

Abstract
Based on density functional theory, structural, electronic, optical and thermoelectric properties of CaO and CaO:F mono-layer compounds have been investigated. Both structures have elastic stability, with Young's and Shear's moduli of 57.78 (N/m) and 23.85 (N/m), respectively, which shows the resistance of these compounds against stress and strain. The reduction of Poisson's ratio with the introduction of F atom indicates the tendency to ionic bonds between atoms, and its magnetic moment reaches to 0.84 μB. The CaO compound is a p-type semiconductor with 2.6 eV, and by adding a F atom to it, it has become a half-metal. In the CaO:F mono-layer, we see a red shift in the real- and imaginary-dielectric function compared to the CaO one. At a temperature of 50 K, the Seebeck coefficient is 32 (μVK−1) and at room temperature it reaches 24 (μVK−1), also the figure of merit coefficient at these temperatures is 1.2 and 0.8, which indicates its thermoelectric capability.
Export citation and abstract BibTeX RIS
1. Introduction
The discovery and synthesis of graphene [1] became a window to the world of two-dimensional materials in order to find new materials with newer structural, electronic, optical and thermoelectric capabilities to make better nano-sensors such as MoS2, WS2, WSe2, and h-BN compounds [2–4]. However, the synthesis of two-dimensional metal oxide monolayers such as MgO, CaO, and CdO compounds still faces difficulties. These materials often prefer to crystallize in three-dimensional close-packed form with ionic bonds rather than in graphite-like layers with van der Waals bonds between layers. In recent years, limited studies have been devoted to the synthesis of two-dimensional metallic materials. In recent years, Nair et al succeeded in synthesizing MgO, CuO, and CaO metal oxide monolayers by trapping an aqueous solution of corresponding salt in graphene nanoenclosures [5–16].
High van der Waals pressure (1 GPa) in these nanoenclosures with light confinement trapped salt solutions were reached with water at room temperature, leading to 2D crystals of corresponding oxides. So now we can discuss and investigate the structural, electronic, optical and other properties of these two-dimensional metal oxide monolayers. Monolayer monoxides with XY stoichiometry have a cubic structure that can be synthesized in thin film form, which gives them the ability to be used in gas sensors, optoelectronic devices, and ceramics.
The CaO film compound is more difficult to make than MgO film, this structure growth on the Mo substrate and along the (001) crystal direction. The electronic and optical properties of CaO film is similar to its bulk case [17]. In addition to the reports of experimental works, other computational studies showed that the CaO mono-layer has elastic and dynamic stability. Also, they proved that this monolayer has a buckling of 0.4 A°, in addition, they showed that it goes out of stability in the flat form. Also, they showed that the CaO mono-layer is a n-type semiconductor by 2.3 eV energy gap with non-magnetic behavior. Also, elastic and phonon calculations indicate the strong stability of this compound [18–28].
Adding impurities to the two dimensional materials has always been a practical solution to create new electronic, optical and thermoelectric properties of materials. The Ca and O atoms have orbital orders [Ar] 4s2, and [He] 2s22p4, respectively. To add impurity, we considered the F atom, with the [He] 2s22p5 orbital structure, which has an extra electron compared to the oxygen atom. We expect that this extra electron in the CaO:F mono-layer will cause fundamental changes in the electronic, optical and thermoelectric behavior.
Therefore, first the mechanical stability of the CaO and CaO:F mono-layers are checked, and then we will check the electronic properties, and then the optical behavior will be compared, and at the end, the thermoelectric properties will be analyzed.
2. Computational methods
Based on the density functional theory (DFT) [29], Kohn–Sham equations, and full-potential linearized augmented plane wave (FP-LAPW) method [30], the mechanical, electronic, and thermoelectric properties of the CaO, CaO:F mono-layers are calculated by the Wien2K and BoltzTraP codes [31, 32]. Also, the exchange–correlation potential is used by the PBE-GGA approximation in spin-polarization mode [33].
The optimized input parameter, RKmax, muffin-tin radii (RMT), lmax, and Gmax, were selected to 7.0, 2.0, 10, and 12, respectively. In addition, the cut off energy of −8 Reyd is considered to separate the core valence electrons, and a (14 × 14 × 2) grid is used in the Brillouin zone. The convergence tolerance of charge and energy less than 10−4 and 10−4 Ry are considered for the self-consistent calculations.
Also, to shows the mechanical stability from dynamic view, the phonon dispersion of these structures calculated by Quantum Espresso software. The KPoint and q-Point are selected to 15 × 15 × 2 and 4 × 4 × 4, respectively. The charge convergence is set to 10−4, and the force optimizations are 10−6 a.u. dyn−1. The degauss parameter is selected to 0.2, and ecut is set to 60.
3. Results
3.1. Structural properties
In figure 1, the unit cell of CaO and CaO:F mono-layers are shown. As we can see in panel (b), this arrangement of atoms is not flat and the atoms have a buckling of 0.22 (A°), after optimization. In panel (c), it can be seen that we have substituted the F atom instead of the oxygen. There are a total of 4 atoms in our unit cell, two atoms related to Ca and two atoms related to O, and in the presence of impurity, we put an F atom instead of one of the O atoms. It can be seen that the type of interatomic bonds are of sp type and by adding F atom, the type of bonds will not change, but an additional electron will remain in the system, which will definitely cause fundamental changes in its electronic and optical behavior.
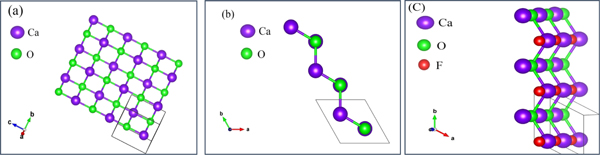
Figure 1. The CaO and CaO:F mono-layers pattern.
Download figure:
Standard image High-resolution imageTo show the mechanical stability from a static point of view, in figure 2, the curve of unit cell energy changes in terms of volume changes (E-V) is drawn based on Birch–Murnaghan equations for the CaO and CaO:F mono-layers. In both (a) and (b) panels, we see the minimum points which indicate the ground state point for both structures, refer to the equilibrium volume for them. The results of these diagrams as the equilibrium volume, lattice constants, bulk modulus, derivative bulk modulus, total energy of unit call, and total magnetic moment are listed in table 1.

Figure 2. The energy-volume diagrams of (a) CaO mono-layer, (b) CaO:F mono-layer.
Download figure:
Standard image High-resolution imageTable 1. The lattice constants (a), (c), equilibrium volume (V), Bulk modulus (B), Derivative of Bulk modulus (B'), Total magnetic moment (Mtot), Total energy (E), and Band Gap of CaO, and CaO:F mono-layers.
| Parameters | a (A0) | c (A0) | V (Bohr3) | B (Gpa) | B/ | Mtot (μB) | E (Ry) | Band Gap (eV) |
|---|---|---|---|---|---|---|---|---|
| CaO | 4.119 | 15.937 | 451.617 | 85.296 | 4.222 | 0.0 | −3023.188 | 2.6 (M-Γ) |
| 3.78 a | 2.59(M-Γ) a | |||||||
| 4.6 b | 2.03 b | |||||||
| CaO-F | 4.105 | 15.921 | 435.329 | 61.154 | 4.263 | 0.84 | −3072.379 | 0.2 (down spin) |
It can be seen the curve slope of the pure state, in smaller volumes and under stress, has a steeper slope than when the unit cell is under strain. Therefore, when the CaO mono-layer is under stress, it gives a greater response to the applied force. With the introduction of F impurity into this compound, it can be seen in panel (b) that the E-V diagram becomes completely symmetrical and this compound shows relatively same responses to external forces under stress and strain. The results of table 1 show, by adding the F impurity, the lattice constant has changed slightly, so it can be said that the inclusion of this element in the composition has made the stronger bonds. Also, the derivative of the bulk modulus in table 1 shows that the bond between atoms are ionic and with the addition of the F atom, these bonds have become stronger. The bulk modulus of both compounds shows the high hardness of them. The CaO:F mono-layer total energy is less than other case, which refer to its stability than CaO mono-layer. The total magnetic moment in the pure case is zero, which is in agreement with other researches and confirms the correctness of our calculations, while with the addition of F atoms, we see an increase in the magnetic moment by 0.84 μB.
In the following, we have examined the phonon band structure curves of CaO and CaO:F mono-layers compounds along the Γ-M-K- Γ in the figure 3. It can be seen that all the phonon branches are positive, which indicates the dynamic stability of these compounds. In the CaO compound, the phonon branches are intertwined and have a greater slope, which means more heat transfer for it. In the CaO:F mono-layer, with the addition of the F atom, two oscillating gaps have been found, which is due to the difference in the atomic weight of F atom compared to the other two atoms. Also, the presence of F atom in the CaO:F compound has reduced the frequency of the optical levels compared to the CaO case.
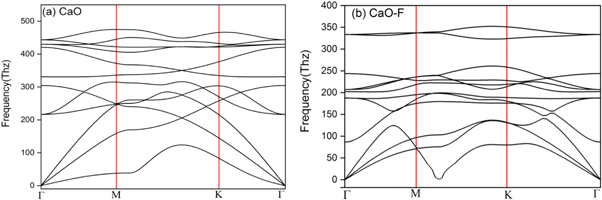
Figure 3. The phonon dispersion of the CaO and CaO:F mono-layers along the Γ-M-K- Γ.
Download figure:
Standard image High-resolution imageOne of the important parameters to determining the mechanical stability of any structure is their elastic behaviour. The elastic constants are the mechanical response of the crystal against stress and strain. In table 2, we have calculated the elastic constants of CaO and CaO:F mono-layers and related parameters such as Young's modulus, Shear modulus and Poisson's ratio. Both mentioned structures have the condition of elastic stability (C11-C12 > 0, C22 > 0, C66 > 0). Positive and relatively large values of Young's and Shear modules show the great resistance to shear stress for these compounds. Next, Poisson's ratio shows that with the addition of F atoms, the bonds have become stronger and tend to be ionic.
3.2. Electronic properties
Investigating the electronic properties of materials is very important for understanding other physical properties, so in the following, we calculated the density of states (DOS), and the band structure of CaO and CaO:F mono-layers with GGA approximation and plotted the results in figures 4 and 5. The DOS curve in figure 4(a) shows that the CaO mono-layer is a p-type semiconductor with 2.6 eV energy gap, which is in agreement with other results [22, 23]. Figure 1 shows that adding the F atom impurity to the CaO mono-layer, we have seen major changes in its electronic behaviour. The CaO:F mono-layer electronic behaviour is converted to a half-metal with a magnetic moment of 0.84 μB and 98% spin polarization at the Fermi level, it has metallic behaviour at up spin and tendency to semiconductor property at down one with about 0.2 eV energy gap. The panel (b) is observed, which is the main cause of the magnetic behaviour is occurred at the Fermi area, due to the presence of an additional electron in F 2p5 than Ca 2p4. These are bonding orbital and are therefore present in the Fermi area. It is observed in panel (d) that in the energy −4 eV to −5.5 eV zone, there are O p orbital electron states that have the least amount of magnetic behavior, and in both up and down spins with 1.7 eV and 2.7 eV are separated from the Fermi level. Electron states in the conduction area in both spins are completely continuous up to 5 eV energies, so when exited electrons, by heat, light or other physical factors, it happen good conduction. Due to the half-metallic behavior of this compound and the presence of two large gaps in the up and down spins, it can be used for the GMR, TMR, optoelectronic, and thermoelectric applications.

Figure 4. (a), (b) The total DOS, (c), (d) The partial DOS of CaO, and CaO:F mono-layers.
Download figure:
Standard image High-resolution image
Figure 5. The bandstructure of (a) CaO mono-layer, (b, c) CaO:F mono-layer in two up and down spins.
Download figure:
Standard image High-resolution imageFigure 5 shows the CaO and CaO:F monolayers bandstructure along the Γ-M-K-Γ path at the Brillouin zone. It can be seen in the pure case that the electron levels of Ca p orbitals are located below the Fermi level with high density, above which there is a large 2.6 eV gap, which makes this compound a suitable option for thermoelectric and optoelectronic purposes. The conduction levels also have a large gradient that allows the excited electrons to high mobility.
With the addition of the F atom to the CaO mono-layer, it can be seen that the bandstructure in the up and down spins are completely different (figures 5(b), (c)). The electronic and magnetic effect of an extra electron of the F atom has caused the p orbital levels of the Ca atom to be shifted to lower energies and at the same time to be split for two spins. In the up spin, an electronic level belonging to the F p orbital has interrupted the Fermi level, while in the down spin we see a small indirect energy gap of 0.2 eV.
3.3. Optical properties
The dielectric function shows the interaction and response of matter to the light emitted on it and represents the electronic structure of the materials. This function is complex and consists of two, real and imaginary parts (ε(ω) = ε1(ω) + iε2(ω)). The real and imaginary parts of dielectric functions follow as Kramers-Kronig relations [25]:
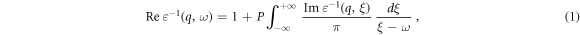
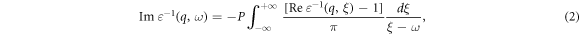
The imaginary part of dielectric function is directly related to the electronic structure of the material so that its peaks represent the electron transitions in two intra- and inter-band transitions and the electronic gap correctly, and derived from it, also, the metallic behavior can be resulted from its graph [36].
In the following, the optical characteristics of these two two-dimensional structures will be investigated. For this purpose, the graphs of real (Re_eps) and imaginary (Im_eps) parts of dielectric function, and energy loss function (eloss) are drawn in two directions of in-plane (x) and perpendicular (z) to it, of light radiation according to the energy of the emitted photon in figure 6. For CaO:F monolayer, based its half-metallic nature, we used from intra-band switch for our calculations. In the (a) panel, it can be seen that the static value of the Re_eps coefficient of CaO mono-layer in both directions has the same value of 3.2, which indicates the strong semiconducting behavior of this compound, and there is no change in the behavior of this compound by changing the angle of rotation. Convergence in optical behavior continues in both x and z directions along with the stability of optical behavior in infrared, visible and ultraviolet edge regions. In the region of 4.8 to 9 eV, the optical response along the x-axis is stronger than along the z-axis, and in this region there are at least three peaks for each direction. These fluctuations can be due to the behavior of excited electrons, which gives metallic behavior to this compound. From the energy of 9.2 eV onwards, the value of the Re_eps coefficient is less than one, which indicates the superluminance behavior for it. At the energy of 9.2 eV, both graphs have a root, which represents the plasmon oscillations, and the response of this compound to the light radiated in both directions becomes zero. By adding the F atom to the CaO mono-layer compound, we have seen a significant change in its optical behavior. The static behavior of the Re_eps coefficient is different in the x and z directions, along the x direction it shows a semiconducting behavior and in the z ones its shifted to the metallic behavior. Along the x-axis, we see optical stability in the infrared region, and in the visible region, it is completely unstable and has a very sharp peak. After three smaller peaks in the ultraviolet region, it has become negative at the energy of 9 eV and has two roots in this region, which has a red shift compared to its pure case. But in the z direction of radiation, we have big instability in the infrared region, and it has shown relatively stable behavior in the visible and ultraviolet regions. In the infrared region, this function has become negative, which has two roots in this region. The presence of these changes in the Re_eps function and bigger responses to the irradiated light compared to the pure case, is due to an extra electron that the F atom has compared to the Ca.

Figure 6. (a), (b) The real parts, (c), (d) The imaginary parts of dielectric function of CaO, and CaO:F mono-layers versus photon energy.
Download figure:
Standard image High-resolution imageIn panels (c) and (d), the Im_eps coefficient along the x and z directions are plotted according to the energy of the radiated photon for CaO and CaO:F monolayers. In the pure case of large optical gap, it confirms the semiconducting behavior in both directions. The peaks of this diagram represent the electron transition, it has been observed that the transitions occurred in the ultraviolet region and the energy range is 5 to 10 eV. In this energy range, the graphs are completely continuous, which indicates the high absorption of light, as a result of which electrons are continuously excited. Two peaks of relatively equal size can be seen along the x-axis and two peaks along the z-axis, but the peak is larger at the energy of 10 eV. In panel (d), it can be seen that the CaO:F mono-layer has different behaviours in the x and z directions, so that along the z direction, the optical gap is zero, and in the x direction, we show the 0.8 eV gap. It can be seen that by adding Fe atom to the composition, we see a red shift for the peaks, which indicates the tendency to metallic behavior, and by changing the direction of the irradiated light, the optical behavior changes and behaves like an optical switch.
At the end of the optical section, the loss function curves of the mentioned structures are drawn. In the pure case of figure 7(a), it can be seen that there is a big gap up to the edge of the ultraviolet region for two mentioned directions, and further up to the range of 10 eV, the values of this diagrams are relatively small, and at 12.5 eV, we see a sharp and big peak, and due to the presence of the Re_eps roots at this energy therefore plasmonic oscillations occurred there. In panel (b), it shows that two case happened when the F atom entered the structure, firstly, all the graphs have a red shift in both directions, and secondly, there is an anisotropy in the x and z directions, and small loss gaps can be seen for both directions. On the other hand, from the infrared region, we see the beginning of the peaks and gradually their magnitude and fluctuations have increased, which indicates that the addition of an electron by F atom to the CaO mono-layer structure has caused the emergence of metallic behavior.

Figure 7. The Eloss function of CaO, and CaO:F mono-layers versus photon energy.
Download figure:
Standard image High-resolution image3.4. Thermoelectric properties
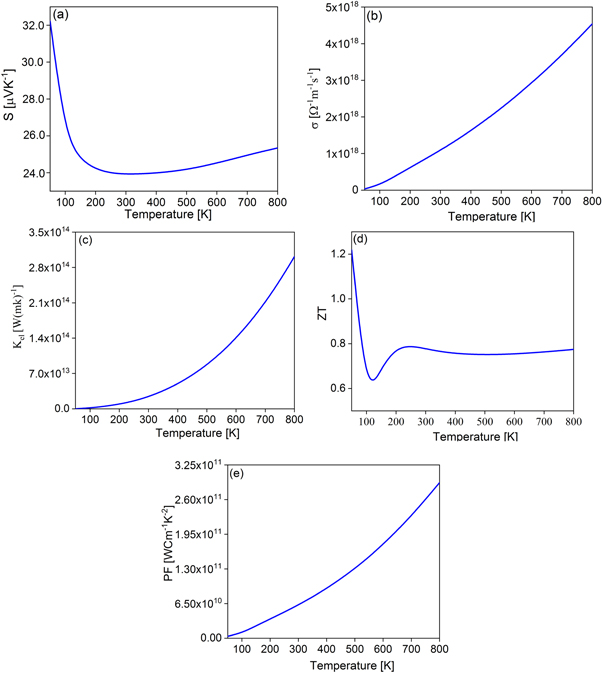
In the following, we will show the thermoelectric behaviour of CaO:F mono-layer compound in down spin under temperature. The thermoelectric property of any material is directly related to its band structure, and this property has higher efficiency quality in semiconductors with a gap less than one eV, with high density levels around the Fermi level. In figure 8, the thermoelectric parameters of CaO:F monolayer compound for down spin are plotted. In the band structure diagrams, it was observed that with the introduction of the F atom into the structure of the pure case, the energy gap has decreased from 2.6 eV to 0.2 eV, on the other hand, we see an increase in the density of the levels around the Fermi level. Panel (a) shows the Seebeck coefficient under temperature, this graph has a maximum at 50 K, which is comparable to other works [23, 24].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. The thermoelectric coefficients of CaO, and CaO:F mono-layers versus temperature.
Download figure:
Standard image High-resolution image{kind=link}
By increasing temperature to room temperature, the Seebeck coefficient is decreased to 24 μVK−1, and it reaches stable conditions at room temperature, so it can be said that this compound can be suitable for thermoelectric purposes at room temperature. The electrical conductivity under temperature has been depicted in panel (b), and it has a zero value at a temperature of 50 K, which is consistent with its semiconductor behaviour. Considering the small band gap and the high density of electron levels, it can be seen that with the increase in temperature, the electrical conductivity increases with a steep slope. In panel (c), we have shown the thermal conductivity caused by the electron contribution under temperature, where we see a thermal gap up to 100 K, and after that, the conductivity increases exponentially. In panel (d), the important thermoelectric parameter named figure of merit (ZT) coefficient under temperature is shown. In thermoelectric materials, the dimensionless coefficient of merit plays an important role in defining their efficiency, this coefficient is calculated from the following equation [31–34]:
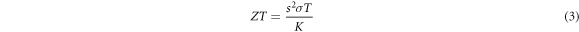
where T is the absolute temperature, S is the Seebeck coefficient, σ is the electrical conductivity, and K is the thermal conductivity value [37–41].
If the values of this parameter are in the range of one, it indicates the thermoelectric quality. This graph has two points, firstly, it has a value of 1.2 at 50 K, and secondly, it has reached a saturation limit at room temperature and has shown a stable behaviour with value of 0.75.
Therefore, CaO:F mono-layer has the desired thermoelectric quality for use in thermoelectric tools at room and upper temperatures. Also, this claim has been proven again in a panel (e), here the power factor (PF) diagram has been drawn and it can be seen that at high temperatures, its values are increasing, so it can be said that this combination is a power generator and is suitable for this application.
4. Conclusion
Based on density functional theory, structural, electronic, optical and thermoelectric properties of CaO and CaO:F mono-layer compounds have been investigated. The energy-volume curves of these monolayers have ground state points, which referred to their static stability from mechanical view. Both structures of CaO and CaO:F have bulk modulus of 85.296 GPa and 61.154 GPa, which indicates their hardness, and, also their derivative of bulk modulus referred to ionic bonds. Elastic coefficients for both structures represent their elastic stability. By adding the F atom to the CaO compound, the elastic coefficients, Young's modulus, Shear modulus and Bulk modulus have decreased, but it still has good elastic stability. So that its Young's modulus and Shear's modulus are 57.78 GPa and 23.85 GPa, respectively. The CaO monolayer is a p-type semiconductor with a gap of 2.6 eV, and adding a F atom to this compound, the half-metallic nature by 0.89 μB magnetic moment was emerged. In the CaO:F monolayer, we saw an indirect energy gap along the M-K path in up spin.
By adding the F atom to the CaO mono-layer, the optical response of this compound has been red-shifted, and the peaks of the imaginary part of dielectric function have moved to the infrared region, and the transitions have become intra-bands. The static responses of the CaO monolayer in the real part of dielectric function to the light emitted in both x and z directions are same, but in the CaO:F case this behaviour has changed and it has a metallic behaviour along the z direction, which are also confirmed in the diagrams of the imaginary part of the dielectric function. Also, the main optical response of CaO to the light was occurred at 5 eV to 10 eV, and for CaO:F monolayer main peak of imaginary part of dielectric function was at infrared region for z direction. The thermoelectric behaviour of CaO:F mono-layer showed that this compound is a suitable option to thermoelectric purposes. At a temperature of 50 K, the figure of merit coefficient has a value of 1.2, and at room temperature, it has reached a stable value of 0.78. The Seebeck coefficient also showed that the thermoelectric response at low temperatures reached 32 μVK−1.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).