mM MnCl2, and 10 µg ml−1 of DNase were each added to the protein samples and incubated for an additional 30 min. Detergent buffer pH 8 consisting of 20 mM Tris HCl, 200 mM NaCl, 1% Triton X-100, and 2 mM EDTA was then added at an equivalent volume to the sample volume and mixed well before centrifuging for 15 min at 5000 rpm. Following removal of the supernatant, an additional 25 ml of detergent buffer was added to each sample, and the samples were again centrifuged. This procedure was repeated until a tight pellet of inclusion bodies was formed at which time 25 ml of extraction buffer was added. Extraction buffer consists of 10 mM Tris HCl, 2 mM EDTA, 8 M urea, 10 mM βME, and 1 protease inhibitor cocktail tablet at a pH of 8, and was used to resuspend the inclusion body pellet. The samples were then centrifuged for 1 h at 16 000 rpm. The resultant supernatant containing the extracted keratin proteins was collected for further purification. rhK31 and rhK81 containing an N-terminal histidine affinity tag were purified using a standard Ni-NTA affinity purification procedure. All buffers used for the purification process also contained 8 M urea to keep proteins in their denatured form until further dialysis.
2.3. Keratin gel electrophoresis and Western blot
SDS-PAGE was used to separate purified protein prior to Western blot analysis. Samples were prepared in a 1:1 ratio of SDS buffer to protein and analyzed on a 10% acrylamide gel. Extracted KTN was diluted at 10 mg ml−1 in sodium phosphate at pH 7.4, and recombinant proteins were prepared at 5 mg ml−1. Following SDS-PAGE, proteins were transferred onto nitrocellulose membranes at 0.35 A for 2 h. The membranes were blocked with 5% non-fat dry milk in Tris Buffered Saline with 0.25% Tween 20 (TBST) for 1 h. Guinea pig anti-human keratin-31 (K31) and guinea pig anti-human keratin-81 (K81) antibodies (Progen Biotechnik, Heidelberg, Baden-Württemberg, Germany) were used as primary probes and were both diluted at 1:2000 in blocking buffer. After 1 h of incubation, the membranes were washed three times with TBST, submerged into a 1:3000 dilution of the rabbit anti-Guinea pig IgG-HRP (Life Technologies) secondary probe for 1 h, then again washed three times with TBST. All incubation periods were conducted at room temperature. Pierce ECL Plus substrate (Thermo Fisher Scientific) mix was added to the membranes 3 min prior to been imaged in a Fujifilm LAS-3000 Imager (General Electric).
2.4. Dialysis
Following affinity purification and molecular weight verification by SDS-PAGE and MS analysis, rhK31 and rhK81 were individually dialyzed out of elution buffer pH 8 containing 300 mM NaCl, 50 mM Tris HCl, 300 mM imidazole, 10 mM βME, and 8 M urea. In the first step of dialysis the protein was dialyzed against buffer pH 8 with 10 mM Na2HPO4, 75 mM NaCl, 5 Mm dithiothreitol (DTT), and 8 M urea. Four additional dialysis steps were completed with decreasing amounts of urea equal to 6, 4, 2, and 0 M. Each of the steps were completed at 3 h intervals except for the last step, which was allowed to equilibrate overnight. Keratin proteins that were previously extracted from human hair fibers and lyophilized were reconstituted and prepared following the same procedure.
2.5. Extraction of natural keratin proteins
Natural keratins used for this study were extracted as previously described [58, 59]. Briefly, a sample of human Chinese hair was obtained from a commercial vendor and used as received. 100 grams of hair was placed into a 2 l solution of 0.5 M thioglycolic acid (TGA) adjusted to a pH of 10.5 and shaken at 100 rpm for 15 h at 37 °C. The hair was recovered by sieve and the extraction solution retained. The hair fibers were then placed in a solution of 4 l of 100 mM tris base and shaken at 100 rpm for 2 h at 37 °C. Hair was again recovered by sieve and placed in a freshly prepared 1 l solution of 0.5 M TGA adjusted to a pH of 10.5 and shaken at 100 rpm for 15 h at 37 °C. The resulting extraction solution was retained and the hair was then placed in 2 l of 100 mM tris and shaken at 100 rpm for 2 h at 37 °C. The hair was then recovered by sieve and discarded. The extraction solution was retained and pooled with extraction solutions obtained in previous steps to form a solution of crude keratin extract. The crude extract was clarified of particulate matter by centrifugation through a solids separator running at 30 000 rpm, followed by filtration through a filter membrane with a 20–25 μm average pore size. Keratin nanomaterials were obtained from this clarified crude keratin extract by ultrafiltration using a 100 kDa NLMWCO polysulfone, tangential flow filtration (TFF) cartridge. TFF was conducted with ten volume washes against a buffer consisting of 10 mM disodium phosphate and 100 mM sodium chloride at pH 9.1, followed by five volume washes against purified water. The purified keratin nanomaterial solution was concentrated, titrated to pH 8.5, frozen and freeze dried to produce a keratin nanomaterial powder.
2.6. Size exclusion chromatography
A Dionex chromatography system with an Ultimate 3000 UV/Vis detector was used for size exclusion chromatography (SEC). Proteins were detected at 280 nm and analyzed with Chromeleon v6.8 chromatography software. Samples were analyzed following each step of the dialysis process. Each sample was passed through a 0.22 µm filter after a 3 h equilibration period in the appropriate dialysis buffer. The mobile phase used for each sample corresponds to the relevant dialysis buffer. Samples were analyzed with a flow rate of 0.5 ml min−1.
2.7. Dynamic light scattering
Dynamic light scattering (DLS) was completed using a Malvern Zetasizer Nano-ZS to analyze the average particle size and size distribution of extracted and recombinant keratin biopolymers in solution. Prior to measurement, samples were filtered using a 0.22 μm filter. Each sample corresponds to steps during the dialysis process, and thus samples contain the corresponding buffer and urea concentration as described in the dialysis section. The Malvern software converts the intensity percent size distribution to volume percent using Mie theory.
2.8. Transmission electron microscopy
Using a Philips EM420 microscope with an accelerating voltage of 120 kV transmission, electron microscopy (TEM) analysis was performed on extracted and recombinant keratin biopolymers. Samples were prepared using 300 mesh carbon-coated grids purchased from Electron Microscopy Science. Following deposition of the sample on the grid, a 1 min drying period was allowed before excess sample was removed. All samples were stained using 2% uranyl acetate, with a 30 s drying period. Excess stain was then removed and samples were allowed to air-dry for 24 h prior to analysis.
2.9. Silane coupling and protein deposition
Microscope slides coated with 5 nm of titanium (Deposition Research Lab Inc, CO, USA) were cut into 0.8 cm by 0.8 cm substrates. These substrates were cleaned with 100% ethanol (EtOH) to remove nominal debris. These pieces were then immersed in silane solutions of 5% 3-Aminopropyltriethoxysilane (a; APTES; TCI America, Portland, OR, USA) in 95:5 EtOH: H2O solution (v v−1) or 10% 3-Isocyanatopropyltriethoxysilane (i; ICPTES; Acros Organics, Geel, Belgium) in 100% EtOH (v/v). The silane solution was filtered with a 0.2 µm pore size filter and the substrates placed in gentle agitation for 3 h, rinsed with 100% EtOH and rinsed with ultrapure water three times. The substrates were subsequently placed into an oven at 110 °C for 30 min each.
Silane coated substrates were placed in either extracted KTN or dimerized rhK31 and rhK81 recombinant human keratin (rhK) solutions at room temperature (RT) overnight. The KTN solutions were at a 1% concentration and dissolved in 10 mM sodium phosphate at pH 7.4; the rhK was at a concentration of approximately 1 mg ml−1 in distilled water. Other silane-coated substrates were immersed in human fibronectin (FN; Corning, Corning, NY, USA) at 5 µg ml−1 in phosphate buffered saline or bovine collagen I (COL; Corning, Corning, NY, USA), which is only used for elemental analysis and was coated at 5 µg cm−2 in 0.01 N HCl. Both FN and COL were coated for 1 h at RT. Gold substrates (not silane-coated) were coated in 1% KTN (w v−1) in 10 mM sodium phosphate at pH 7.4 overnight at RT. All substrates were rinsed with ultrapure water, air dried, and exposed to ultraviolet (UV) light for 1 h. In subsequent sections, the coatings are noted as iKTN for ICPTES-coupled KTN, AuKTN for gold-coupled KTN, iRhK for ICPTES-coupled recombinant dimer, aFN for APTES-coupled fibronectin, aCOL for APTES-coupled collagen, and pTi for plain titanium. From a previous study conducted in our lab, silane optimization was performed [58], silane coupling selection for FN and COL was adapted from published studies [60–62].
2.10. Atomic force microscopy (AFM)
Dry substrates were examined in tapping mode on the Veeco BioScope II (Oyster Bay, NY) at RT. Five 2 µm by 2 µm spots were analyzed per substrate under a silicon tip (Nanosensors, Switzerland) with curvature of 10 nm and a force constant ranging between 10 and 130 N m−1. Surface roughness and rendered images were created and analyzed through with the Bruker's Nanoscope software.
2.11. Cell static adhesion immunochemistry
Human neonatal primary dermal fibroblast (PCS-201-010; ATCC, Manassas, VA, USA) were grown in RPMI 1640, L-Glutamine, with no sodium pyruvate (Gibco Life Technologies Carlsbad, CA, USA) with an additional 10% fetal bovine serum (FBS; Gibco Life Technologies Rockville, MD, USA) and 1% penicillin streptomycin (P/S) added. Human keratinocytes HaCaT cells (Catalog #T0020001; AddexBio Technologies,San Diego, CA, USA) were cultured in Dulbecco's Modified Eagle's Medium (DMEM; ThermoFisher, Waltham, MA, USA), with 10% FBS, 1% P/S and 1.5 mM sodium pyruvate (ThermoFisher, Waltham, MA, USA). All cells were split 1 to 3 every 2–3 d and incubated at 37 °C, 5% CO2.
For immunochemistry assays, fibroblasts and HaCaT cells were seeded at 10 000 cells cm−2 in a 40 µl serum-free droplet placed onto the substrates and incubated at 37 °C for 3 h, then fixed with 4% paraformaldehyde. Cells were permeabilized, washed, blocked, and focal adhesions were stained with the FAK 100 kit according to the manufacture's protocol (EMD Millipore, Billerica, MA, USA). Dilutions of antibodies were as follows: primary antibody Anti-Vinculin (1:350), the secondary Alexa Flour 488 (1:300), Phalloidin (1:350) and DAPI (1:1000). Cells were imaged at 10x using a Zeiss LSM 800 confocal microscope.
2.12. Involucrin detection
HaCaT cells were seeded at 10 000 cells cm−2 in a 40 µl droplet onto the substrates and incubated at 37 °C initially for 3 h. After 3 h, a 1 ml solution of media was added and replaced daily for 7 d. For a positive control, HaCaT cells were seeded in high calcium media (2.8 mM Ca2+). After 7 d of culture, cells were removed using a cell scraper and RIPA buffer. Each whole cell lysate sample and capillary Western blot assay was prepared according to the manufacturer's instructions for the Wes™ (ProteinSimple, San Jose, CA, USA). Briefly, cell lysates were prepared with 1 part 5x Fluorescent master mix and 4-part cell lysate, which could be diluted with 0.1x sample buffer, if needed. Involucrin Human Recombinant protein (Thermo Fisher, Waltham, MA, USA) in buffer was used as an analytical control. The ladder is provided but additional sample buffer and dithiothreitol is added according to the preparation guidelines. Biotinylated ladder and samples were denatured at 95 °C for 5 min. Samples, blocking reagent, antibody diluent, primary antibodies, secondary antibodies, streptavidin HRP, wash buffer, and Luminol-peroxide were placed in the provided ProteinSimple well plate. The manufacturer's anti-mouse secondary antibody was used in conjunction with both primary antibodies tested, which included the primary antibody, involucrin (Thermo Fisher, Waltham, MA, USA) (1:1000) and the secondary antibody goat anti-mouse IgG-HRP (1:5000) (Santa Cruz Biotechnology, Dallas, TX, USA). The well plate and one time use capillary insert were placed in the WesTM instrument, where the fully automated process begins. Upon completion, digital images and area peaks were analyzed in ProteinSimple's Compass software.
2.13. Smooth muscle actin detection
Fibroblasts were seeded at 10 000 cells cm−2 in a 40 µl droplet onto the substrates and incubated at 37 °C initially for 3 h. After 3 h, a 1 ml solution of media was added and replaced daily for 7 d. For the smooth muscle actin (SMA) positive control, HeLa cells CCL-2 (ATCC, Manassas, VA, USA) were used on a tissue culture substrate. The same procedure for the Wes system, as described above for involucrin, was followed, except that antibodies used included primary antibody, alpha-SMA (1:500) and secondary antibody, goat anti-mouse IgG-HRP (1:5000) (Santa Cruz Biotechnology, Dallas, TX, USA).
2.14. Statistics
Replicates of ≥3 were used in all experiments. Graphs were created in either Prism™ (GraphPad Software, San Diego, CA) or Microsoft Excel. Image J and Photoshop (Adobe, San Jose, CA) were used for image processing and analysis. Quantitative data is expressed as mean ± standard deviation. Differences between experimental groups were assessed by analysis of variance (ANOVA) and a Tukey's post-hoc test for significance using Prism software.
3. Results and discussion
Recombinant DNA technology and protein engineering are greatly influencing the next-generation biomaterials landscape. Recombinant protein-based biomaterials provide the structural and mechanical properties of their natural counterparts while offering the potential for creating materials with tunable sequences, and thus tailored and improved characteristics. Cellular binding motifs, degradation sites, and protein fusions exemplify some of the benefits afforded from recombinantly expressed biopolymers [63–67]. Indeed, many protein-based biomaterials, including silk [64, 66, 68, 69], elastin [66, 69–71], collagen [70, 71], and resilin [72] have benefited from recombinant DNA technology. In addition to providing a path to increased structural and functional complexity of biomaterials, recombinant biopolymers are indispensable in structure-property relationships studies.
3.1. Composition and homogeneity of recombinant and extracted keratin samples
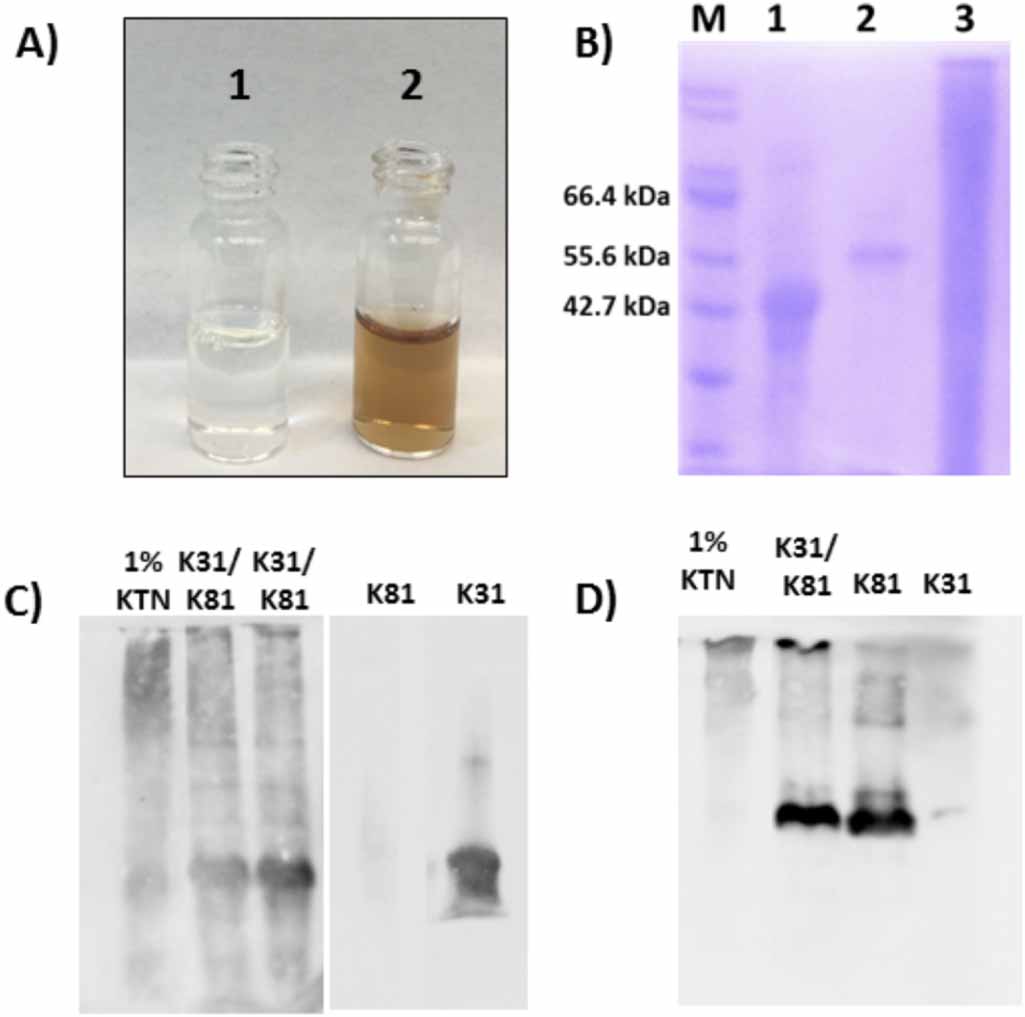
In addition to K31 and K81, hair fibers contain different types of keratin proteins, including gamma-, alpha-, and beta-keratins [43]. Beta-keratins provide protection to the hair fiber, gamma-keratins serve as a crosslinking agent, and alpha-keratins function as the main structural component [43]. As such, the desired component for fabrication of biomaterials is the alpha-keratin due to its important structural properties. However, through the extraction and purification process it is often difficult to remove the other types of keratin proteins, as well as additional by-products, which results in a heterogeneous mixture following extraction and purification. On the other hand, recombinant proteins are expressed and purified individually. The N-terminal histidine tag enables metal affinity purification allowing for efficient removal of all other proteins and by-products not containing the specific affinity tag. In figure 1(A) we first visually compare solutions of extracted and recombinant proteins. The purified recombinant protein solution is labeled '1' and the extracted protein solution is labeled '2'. The recombinant protein solution is clear while the extracted solution, even after purification, appears brown. The observed color is from melanin not removed during purification, demonstrating the difficulty of removing compounds that may be tightly complexed to the larger keratin aggregates. In addition, SDS-PAGE analysis of the purified proteins further indicates the improved sample homogeneity of the recombinant keratins. In figure 1(B), lanes 1 and 2 represent affinity purified rhK31 (lane 1) and rhK81 (lane 2). A single protein band is present in each, showing that both recombinant proteins have been successfully purified with no observable by-products or unwanted contaminants. However, the extracted KTN sample in lane 3 has many protein components present, indicating either the difficulty in removing residual hair fiber components or protein degradation, or both.
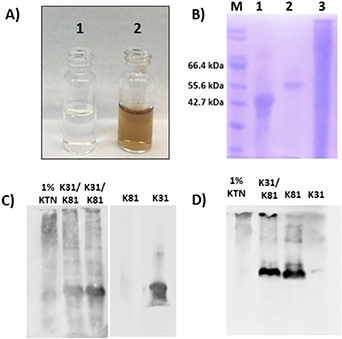
Figure 1. Comparison of purified recombinant and extracted keratins. (A) Picture of purified recombinant (1) and extracted (2) solutions, (B) SDS-PAGE fractions: M-marker, 1-rhK31, 2-rhK81, 3-Extracted KTN, (C) Western blot with K31 antibody and (D) Western blot with K81 antibody.
Download figure:
Standard image High-resolution imageTo verify the identity of the proteins observed in SDS-PAGE, we conducted a Western blot analysis, which confirmed that the predominant proteins in the recombinant materials preparations were K31 and K81 (figures 1(C) and (D), respectively), as expected. Interestingly however, in the extracted sample, the stained bands correspond to higher molecular weights even under denaturing and reducing gel conditions. This suggests the existence of irreversible higher order oligomers in the extracted samples at the same solution conditions in which the recombinant sample is monomeric. The exception is the 1% KTN sample where a band corresponding to the molecular weight of monomeric K31 is observed (figure 1(C)). Furthermore, not all bands present in SDS-PAGE of the KTN also appear in the Western blot, signifying the existence of additional proteins in the extracted sample. From the results obtained from SDS-PAGE and Western blot, it appears that recombinant protein production and purification methods provide starting materials of improved homogeneity over that of extracted keratins. However, when rhK31 and rhK81 are combined, bands corresponding to higher molecular weights than monomers appear in Western blots, analogous to extracted KTN sample. Thus, the recombinant heterodimer is capable of heteropolymerization and formation of higher order structures. To further investigate heteropolymerization of rhK31 and rhK81 we utilized SEC and DLS.
3.2. Oligomerization states of recombinant and extracted keratins
To prepare samples for SEC analysis, rhK31 and rhK81 were mixed in 8 M urea buffer (Materials and Methods). KTN samples were resuspended in buffer of the same composition as the recombinant samples. SEC data obtained in buffer containing 8 M urea shows that both the extracted and recombinant keratin proteins contain structures that are larger than the K31/K81 heterodimer (figure 2). Interestingly, the higher order oligomers are present in both samples even in the presence of a denaturant (urea) and reducing agent (DTT). It is important to note that all samples are passed through a 0.22 µm filter before SEC analysis. Therefore, all structures larger than the filter cut-off will be excluded from this method of analysis.

Figure 2. Chromatogram of recombinant (blue) and extracted (black) keratin in 8 M urea obtained from SEC. Red numbers correspond to peak labels listed in table 1.
Download figure:
Standard image High-resolution imageTable 1. Estimated oligomeric states from SEC analysis.
| Peak # | rhK31/K81 | KTN |
|---|---|---|
| 1 | Octamer | N/A a |
| 2 | Tetramer | Dimer |
| 3 | Dimer | — |
| 4 | Monomer | — |
While five peaks are observed in the recombinant sample chromatogram, the extracted heteropolymer chromatogram contains two broader peaks shifted toward shorter elution times (figure 2). The estimated oligomeric states for each peak (labeled by the red numbers in the figure 2) are shown in table 1.
Higher molecular weight peaks 1, 2, and 3 in the recombinant sample overlap with peaks 1 and 3 in the extracted sample suggesting that the K31/K81 octamer, tetramer, and dimer are present in both the extracted and recombinant samples. Peak 1 in the extracted sample, not observed in the recombinant proteins sample, elutes at the time corresponding to the column void volume. Thus, sample components too large to be retained on the column are already present in the extracted sample. At the same time, there is no protein eluting at retention times corresponding to K31 and K81 monomers in the extracted sample. From the SEC analysis we conclude that in the recombinant sample under denaturing conditions the major fraction of the solution is monomeric rhK31 and rhK81 in equilibrium with higher order oligomers. In contrast, the major fraction of the KTN solution corresponds to oligomers larger than octamers and the smallest observable component is a dimer. Dimer observation in the KTN sample is consistent with the formation of the obligate keratin dimer in nature. However, it is interesting that this dimer is resistant to reducing and denaturing conditions.
The presence of larger heteropolymers in the extracted KTN sample, as observed in the SEC, SDS-PAGE, and Western blot analyses, is consistent with the extraction procedure that relies on breaking down preformed, durable keratin-based structures. In order to efficiently extract the desired keratin biopolymers, the extensive network of intermolecular disulfide bonds must be reduced. Thus, the size of sample components acquired from extraction is dependent on the efficiency with which this network is disrupted, and the resulting higher order structures persist due to covalent interactions that are not affected by the solution conditions. Conversely, recombinant protein production facilitates assembly from each individual component. To further probe the solution behavior of the recombinant and extracted keratins, we used DLS to monitor changes in oligomerization equilibrium as the concentration of denaturant in the sample was decreased.
We performed DLS on aliquots of samples obtained during each dialysis step. As the sample is dialyzed, the concentration of urea in the buffer is reduced in a step-wise manner in order to allow the proteins to return to their native state, and in the case of keratin proteins, to allow for self-assembly to occur. Thus, as the urea concentration decreases, we expect the oligomerization equilibrium to shift towards higher order oligomers and finally fibers. However, all samples are filtered prior to analysis using a 0.22 µm filter. Consequently, any sample components larger than 0.22 µm will not be observed, similar to the SEC. Figure 3 shows recombinant (figure 3(A)) and extracted (figure 3(B)) heteropolymer samples at 8 M (blue), 4 M (red), and 0 M (black) urea concentrations. Consistent with SEC data, nanostructures present in the KTN sample are larger than in the recombinant sample at the same solution conditions (table 2, figure 3).

Figure 3. DLS from (A) recombinant keratin urea series, (B) extracted keratin urea series and (C) recombinant (solid line) and extracted (dotted line) keratin samples in 0 M urea.
Download figure:
Standard image High-resolution imageTable 2. Hydrodynamic radius of species present in solution at decreasing urea concentrations.
| Urea (M) | Peak # | rhK31/K81 (nm) | KTN (nm) |
|---|---|---|---|
| 8 | 1 | 18 | 120 |
| 8 | 2 | 225 | 1100 |
| 4 | 1 | 13 | 110 |
| 4 | 2 | 120 | 900 |
| 4 | 3 | 460 | — |
| 0 | 1 | 8 | 50 |
| 0 | 2 | 60 | 295 |
An overlay of the volume percent of recombinant and extracted keratins in 0 M urea is shown in figure 3(C). The volume percent distribution provides the relative proportion of the different sample components. The populations detected in the DLS at 0 M urea corresponds to the keratin that has not been incorporated into IF after all denaturant has been removed from the system. The major scattering species in the recombinant keratin sample has an 8 nm hydrodynamic radius, consistent with K31 and/or K81 monomers [73]. The extracted keratin sample is mostly composed of 50 nm oligomers. This is consistent with measurements obtained at each urea concentration, as well as the SEC and Western blot analysis, and further indicates that upon extraction the starting material is not completely reduced to individual proteins.
3.3. Nanostructure of recombinant and extracted keratins
One of the essential features of keratin protein materials is their intrinsic capacity for self-assembly. Retaining this important biological feature provides the ability for generation of materials such as films [39], hydrogels [37], and sponges [44]. Self-assembly of keratin IFs is a well-studied process [28, 74]. During IF assembly a dimer composed of one type I (acidic) and one type II (basic) keratin is formed. Following dimer formation, tetramers form through antiparallel alignment of two heterodimers to create a staggered conformation. Subsequent parallel head to tail stacking of tetramers results in protofilaments, which further assemble to form 10 nm diameter IF [28, 75].
In order for recombinant keratin proteins to be viable for use as biomaterials, they too must possess the ability to self-assemble into fibers after expression and purification. Results from solution characterization point to the formation of higher order structures in both the recombinant and extracted samples.
Figures 4(A) and (B) are representative TEM images of rhK31/K81 nanostructures. The recombinant proteins do self-assemble into standard 10 nm diameter keratin IFs, and further form large bundles through additional IF interactions. These structures are several microns long and average 150 nm wide. Figures 4(C) and (D) are representative TEM images of the extracted keratin nanostructures. At a 5 mg ml−1 concentration (same as rhK), the extracted materials readily formed films on the TEM grid. These films appeared featureless and individual fibers or fiber bundles were not observed. Therefore, the extracted samples were diluted to 0.5 mg ml−1. This sample concentration resulted in fibers observable by TEM, but the overall number of fibers, in comparison to the recombinant sample, was low. Moreover, these fibers do not have the typical IF morphology. The width of fibers was estimated to be between 70 and 100 nm, with lengths of a few microns (figures 4(C) and (D)). However, similar to the recombinant sample, some bundling of fibers is apparent (figure 4(C)).
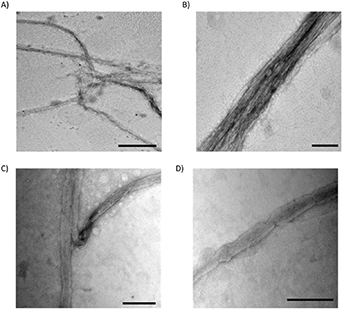
Figure 4. TEM images of recombinant (A)–(B) and extracted (C)–(D) keratin proteins. Scale bars are (A) 500 nm and (B)–(D) 300 nm. All images are stained with 2% uranyl acetate.
Download figure:
Standard image High-resolution image3.4. Topology of keratin materials
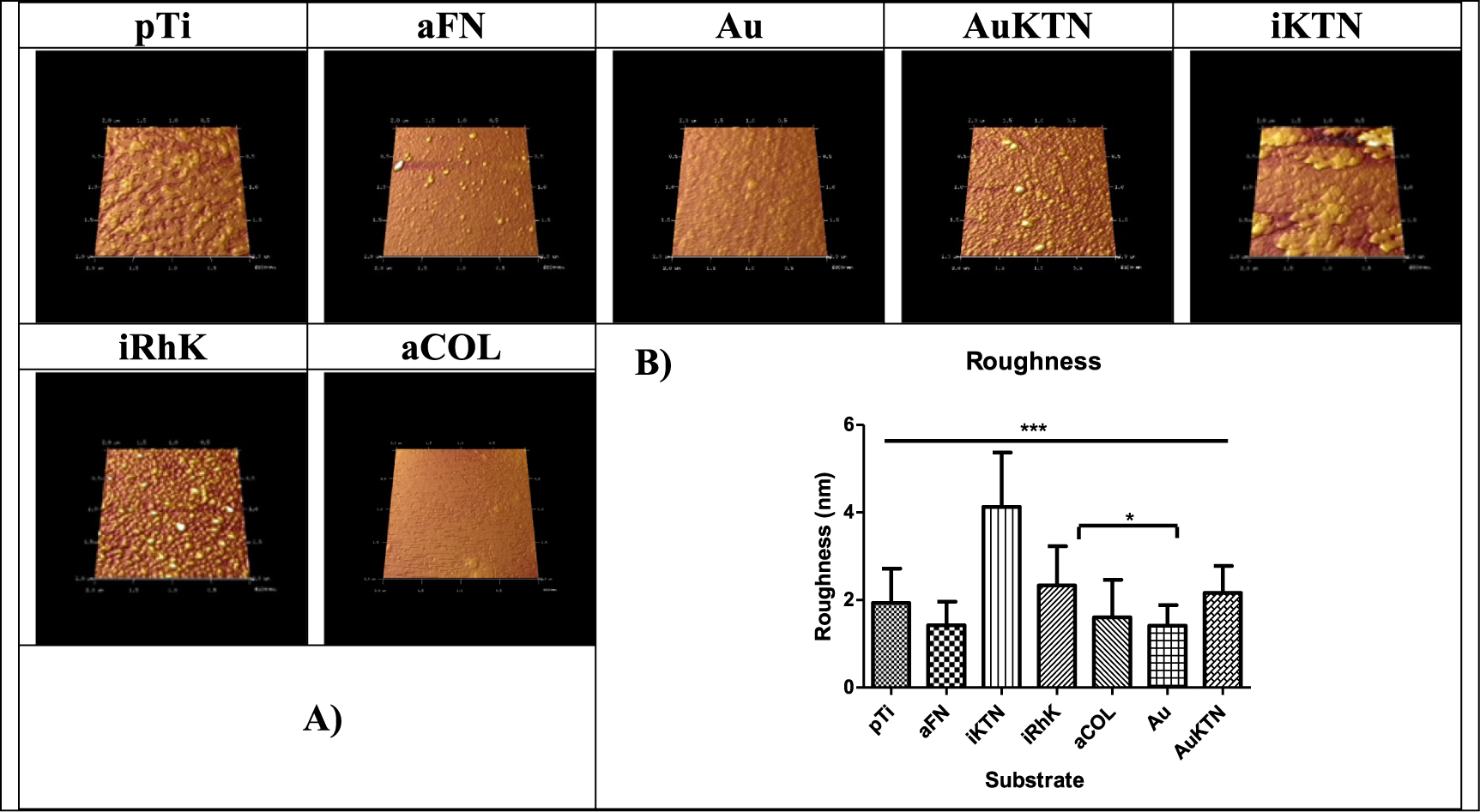
AFM is a commonly used technique with high resolution of approximately 1 Å to characterize topography, which is only limited by thermal and electrical noise [76]. This resolution allows for visualization of molecular structures. AFM confirms that recombinant keratins are able to create a uniform coating, although the roughness is not significantly different than pTi. When observing the topography visually, KTN on gold and recombinant keratins display similarly homogenous surfaces with limited appearance of aggregated features (figure 5(A)). Here, KTN on gold is used as a positive control as it has been previously been shown that cysteine readily bonds to gold surfaces, a phenomenon that has been used for the formation of self-assembled monolayers or SAMs [77]. KTN attached through a silane coupling layer appears more irregular, suggesting that protein aggregates are present, which is also suggested by the SDS-PAGE, SEC and DLS data. The images are confirmed by roughness measurements that show the KTN on silane is significantly rougher than any of the other test substrates (figure 5(B)).
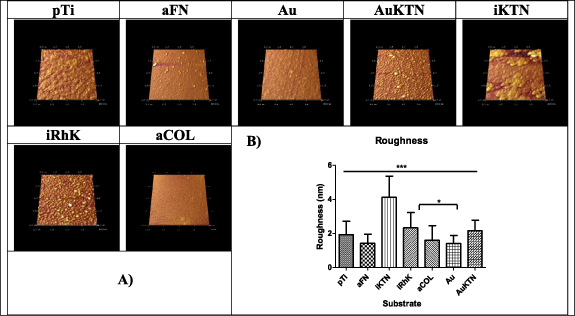
Figure 5. Rendered images of substrates examined by AFM.
Download figure:
Standard image High-resolution image3.5. Cellular adhesion immunochemistry
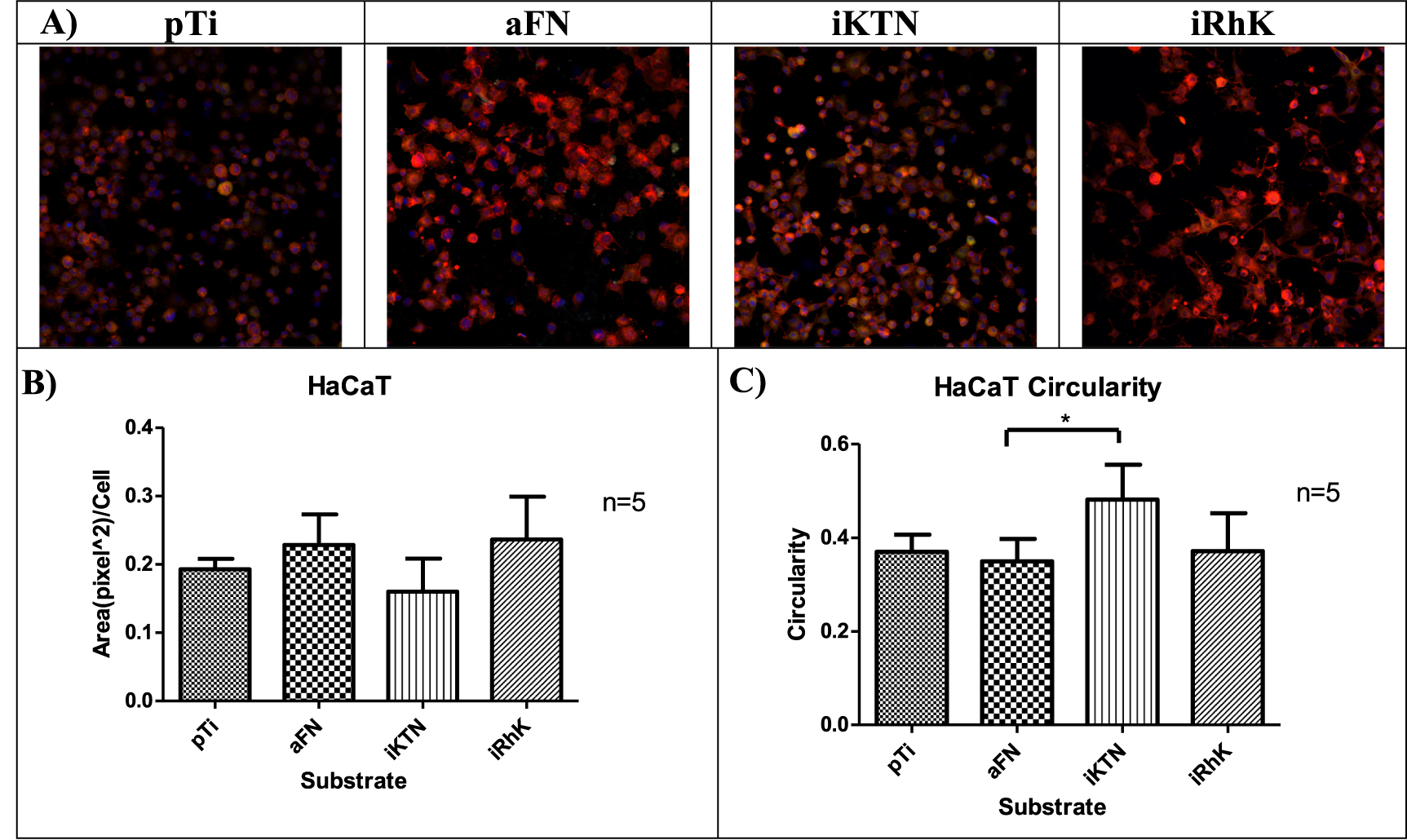
A cells' biological interactions with coatings can be partially characterized by the growth, maturation and mechanism of attachment. An epithelial cell line (HaCaT) and connective tissue cells (fibroblasts) were used in this study to observe cell attachment. HaCaT cells are immortalized keratinocytes, with their primary in vivo cell attachment mediated to the basement membrane [78]. Fibroblasts serve as a connective tissue cell model and attach to the ECM, a non-cellular structure that contains fibrous proteins [79]. Focal adhesions create transmembrane communication channels that can activate cell proliferation [80, 81], phenotypic changes [82, 83], and cell signaling cascades [84]. Actin fibers are an indication of focal adhesion formation [81]. Figures 6 and 7 show the areas of positive actin staining for HaCaT and fibroblast cells, respectively. Although significant differences are not present between the various substrates, the morphologies described through circularity measures exhibit significant changes. In both HaCaT and fibroblast cells, plain Ti, fibronectin, and recombinant keratins allow for the cells to adopt a spread morphology. Cell spreading can indicate that cells perceive the underlying substrate through trans-membrane receptors, even though they may have similar actin protein areas upon staining.
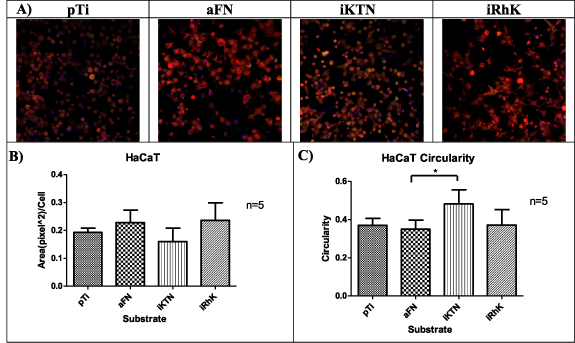
Figure 6. (A) HaCaT cells focal adhesions represented by red (actin), green (vinculin), blue (nucleus). (B) Area of actin staining quantified by Image J (n = 5). (C) Circularity observed by Image J is graphical represented (n = 5). Significance is identified by * for p ≤ 0.05, ** for p ≤ 0.01, and *** for p ≤ 0.001. *** 0.001.
0.001.
Download figure:
Standard image High-resolution image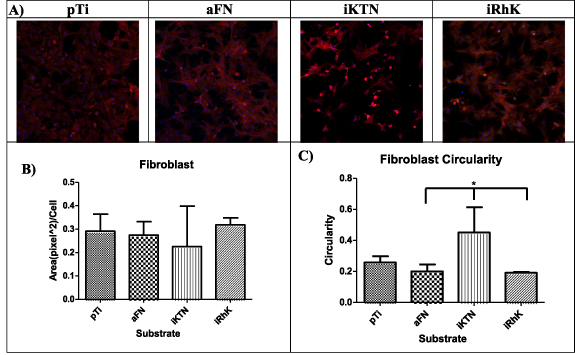
Figure 7. (A) Fibroblasts focal adhesions represented by red (actin), green (vinculin), blue (nucleus). (B) Area of actin staining quantified by Image J. (C) Circularity observed by Image J is graphical represented. Circularity ranges from 0 to 1, where 1 indicates a perfect circle. Significance is identified by * for p ≤ 0.05, ** for p ≤ 0.01, and *** for p ≤ 0.001. *** 0.001.
0.001.
Download figure:
Standard image High-resolution image3.6. Involucrin and SMA detection
Upregulation of involucrin is an early marker of terminal differentiation for keratinocytes [85], which is defined as the progression of keratinocytes towards a stratified epidermis structure. HaCaT cells exposed to the recombinant keratin coating expressed a higher concentration of the involucrin protein in comparison to other experimental groups (figure 8). During this relativity short-term culture period, the recombinant keratin provokes a response in the HaCaT cells indicating the cells are beginning terminal differentiation. When these cells are transforming from spinal cells to granular cells, an upregulation of involucrin occurs, denoting the beginning of keratinization. Other studies have examined involucrin upregulation as a defining test for artificial skin equivalents [86, 87]. These data suggest that the recombinant keratin has a higher propensity for inducing involucrin and hence, maturation in terms of potential skin cell differentiation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. (A) Involucrin. A digitized Western blot lane view from the ProteinSimple Wes instrument for involucrin including Ca2+ induced HaCaT cells, which represent the positive control. (B) SMA. Digitized Western blot for SMA in fibroblasts.
Download figure:
Standard image High-resolution image{kind=link}
SMA is a definitive characteristic of myofibroblast, which can be induced through mechanotransduction and biochemical cues. Fibroblasts are observed to express SMA on all substrates. Although SMA is a marker for myofibroblasts present in granulation tissue during wound healing [88], substrate stiffness can induce the SMA phenotype and cell contractility [89–91]. HeLa cells served as the positive control but were seeded on a tissue culture plate, which does not have an equal stiffness to titanium. This could provide an explanation to why band area and intensity for SMA in HeLa cells is less than all other substrates.
4. Conclusions
Keratin-based biomaterials have been successfully used as scaffolds in regenerative medicine and tissue engineering [42, 52, 92, 93]. However, structure-property studies were impeded by the lack of homogeneity and consistent samples. We have shown here that trychocytic keratins K31 and K81 can be produced recombinantly, resulting in samples of increased homogeneity. Not surprisingly, the composition of extracted versus recombinant materials is shifted towards higher molecular weight oligomers. The distribution of oligomers and their sizes are dependent on the efficiency with which the extensive network of intermolecular disulfide bonds is disrupted in hair fibers, further contributing to the sample heterogeneity.
The recombinant heteropolymer forms standard IF that further associate to create large bundled structures, while extracted keratins fiber did not appear to have the typical IF morphology. Nonetheless, extracted keratins have a remarkable propensity for self-assembly in films that was not observed for recombinant samples under the same conditions. Functional differences in recombinant and extracted keratin composition can also be observed in topography and cellular responses. This further reinforces the notion that recombinant and extracted samples have different compositions. It appears that recombinant keratins, probably due to their relatively higher level of homogeneity and lack of low molecular weight contaminants, form more uniform coatings, which in turn leads to increased biological activity when compared to KTN.
Design flexibility afforded by recombinant technology will result in multifunctional, dynamic keratin materials. Tailoring of the function, composition, and nanostructure can now be achieved at the amino acid sequence level. Both extracted and recombinant keratin biopolymers provide a promising tool for engineering novel biomaterials with controlled chemical and physical properties, and future work on the fabrication and characterization of these materials will allow for new advances in regenerative medicine, medical device coatings, and tissue engineering.
Acknowledgments
The authors would like to thank Virginia Tech, the VT Chemistry Department, the Department of Biomedical Engineering and Mechanics, the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) (Grant No. W81XWH-15-1-0343), and Virginia Tech-Initiative for Maximizing Student Development (IMSD) (Grant No. R25GM072767-10) for funding. We also thank Dr Sujee Jeyapalina for the gift of the HaCaT cells and for helpful discussions regarding culture and assays for these cells. The authors are also thankful to the Grove and Van Dyke labs for discussions and careful reading of the draft manuscript.