



Abstract
The relationship between the magnetic interaction and photoinduced dynamics in antiferromagnetic perovskites is investigated in this study. In La1/3Sr2/3FeO3 thin films, commensurate spin ordering is accompanied by charge disproportionation, whereas SrFeO3−δ thin films show incommensurate helical antiferromagnetic spin ordering due to increased ferromagnetic coupling compared to La1/3Sr2/3FeO3. To understand the photoinduced spin dynamics in these materials, we investigate the spin ordering through time-resolved resonant soft x-ray scattering. In La1/3Sr2/3FeO3, ultrafast quenching of the magnetic ordering within 130 fs through a nonthermal process is observed, triggered by charge transfer between the Fe atoms. We compare this to the photoinduced dynamics of the helical magnetic ordering of SrFeO3−δ. We find that the change in the magnetic coupling through optically induced charge transfer can offer an even more efficient channel for spin-order manipulation.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
In the last century, the challenges in material science have mainly included the observation and manipulation of charges in materials, for application in semiconductor electronics. In the past decades, the utilization of spins, i.e., spintronics, has attracted considerable interest. The faster and energy-efficient manipulation of spins is one of the main issues in spintronics. Optical control of spins with ultrashort laser pulses is an important research topic for the ultrafast control of magnetization [1]. The first reported ultrafast control involved photoinduced demagnetization in ferromagnetic Ni within 1 ps [2]. Subsequently, the photoinduced charge and spin dynamics have been studied extensively through diffraction and spectroscopy in ferro-, antiferro-, and ferrimagnetic materials [3–13].
Antiferromagnets are expected to be the key material for the photocontrol of magnetic orderings. In the case of ferromagnetic materials, the magnetic moments need to be transferred from the spin to the other degrees-of-freedom, such as the lattice, when the magnetic moments are changed. However, due to the quenched total magnetic moments of antiferromagnets, angular momentum transfer between the spin and other systems is not necessary in antiferromagnets, enabling ultrafast magnetization changes. This was clarified by comparing the ferro- and antiferromagnetic phases of Dy [14].
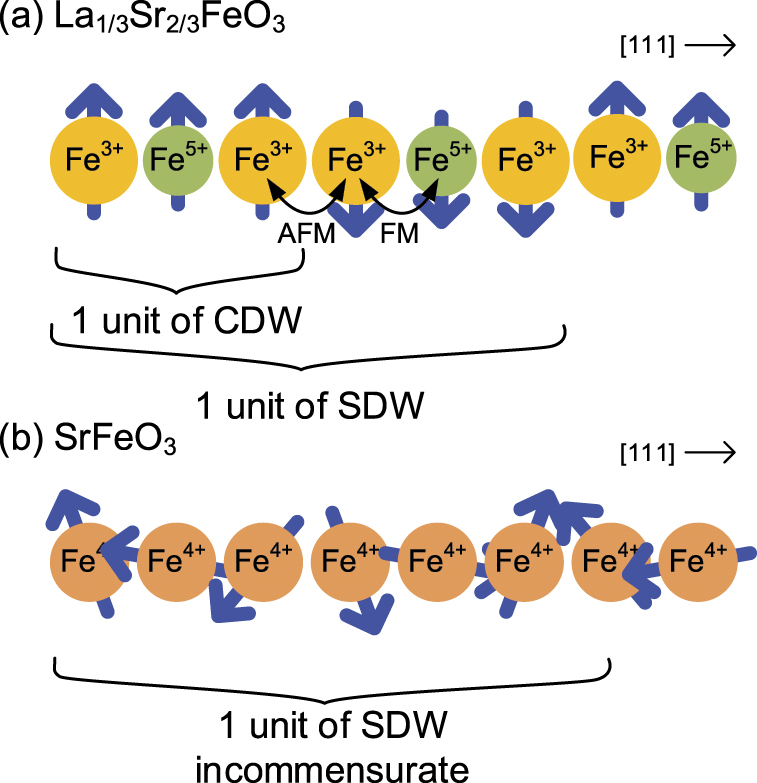
Several perovskite oxides with antiferromagnetic orderings have been discovered, whose properties can be controlled through elemental substitution and doping because of the strong interactions between the electrons, lattice, and spin degree-of-freedom [15]. Taking advantage of this feature, perovskite oxide materials suitable for the fast optical control of magnetism have been discovered [9, 10, 16, 17]. La1/3Sr2/3FeO3 and SrFeO3−δ perovskite thin films contain high valence Fe ions and oxygen holes. These features are clearly different from ordinary metals and insulators. In particular, La1/3Sr2/3FeO3 has charge and spin ordered structure with three and six Fe ions period and ordered structures such as this are realized in a few perovskites. We focused on antiferromagnetic orderings of Fe perovskite oxides and surveyed the photoinduced dynamics of these orderings directly.
The equilibrium magnetic properties of La1−x
Srx
FeO3 perovskites can be controlled based on the La/Sr composition. La1/3Sr2/3FeO3 thin films exhibit a charge disproportionation (CD) of 3Fe3.67+ → Fe5+ + 2Fe3+ accompanying antiferromagnetic ordering below TN = TCD ≈ 190 K [18, 19]. The high valence state of Fe5+ is realized as  , where
, where  denotes an oxygen 2p hole [20]. This CD phase results in a charge density wave with a period comprising three Fe ions as that of the crystal lattice, and a spin density wave of six Fe ion period along the 111 directions, as revealed by a neutron diffraction study (see figure 1(a)) [15]. SrFeO3−δ
includes the Fe4+ ion and shows a helimagnetic phase with incommensurate periodicity, which occurs because of the competition between the nearest neighbor ferromagnetic and the next-nearest neighbor antiferromagnetic interaction of the conducting 3d electrons, as indicated by neutron scattering results [21]. Fe4+ is realized as the
denotes an oxygen 2p hole [20]. This CD phase results in a charge density wave with a period comprising three Fe ions as that of the crystal lattice, and a spin density wave of six Fe ion period along the 111 directions, as revealed by a neutron diffraction study (see figure 1(a)) [15]. SrFeO3−δ
includes the Fe4+ ion and shows a helimagnetic phase with incommensurate periodicity, which occurs because of the competition between the nearest neighbor ferromagnetic and the next-nearest neighbor antiferromagnetic interaction of the conducting 3d electrons, as indicated by neutron scattering results [21]. Fe4+ is realized as the  in SrFeO3 perovskites [20, 22]. TN was reported to be 134 K [21] (bulk),
in SrFeO3 perovskites [20, 22]. TN was reported to be 134 K [21] (bulk),  K [23],
K [23],  K [24] (thin films). A multiple Q helimagnetic phase in SrFeO3−δ
was argued [23, 25], which could possibly host skyrmion crystals [26].
K [24] (thin films). A multiple Q helimagnetic phase in SrFeO3−δ
was argued [23, 25], which could possibly host skyrmion crystals [26].

Figure 1. Schematic of the spin and charge ordering in (a) La1/3Sr2/3FeO3 and (b) SrFeO3−δ (only Fe ions along the 111 direction are depicted).
Download figure:
Standard image High-resolution imageResonant soft x-ray scattering (RSXS) [27] is a powerful tool for revealing the ordered structures in solids, such as the magnetic, charge, and orbital ordering [27–33]. RSXS can be performed with the core level absorption process, and the interaction between x-rays and a specific element can be enhanced. Hence, RSXS can be applied to thin films with small sample volumes. X-ray polarization is sensitive to the orbitals of the valence electrons and RSXS can detect orbital and spin ordering, which are coupled through spin-orbital interaction. The magnetic ordering of irons in La1/3Sr2/3FeO3 [33, 34] and SrFeO3−δ
[23] have been observed through RSXS at the Fe L2,3 absorption edge (2p → 3d,  eV).
eV).
In this study, we report the photoinduced magnetic dynamics of La1/3Sr2/3FeO3 and SrFeO3−δ thin films determined through time-resolved RSXS measurements. We ascertain the ultrafast melting of the La1/3Sr2/3FeO3 magnetic ordering through charge transfer between Fe ions, which can be attributed to the strong coupling between the charge and spin in the system. Based on the comparison between La1/3Sr2/3FeO3 and SrFeO3−δ , the effect of the electronic properties on the dynamics of the photoinduced quenching of the magnetic orderings is examined.
2. Experimental
La1/3Sr2/3FeO3 thin films with ≈40 nm thicknesses were grown epitaxially on SrTiO3 (111) substrates using the pulsed laser deposition method. The details of the synthesis of La1/3Sr2/3FeO3 can be found in reference [35]. The magnetic ordering along [111] direction of La1/3Sr2/3FeO3 was more stabilized than the other ⟨111⟩ direction owing to the rhombohedral distortion [34]. By oxidizing SrFeO2.5 grown epitaxially on SrTiO3 (100) substrates using the pulsed laser deposition method, SrFeO3−δ
thin films with  nm thicknesses were fabricated on SrTiO3(100). SrFeO3−δ
was obtained by annealing SrFeO2.5 in an ozone atmosphere at 300 °C for 6 h with the sample exposed to UV light in a UV/O3 DRY CLEANER UV-1 (SAMCO Inc.). While, SrFeO3 is metallic, SrFeO3−δ
is semiconductive and this introduced δ of our samples enables the comparison to the insulating La1/3Sr2/3FeO3. In SrFeO3−δ
thin films, all ⟨111⟩ directions are equivalent and we observed one of four equivalent ⟨111⟩ directions.
nm thicknesses were fabricated on SrTiO3(100). SrFeO3−δ
was obtained by annealing SrFeO2.5 in an ozone atmosphere at 300 °C for 6 h with the sample exposed to UV light in a UV/O3 DRY CLEANER UV-1 (SAMCO Inc.). While, SrFeO3 is metallic, SrFeO3−δ
is semiconductive and this introduced δ of our samples enables the comparison to the insulating La1/3Sr2/3FeO3. In SrFeO3−δ
thin films, all ⟨111⟩ directions are equivalent and we observed one of four equivalent ⟨111⟩ directions.
In order to elucidate the photoinduced dynamics of the antiferromagnetic ordering in La1/3Sr2/3FeO3 and SrFeO3−δ
, we performed time-resolved RSXS measurements using the pump-probe method at the slicing facility UE56/1-ZPM Helmholtz-Zentrum, Berlin [36]. The experimental setup is shown in figure 2. A Ti:sapphire laser (λ = 800 nm, 1.5 eV) was employed for photoexcitation, with a pulse duration of 50 fs. The spot size of the pump laser was  , and that of the probe x-ray was
, and that of the probe x-ray was  [36]. The separation angle of both beams was smaller than 2° and this separation angle had a negligible effect on the delay time error within the overall temporal resolution of the experiment. The laser and x-ray pulse frequencies were 3 and 6 kHz, respectively, and signals with and without pump laser excitation were obtained alternately. The signals without excitation were used for normalizing the pumped signals. The scattered x-rays were detected using an avalanche photodiode. The laser slicing facility at BESSY has two operation modes: one with 50 ps temporal resolution and high pulse energy and one with 130 fs temporal resolution and low pulse energy. We used 50 ps x-ray pulses to obtain the outline of the photo-induced dynamics of La1/3Sr2/3FeO3 and SrFeO3−δ
and then investigated the detailed dynamics of La1/3Sr2/3FeO3 using 100 fs x-ray pulses generated using the laser slicing technique [36]. The total time resolution was
[36]. The separation angle of both beams was smaller than 2° and this separation angle had a negligible effect on the delay time error within the overall temporal resolution of the experiment. The laser and x-ray pulse frequencies were 3 and 6 kHz, respectively, and signals with and without pump laser excitation were obtained alternately. The signals without excitation were used for normalizing the pumped signals. The scattered x-rays were detected using an avalanche photodiode. The laser slicing facility at BESSY has two operation modes: one with 50 ps temporal resolution and high pulse energy and one with 130 fs temporal resolution and low pulse energy. We used 50 ps x-ray pulses to obtain the outline of the photo-induced dynamics of La1/3Sr2/3FeO3 and SrFeO3−δ
and then investigated the detailed dynamics of La1/3Sr2/3FeO3 using 100 fs x-ray pulses generated using the laser slicing technique [36]. The total time resolution was  . The scattering intensity from SrFeO3−δ
was too small for experiments with the weak 100 fs x-ray pulses. So we restricted ourselves to the lower temporal resolution of about 50 ps. SrFeO3−δ
was mounted on a wedge-shaped jig at an angle of 55 deg. in order to orient the [111] direction in the scattering plane, and the temperature was set to 35 K. We set the x-ray photon energy and polarization to the Fe L3 edge (
. The scattering intensity from SrFeO3−δ
was too small for experiments with the weak 100 fs x-ray pulses. So we restricted ourselves to the lower temporal resolution of about 50 ps. SrFeO3−δ
was mounted on a wedge-shaped jig at an angle of 55 deg. in order to orient the [111] direction in the scattering plane, and the temperature was set to 35 K. We set the x-ray photon energy and polarization to the Fe L3 edge ( eV) and horizontal, respectively, for obtaining the magnetic signals. Static La1/3Sr2/3FeO3 measurements were performed with a diffractometer at the soft x-ray beamline BL-16A, Photon Factory, KEK, Japan [37]. The experimental geometry and temperature were equivalent to the time-resolved measurement, and a silicon drift detector was used.
eV) and horizontal, respectively, for obtaining the magnetic signals. Static La1/3Sr2/3FeO3 measurements were performed with a diffractometer at the soft x-ray beamline BL-16A, Photon Factory, KEK, Japan [37]. The experimental geometry and temperature were equivalent to the time-resolved measurement, and a silicon drift detector was used.

Figure 2. (a) Experimental setup for time-resolved RSXS using the pump-probe method and (b) geometry for the SrFeO3−δ RSXS measurements.
Download figure:
Standard image High-resolution image3. Results and discussion
Figure 3 depicts the Q = (q, q, q) antiferromagnetic ordering peaks of (a) La1/3Sr2/3FeO3 and (b) SrFeO3−δ
. The diffraction peaks were scanned along the [111] direction. The temperature dependence of q and the peak intensities of La1/3Sr2/3FeO3 and SrFeO3−δ
are displayed in figures 3(c) and (d) respectively. The temperature evolution of RSXS intensity reproduced results of the previous study Néel temperature TN was determined via power-law fit  of peak intensity, as shown in figures 3(c) and (d). TN of La1/3Sr2/3FeO3 and SrFeO3−δ
was estimated to be 187 K and 110 K, respectively. For the La1/3Sr2/3FeO3 thin films, the peak position is fixed at (1/6, 1/6, 1/6) in the entire temperature range below TN, as shown in figure 3(a). The SrFeO3−δ
diffraction peak appears below TN ≈ 110 K, and the peak position shifts according to the temperature. Our SrFeO3−δ
thin films show the helimagnetic ordering below TN = 110 K with q = 0.126 r.l.u. along with insulating feature, which implies that δ of our SrFeO3−δ
samples is
of peak intensity, as shown in figures 3(c) and (d). TN of La1/3Sr2/3FeO3 and SrFeO3−δ
was estimated to be 187 K and 110 K, respectively. For the La1/3Sr2/3FeO3 thin films, the peak position is fixed at (1/6, 1/6, 1/6) in the entire temperature range below TN, as shown in figure 3(a). The SrFeO3−δ
diffraction peak appears below TN ≈ 110 K, and the peak position shifts according to the temperature. Our SrFeO3−δ
thin films show the helimagnetic ordering below TN = 110 K with q = 0.126 r.l.u. along with insulating feature, which implies that δ of our SrFeO3−δ
samples is  by comparing it with the results of previous studies [23, 24, 38, 39]. q of the SrFeO3−δ
helimagnetic ordering was reported to be 0.128–0.112 (SrFeO2.87) [21, 40], 0.13 (SrFeO3−δ
) or 0.125 (Sr0.99Co0.01FeO3) [23]; our observed q = 0.126 is in a similar range. In the heating and cooling cycle, thermal hysteresis was observed for the peak intensity and peak positions. The hysteresis has been reported to be related to the phase separation of paramagnetic and antiferromagnetic phases [41]. Similar phase separation was pointed out in La1/3Sr2/3FeO3 [34]. According to previous resistivity measurement reports [23, 24], thermal hysteresis was observed at T = 46–71 K, and our observed thermal hysteresis reflects this phenomenon.
by comparing it with the results of previous studies [23, 24, 38, 39]. q of the SrFeO3−δ
helimagnetic ordering was reported to be 0.128–0.112 (SrFeO2.87) [21, 40], 0.13 (SrFeO3−δ
) or 0.125 (Sr0.99Co0.01FeO3) [23]; our observed q = 0.126 is in a similar range. In the heating and cooling cycle, thermal hysteresis was observed for the peak intensity and peak positions. The hysteresis has been reported to be related to the phase separation of paramagnetic and antiferromagnetic phases [41]. Similar phase separation was pointed out in La1/3Sr2/3FeO3 [34]. According to previous resistivity measurement reports [23, 24], thermal hysteresis was observed at T = 46–71 K, and our observed thermal hysteresis reflects this phenomenon.

Figure 3. Temperature dependence of the diffraction peak of (a) La1/3Sr2/3FeO3 and (b) SrFeO3−δ ; (c) La1/3Sr2/3FeO3 peak position q and intensity I, and (d) SrFeO3−δ plotted as a function of the temperature.
Download figure:
Standard image High-resolution imageWe first discuss the La1/3Sr2/3FeO3 dynamics. The time evolution of the magnetic peak intensity after laser pumping, revealed in the laser slicing mode, is shown in figure 4(a). Rapid reduction in the diffraction intensity is observed within a time resolution of 130 fs. The diffraction intensities decrease to 50% of the initial values with the fluence of 0.5 mJ cm−2. This fluence corresponds to  photons/unit cell assuming absorption coefficient of 1.7 × 105 cm−1 [17] and unit cell volume of (0.39 nm)3. We confirmed that the intensity of unpumped signal intensity remained unchanged due to the pump laser fluence, which implies that the base temperature was constant. The decreased diffraction intensity recovers 30 ps after laser excitation in the lower fluence regime; however, the recovery is slower at higher fluence. In order to discuss the results quantitatively, we fitted them using the function depicted by equation (1) for the decay and recovery processes. The fitting function is as follows:
photons/unit cell assuming absorption coefficient of 1.7 × 105 cm−1 [17] and unit cell volume of (0.39 nm)3. We confirmed that the intensity of unpumped signal intensity remained unchanged due to the pump laser fluence, which implies that the base temperature was constant. The decreased diffraction intensity recovers 30 ps after laser excitation in the lower fluence regime; however, the recovery is slower at higher fluence. In order to discuss the results quantitatively, we fitted them using the function depicted by equation (1) for the decay and recovery processes. The fitting function is as follows:

where τdecay describes the time constant for photo-induced reduction, τfast and τ describe the time constants for the fast and slower components of recovery with an amplitude of Rfast and R. To consider time resolution, the fitting function was convoluted by a Gaussian with a full-width-at-half-maximum of τresolution = 130 fs. The parameters extracted from the experimental delay scans are summarized in table 1. The τdecay time scale was estimated to be 0.1 ± 0.05 ps.

Figure 4. (a) Time evolution of the magnetic peak intensity of La1/3Sr2/3FeO3 at τresolution = 130 fs for various pump laser fluence obtained in the laser slicing mode (the solid lines are fitted to the data using equation (1)) and (b) illustration of the photoinduced transient states of La1/3Sr2/3FeO3.
Download figure:
Standard image High-resolution imageTable 1. Fitting parameters for the delay scans of the La1/3Sr2/3FeO3 RSXS intensities in figure 4(a) observed in the slicing mode. τresolution = 130 fs. τdecay is less than time resolution τresolution.
| Fluence (mJ cm−2) | R + Rfast | τfast (ps) | τ (ps) |
|---|---|---|---|
| 0.39 | 0.51 ± 0.05 | 1.7 ± 0.3 | 60 ± 11 |
| 0.52 | 0.72 ± 0.03 | 3.5 ± 1.0 | 210 ± 36 |
| 0.65 | 0.78 ± 0.08 | — | 550 ± 100 |
For the nonthermal La1/3Sr2/3FeO3 process with an ultrashort time scale of τdecay ⩽ 0.1 ps, we consider that the ultrafast magnetization dynamics occurs because of the ultrafast photo-melting of the charge ordering, which destroys the magnetic ordering due to the strong coupling between the charge and spin. Demagnetization due to the photo-melting of the charge ordering has also been observed, for example, in NdNiO3 thin films [9]. Figure 4(b) illustrates the origins of the ultrafast melting of the magnetic ordering. Due to the strong onsite Hund's rule coupling, excitation with linearly polarized light at 1.5 eV can result only in transitions between the ferromagnetically coupled sites [42]. Optical conductivity measurement has demonstrated that the optical gap of 0.15 eV has a strong interatomic d–d transition characteristic [42]. Therefore, the excited electron would be transferred only in the ferromagnetically coupled Fe3+–Fe5+–Fe3+ sites, and the observation in this study suggests that the excitation induces charge transfer in a unit of the charge-ordering site of Fe3+–Fe5+–Fe3+ into Fe3+–Fe4+–Fe4+, as shown in figure 4(b). It has been suggested that this charge transfer is a metastable state accompanying Fe–O bond-length change, in a previous report on the photoinduced transient states of La1/3Sr2/3FeO3 [43]. Transfer in the charge ordering induces changes in the magnetic interactions. Fe3+–Fe4+ ions have ferromagnetic interactions, whereas the Fe4+–Fe4+ sites have antiferromagnetic ones, as shown in figure 4(b).
Furthermore, time-resolved RSXS measurements were performed for 35 and 80 K SrFeO3−δ thin films as shown in figure 5(a) with a time resolution of τresolution ≈ 50 ps; the experimental results for La1/3Sr2/3FeO3 with a similar time resolution are also shown in the right panel for comparison. Due to τresolution ≈ 50 ps, the fast decay process cannot be resolved in figure 5(a), and we focus on the changes in the magnetic ordering peak intensity. We fitted the results using equation (1) with setting the values of Rfast, τdecay, and τfast to 0 because fast component cannot be observed due to the lower time resolution. The function for fitting is

The degrees of quenching R, defined by equation (2), of the La1/3Sr2/3FeO3 and SrFeO3−δ magnetic ordering are plotted as a function of the laser fluence in figure 5(b). For La1/3Sr2/3FeO3, the quenching of the peak intensity reaches ≈70% at a fluence of approximately 0.75 mJ cm−2; however, the SrFeO3−δ diffraction intensity decreases by less than 60% of the initial intensity below a fluence of 2.8 mJ cm−2. This suggests that the antiferromagnetic ordering of La1/3Sr2/3FeO3 is quenched with less excitation energy compared to SrFeO3−δ . It should be noted that we observed ultrafast reduction of peak intensity of La1/3Sr2/3FeO3 with the time resolution of 130 fs and we compared La1/3Sr2/3FeO3 and SrFeO3−δ results based on the experiments of which time resolution is 50 ps. The electron temperature increase is estimated to be about several hundred K, which is in the similar range of previous studies [2, 5]. The peak positions of the photoinduced transient states and the heating process are plotted as a function of the peak area intensity in figure 5(c). The diffraction peaks of the transient states are shown in figure 5(d). Area intensities are obtained as the integration of the diffraction peak scan. Both curves are similar, indicating that the observed photoinduced quenching of the SrFeO3−δ helimagnetic ordering is triggered by the same mechanism as the temperature-induced change (table 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. (a) Pump-probe delay scans of SrFeO3−δ (right panel) and La1/3Sr2/3FeO3 (left panel) τresolution = 50 ps, (b) degree of photoinduced quenching of the diffraction peak intensity as a function of the laser fluence, (c) magnetic peak position of the photoinduced transient states and static states of SrFeO3−δ as a function of the peak area intensity and (d) the diffraction peaks of photo-induced transient states. Solid lines indicate the results of fitting. (e) Illustration of the photoinduced transient states of SrFeO3−δ .
Download figure:
Standard image High-resolution image{kind=link}
Table 2. Fitting parameters for the delay scans of the La1/3Sr2/3FeO3 and SrFeO3−δ RSXS intensities in figure 5 with the τresolution = 130 fs. τdecay is less than time resolution τresolution. τ was not suitably determined due to the small temporal scan range.
| Fluence (mJ cm−2) | R | τ (ps) | |
|---|---|---|---|
| La1/3Sr2/3FeO3 | 0.26 | 0.070 ± 0.002 | — |
| 0.39 | 0.169 ± 0.003 | — | |
| 0.46 | 0.300 ± 0.004 | — | |
| 0.52 | 0.514 ± 0.004 | — | |
| 0.65 | 0.701 ± 0.003 | — | |
| 0.78 | 0.759 ± 0.004 | — | |
| SrFeO3−δ | 0.71 | 0.14 ± 0.02 | 542 ± 196 |
| 1.41 | 0.16 ± 0.03 | 335 ± 76 | |
| 2.12 | 0.44 ± 0.03 | 308 ± 41 | |
| 2.82 | 0.54 ± 0.03 | 357 ± 42 |
SrFeO3 is metallic and 1.5 eV laser excitation corresponds to the interband transition [44]. The photoinduced quenching of the helimagnetic ordering in SrFeO3−δ can be interpreted as the result of the increase in the spin and electron temperatures, as depicted in figure 5(e). On the other hand, La1/3Sr2/3FeO3 is insulating and its magnetic orderings disappear because of the charge transfer between Fe ions induced locally by photoexcitation, as discussed above. The photoinduced dynamics of the antiferromagnetic orderings reflect the versatile electronic feature of perovskite oxides. Owing to the Sr substitution dependence of the absorption coefficient [17], the absorption coefficient becomes larger, which indicates that the observed trend does not originate from the strong absorption.
4. Conclusion
In this study, we examined the photoinduced dynamics of the antiferromagnetic orderings in two perovskites, La1/3Sr2/3FeO3 and SrFeO3−δ , through time-resolved RSXS. The magnetic ordering in La1/3Sr2/3FeO3 thin films was quenched within 130 fs. This ultrafast dynamics can be explained based on the photoinduced charge transfer between Fe ions, which induces a change in the strong magnetic interactions. The process is nonthermal. The spin moment can be canceled with the nearest neighbors in antiferromagnetic La1/3Sr2/3FeO3 through this ultrafast process, leading to an ultrafast photoinduced change in the magnetic ordering. Compared to helimagnetic SrFeO3−δ , the magnetic ordering of La1/3Sr2/3FeO3 was destroyed at lower fluence. Spin manipulation in helical systems has been found to be more energy efficient than in ferromagnets. An even more energy-efficient process can be realized in La1/3Sr2/3FeO3. Ultrafast changes in the magnetic ordering of antiferromagnetic perovskite thin films were discovered, and the photoinduced dynamics was shown to be controlled by tuning the electronic feature through perovskite doping. However, this comparison does leave aside the possibility that a strong reduction of the SrFeO3−δ antiferromagnetic peak intensity was neglected due to the lower time resolution compared to La1/3Sr2/3FeO3 case. Further investigations with better time resolution are needed in order to determine the time evolution of antiferromagnetic orderings completely. Changes of substrates or thickness affect the nature of thin films and such parameters characteristic of thin films are important to control the photo-induced magnetization dynamics. The photoinduced dynamics of the spin order in antiferromagnetic perovskite thin films is important for integrating functional oxides into spintronic device architectures.
Acknowledgments
We thank Yasuyuki Hirata for the productive discussions and HZB for the allocation of the synchrotron radiation beamtime, Karsten Holldack and Rolf Mitzner for experimental support. This work was performed under the approval of the Photon Factory Program Advisory Committee (Proposal Nos. 2016PF-BL-19B, 2015G556, 2015S2-007, 2013G058, 2013G661). This work was partially supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Nos. 19H05824, 19H01816, 19K23430, and 17K14334, and the MEXT Quantum Leap Flagship Program (MEXT Q-LEAP) Grant No. JPMXS0118068681. KY acknowledges the support from the ALPS program of the University of Tokyo.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.