

Abstract
Magnesium-aluminum (Mg-Al) intermetallic compounds that form as precipitates can significantly influence the mechanical properties of Mg-Al alloys. A computational evaluation of known and unknown Mg-Al intermetallic compounds could help design new Mg-Al alloy microstructures with optimal properties. Here, we employ the cluster-expansion method with energies efficiently calculated with orbital-free density functional theory (OFDFT) and predict a new, metastable intermetallic compound Mg3Al with a D019 hexagonal structure that is slightly more stable than an alternative L12 cubic structure. We apply Kohn-Sham DFT (KSDFT) to accurately evaluate various metastability criteria for D019 and L12 Mg3Al, including Born's criterion and phonon dispersion. We show that both Mg3Al crystalline phases satisfy the metastability criteria and hence should be at least metastable. We further compare ductility metrics for D019 and L12 Mg3Al to that of hexagonal-close-packed Mg by computing Pugh's ratio and generalized stacking fault energies. The ductility is predicted to follow the order: D019 Mg3Al > L12 Mg3Al > Mg, based on the highest Pugh's ratio and the lowest unstable stacking and twinning fault energies of D019 Mg3Al compared to that of Mg. We also predict a very low antiphase boundary energy for Mg3Al and therefore expect D019 Mg3Al to be beneficial for improving the ductility of Mg-rich Mg-Al alloys. A computational design of Mg-Al alloy microstructures may become possible by combining the strengths of both OFDFT and KSDFT, i.e., the efficiency of the former and the accuracy of the latter, as demonstrated here.
Export citation and abstract BibTeX RIS
1. Introduction
Magnesium-aluminum (Mg-Al) alloys are lightweight and exhibit excellent mechanical properties, enabling a wide range of applications in automotive, aerospace, and electronic device industries [1]. However, the mechanical properties of Mg-Al alloys strongly depend on various precipitated Mg-Al intermetallic compounds. For example, the presence of brittle Mg2Al3 and Mg17Al12 compounds leads to welding cracks between Mg and Al interfaces that are susceptible to fracture [2]. As such, studying structure-property relationships of Mg-Al intermetallic compounds serves as a critical step in providing guidance for optimizing Mg-Al alloy mechanical properties.
Computational tools based on density functional theory (DFT) are reliable for characterizing the structures and mechanical properties of Mg-Al intermetallic compounds [3–7]. Here, we utilize two different DFT methods. The first is Kohn-Sham DFT (KSDFT) [8, 9], which uses both orbitals and electron density to evaluate properties. The second is orbital-free DFT (OFDFT) [10], which employs electron density as its only variable. KSDFT is more accurate than OFDFT because the kinetic energy of non-interacting electrons can be evaluated exactly via orbitals in the former, whereas an approximate kinetic energy density functional (KEDF) must be used in the latter. However, the number of metal atoms treatable with conventional KSDFT is dramatically smaller than with OFDFT, mainly due to the cubic scaling algorithm required to perform time-consuming orthonormalization of the KS orbitals. Instead, use of a KEDF enables OFDFT to scale quasi-linearly with system size. The tradeoff between efficiency and accuracy motivates use of both DFT methods when studying Mg-Al intermetallics.
Mg and Al can form a number of intermetallic compounds, as documented in well-known databases such as the American Society for Metals (ASM) Alloy Phase Diagram Database [11] and the Inorganic Crystal Structure Database (ICSD) [12]. These databases play important roles in the design and selection of Mg-Al alloys. The crystal structures archived in the databases are also useful as starting geometries for simulations. However, only a limited number of compounds are listed in the databases. Going beyond known Mg-Al intermetallic compounds to search for new ones with promising mechanical properties requires the predictive power of quantum mechanics calculations such as those employed here.
In this work, we combine the abovementioned strengths of OFDFT and KSDFT to explore other possible Mg-Al intermetallic compounds. We are specifically interested in Mg-rich Mg-Al alloys to maximize the light-weighting of metal alloys and furthermore are interested in precipitates that could form within the hexagonal-close-packed (hcp) Mg matrix. Those precipitates are likely to be subject to the hcp host lattice type constraint in order to avoid large interface energies. We first use the OFDFT formation energy as a screening parameter to find stable structures subject to the hcp host lattice constraint. We identify a new Mg-Al intermetallic compound Mg3Al with the D019 hexagonal structure that has a small formation energy, slightly lower in energy than a hypothetical L12 face-centered cubic (fcc) structure previously used to fit an interatomic potential [7] that we also compare against. We then turn to KSDFT for more accurate characterization of Mg3Al properties. Both D019 and L12 Mg3Al are found to obey common criteria for metastability. We further predict that D019 Mg3Al should exhibit superior ductility compared to hcp Mg and L12 Mg3Al. The prediction and characterization of lightweight engineering alloys is thus facilitated via OFDFT for screening and KSDFT for the assessment of mechanical properties, as demonstrated here.
2. Methodology
We adopted the cluster-expansion (CE) method as implemented in the Alloy Theoretic Automated Toolkit (ATAT) [13] in search of stable Mg-Al intermetallic structures. The energy of a system within the CE method can be written as [14]
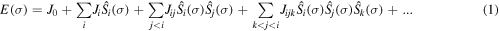
where Ji, Jij, and Jijk refer to the CE coefficients for the clusters consisting of one, two, and three atoms, respectively.  denotes that a site is occupied by either an Mg (+1) or Al (−1) atom. Together with the constant J0, these CE coefficients were determined from OFDFT formation energies of 101 different Mg-Al compounds calculated with the PROFESS 3.0 package [15]. Specifically, 55 clusters up to quadruplets were used in the least square fitting process in order to minimize the difference between the CE and OFDFT energies. Originally proposed by Collony and Williams [16], this fitting procedure is called the Collony-Williams method or the structure inversion method [13]. We implemented a scripting interface between ATAT and PROFESS 3.0, whereby the OFDFT formation energies are automatically utilized by ATAT. All OFDFT calculations used periodic boundary conditions, the Wang-Teter (WT) KEDF [17], and bulk-derived local pseudopotentials (BLPSs) [18, 19] for Mg and Al [20] to describe the electron-ion interactions. We employed the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) functional [21] for electron exchange-correlation (XC) and a planewave basis kinetic energy cutoff of 1200 eV. OFDFT with the WT KEDF, these GGA BLPSs [20], and the PBE XC functional yields properties in reasonable agreement with experiment and KSDFT for hcp Mg, fcc Al, and four Mg-Al intermetallic compounds such as Mg17Al12 [22].
denotes that a site is occupied by either an Mg (+1) or Al (−1) atom. Together with the constant J0, these CE coefficients were determined from OFDFT formation energies of 101 different Mg-Al compounds calculated with the PROFESS 3.0 package [15]. Specifically, 55 clusters up to quadruplets were used in the least square fitting process in order to minimize the difference between the CE and OFDFT energies. Originally proposed by Collony and Williams [16], this fitting procedure is called the Collony-Williams method or the structure inversion method [13]. We implemented a scripting interface between ATAT and PROFESS 3.0, whereby the OFDFT formation energies are automatically utilized by ATAT. All OFDFT calculations used periodic boundary conditions, the Wang-Teter (WT) KEDF [17], and bulk-derived local pseudopotentials (BLPSs) [18, 19] for Mg and Al [20] to describe the electron-ion interactions. We employed the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) functional [21] for electron exchange-correlation (XC) and a planewave basis kinetic energy cutoff of 1200 eV. OFDFT with the WT KEDF, these GGA BLPSs [20], and the PBE XC functional yields properties in reasonable agreement with experiment and KSDFT for hcp Mg, fcc Al, and four Mg-Al intermetallic compounds such as Mg17Al12 [22].
All KSDFT calculations were performed with the Vienna Ab-Initio Simulation Package (VASP, version 5.3.5) [23]. Projector-augmented-wave (PAW) [24, 25] potentials for Mg, Al, and Ni were used to account for the nuclei and frozen core electrons, with the 3 s states of Mg, 3s3p states of Al, and 3d4s states of Ni optimized self-consistently. We used a 500 eV planewave basis kinetic energy cutoff. Within the Monkhorst-Pack scheme [26], we used k-point grids of 12 × 12 × 12, 16 × 16 × 16, 16 × 16 × 16, 16 × 16 × 16, 8 × 8 × 8, 18 × 18 × 12, and 18 × 18 × 18 for eight-atom D019 Mg3Al, four-atom D03 Mg3Al, four-atom L12 Mg3Al, four-atom L12 Ni3Al, 29-atom Mg17Al12, two-atom hcp Mg, and one-atom fcc Al primitive cells, respectively. The elastic constants were calculated using a symmetry-general strain-stress method [27] as implemented in VASP. Increasing the k-point mesh density by up to 1.5 times converges the calculated elastic constants to within a range of 1 to 3 GPa (see supplemental material, table S1). Pugh's ratio (vide infra) is rather insensitive to the k-point density. The selected planewave kinetic energy cutoff and k-point meshes converge the total energy to within 1.0 meV/atom. The Brillouin-zone integration follows the Methfessel-Paxton method [28] with a smearing width of 0.2 eV. All lattice parameters and atomic coordinates were fully optimized until a force threshold of 0.01 eV/Å is reached.
We performed Monte Carlo (MC) simulations using the OFDFT-derived CE energy expression to calculate the order-disorder transition temperature for D019 and L12 Mg3Al using the ATAT package. A cutoff radius of 15 Å was used to determine the size of the MC supercell. The simulation systems were cooled down from 800 K to 300 K with a step of 20 K. At each temperature, 10 000 MC steps were run before another 10 000 steps were used for averaging the total energy. The constant volume heat capacity was then calculated as the first derivative of the temperature-dependent energy.
We computed the phonon spectra of D019 and L12 Mg3Al via PHONOPY [29], which post-processes the KSDFT force constants obtained from VASP. We used 3 × 3 × 3, 4 × 4 × 4, 4 × 4 × 4, 2 × 2 × 2, 6 × 6 × 6, and 6 × 6 × 6 supercells to calculate the force constants of D019 Mg3Al, D03 Mg3Al, L12 Mg3Al, Mg17Al12, hcp Mg, and fcc Al, respectively. We calculated the temperature-dependent formation energies of both Mg3Al structures based on the quasi-harmonic approximation [30], which requires volume-dependent phonon spectra data and total energies. 17 different Mg3Al primitive cells with equilibrium lattice constants scaled by 0.98 to 1.02 with an incremental step of 0.0025 were used.
We employed KSDFT to calculate the generalized stacking fault energies (GSFEs) γGSFE of D019 and L12 Mg3Al using surface slab models containing 16 atomic layers. A vacuum spacing of 15 Å was used to ensure that interactions between periodic images are negligible. To obtain a full γGSFE curve, we successively displace the atoms in the top eight layers of a relaxed surface slab and optimize only the out-of-plane coordinates of these atoms. Each surface slab has only one stacking fault and γGSFE therefore is determined as

where Edisp is the energy of the slab with displaced atoms, E0 is the energy of the reference slab before the atomic displacements, and A0 is the cross-sectional area of the lateral unit cell.
3. Results and discussion
We first search for potential stable Mg-Al intermetallic structures with convex hulls constructed on-the-fly by ATAT (figure 1(a)). We use the formation energy Ef of an MgxAl1−x compound as a descriptor of structural stability. Ef is calculated as
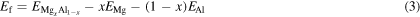
where  EMg, and EAl refer to the total energy per formula unit of hcp MgxAl1−x, hcp Mg, and hcp Al cells, respectively, with x denoting the molar composition of a MgxAl1−x compound. ATAT conventionally uses the same (parent) crystal structure for the compounds with compositions of x = 0 and 1. We follow this convention by using the energy of structurally relaxed Al initially adopting the same structure as hcp Mg, with the latter considered to be the parent structure (equation (3)). The relaxed Al structure here is a local minimum that remains hcp but has lattice vectors optimized for Al. The OFDFT total energy of ground-state fcc Al is lower than that of hcp Al by 4.4 meV/atom.
EMg, and EAl refer to the total energy per formula unit of hcp MgxAl1−x, hcp Mg, and hcp Al cells, respectively, with x denoting the molar composition of a MgxAl1−x compound. ATAT conventionally uses the same (parent) crystal structure for the compounds with compositions of x = 0 and 1. We follow this convention by using the energy of structurally relaxed Al initially adopting the same structure as hcp Mg, with the latter considered to be the parent structure (equation (3)). The relaxed Al structure here is a local minimum that remains hcp but has lattice vectors optimized for Al. The OFDFT total energy of ground-state fcc Al is lower than that of hcp Al by 4.4 meV/atom.
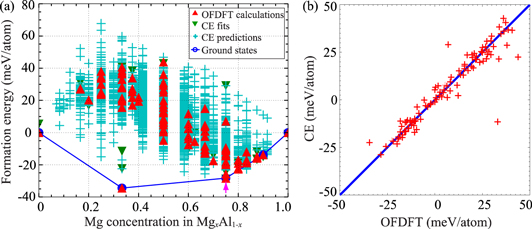
Figure 1. (a) Formation energies of Mg-Al compounds as a function of the concentration of Mg, from OFDFT (red triangles), CE fits (green triangles), and CE predictions (light blue crosses). Dark blue circles and lines form the convex hull of ground states. The magenta arrow highlights the Mg3Al compound that is the focus of this work. (b) Comparison (red crosses) of the formation energies of Mg-Al compounds obtained from CE and OFDFT methods. A perfect match between the CE and OFDFT formation energies is represented by the solid blue line. The number of green triangles and red triangles in figure 1(a) are equal. Most of the green triangles cannot be seen because they are underneath the red ones due to the overall excellent fit of the OFDFT formation energies to the CE method.
Download figure:
Standard image High-resolution imageFigure 1(a) displays four sets of data points. The first data set denoted 'OFDFT calculations' contains the formation energies of 101 Mg-Al intermetallic compounds calculated according to equation (3). The structures of these compounds are automatically generated by ATAT based on an algorithm [31] that is capable of efficiently enumerating superlattices and atomic configurations conforming to the symmetry of the parent structure. The lattice vectors and atomic coordinates of the structures are completely relaxed to compute the formation energies within the constraint of the parent structure symmetry. The energies of these compounds computed with the CE method are assembled in the second data set with an equal number of (101) data points, denoted 'CE fits.' The excellent agreement between these two data sets is shown in figure 1(b), validating the accuracy of the fitted CE coefficients. ATAT then uses the CE coefficients to predict the formation energies of 5675 other Mg-Al intermetallic compounds with different structures and stoichiometries, as represented by the 'CE predictions' data set (figure 1(a)). These structures are also generated by the same abovementioned algorithm. The final results obtained (figure 1(a)) indicate that there are three intermediate 'ground state' structures located at the convex hull, corresponding to the chemical formulae: MgAl2, Mg3Al, and Mg9Al.
Because we employ hcp Mg as the parent structure, the structure search is limited to hcp structures. As a result, the structural energies should only be considered useful for considering relative energies of precipitates constrained by the matrix to have an hcp structure. We therefore restrict our attention to the two compounds (Mg3Al and Mg9Al) with Mg concentrations on the Mg-rich side (x > ½) of the convex hull, which could maintain a hexagonal lattice. (One could use other structure search methods, such as the evolutionary search approach as implemented in, e.g., the USPEX package [32] to identify possible Mg-Al intermetallic compounds with different lattice types on the Al-rich side of the convex hull, if this was of interest.) In this work, however, we ended up studying only the properties of the Mg3Al intermetallic compound for the following reasons. In addition, predicting that Mg3Al has a lower formation energy than Mg9Al, we find that disproportionation of Mg9Al into D019 Mg3Al and hcp Mg is thermodynamically favorable. In particular, we predict  to be exoergic by −8.0 meV/atom, suggesting that D019 Mg3Al precipitates in hcp Mg could be at least metastable. We leave the study of mechanical properties of Mg9Al to future work, as its structure is more complicated than either of the possible Mg3Al structures, as can be seen in figure S2 in the supplemental material, available at stacks.iop.org/MSMS/25/075002/mmedia. More specifically, unlike Mg3Al, Mg9Al is not a simple superlattice structure of Mg but has a much lower symmetry. As a result, larger supercells with hundreds of atoms are required to calculate the generalized stacking fault energies to the same degree of accuracy done here (vide infra). We therefore focus first on the simpler D019 Mg3Al and its potentially competing L12 phase.
to be exoergic by −8.0 meV/atom, suggesting that D019 Mg3Al precipitates in hcp Mg could be at least metastable. We leave the study of mechanical properties of Mg9Al to future work, as its structure is more complicated than either of the possible Mg3Al structures, as can be seen in figure S2 in the supplemental material, available at stacks.iop.org/MSMS/25/075002/mmedia. More specifically, unlike Mg3Al, Mg9Al is not a simple superlattice structure of Mg but has a much lower symmetry. As a result, larger supercells with hundreds of atoms are required to calculate the generalized stacking fault energies to the same degree of accuracy done here (vide infra). We therefore focus first on the simpler D019 Mg3Al and its potentially competing L12 phase.
We consider here the lowest energy hcp structure determined by our ATAT search, D019, as well as two hypothetical cubic structures, D03 and L12 [7], for Mg3Al. Figure 2 displays top and side views of these Mg3Al primitive unit cells. The D019 structure, according to the Strukturbericht notation [33], has six Mg and two Al atoms in its primitive cell. A typical feature of this superstructure is that the in-plane lattice constant a0 is nearly twice that of hcp Mg (table 1). The D019 structure is adopted by many A3B-type intermetallic compounds with Ni3Sn as the prototype [33]. Several Mg-based intermetallic compounds such as Mg3Cd also crystallize in this D019 structure [34]. The D03 and L12 structures consist of three Mg and one Al atoms in their primitive cells. These two postulated structures were used in [7] to generate a Mg-Al interatomic potential.

Figure 2. Schematic top-view (left panel) and side-view (right panel) representation of the primitive unit cells of (a) D019, (b) D03, and (c) L12 Mg3Al.
Download figure:
Standard image High-resolution imageTable 1. Structural properties including lattice constants a and c (Å), independent elastic constants Cij (GPa), bulk modulus B (GPa), shear modulus G (GPa), Young's modulus E (GPa), and Pugh's ratio (B/G) of D019 and L12 Mg3Al calculated with KSDFT. The bulk and shear moduli calculated with the Voigt and Reuss expressions [35] are denoted with additional subscripts V and R, respectively. The same properties of hcp Mg, calculated in [22], are also shown for comparison.
| a | c | C11 | C12 | C13 | C33 | C44 | BV | BR | B | GV | GR | G | E | B/G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D019 Mg3Al | 6.19 | 5.04 | 73 | 33 | 30 | 73 | 13 | 45 | 45 | 45 | 17 | 17 | 17 | 45 | 2.65 |
| L12 Mg3Al | 4.38 | 4.38 | 71 | 32 | 32 | 71 | 26 | 45 | 45 | 45 | 23 | 23 | 23 | 59 | 1.96 |
| hcp Mg | 3.19 | 5.18 | 66 | 25 | 19 | 70 | 20 | 37 | 37 | 37 | 21 | 21 | 21 | 54 | 1.76 |
Considerations of strain and electronegativity lend some physical insight into formation of the Mg3Al phase. First, the atomic radii of Mg (1.50 Å) and Al (1.25 Å) [36] differ by more than 15%, a mismatch strain that disfavors solid solutions and favors intermetallic compounds instead [37]. Second, the electronegativities of the two species (Mg: 1.31; Al: 1.61 by the Pauling scale [38]) are somewhat different, which also drives formation of intermetallic compounds [37], via polar/metallic bonding. OFDFT predicts Ef values for D019, D03, and L12 Mg3Al of −23.9, 16.2, and −31.4 meV/atom, respectively, upon replacing the energy of hcp Al in equation (3) with that of ground-state fcc Al. By contrast, KSDFT yields values for Ef of 3.9, 43.0, and 5.4 meV/atom for D019, D03, and L12 Mg3Al, respectively. The accuracy limits of the WT KEDF used here for screening may cause discrepancies in the sign of Ef that can be remedied by using the more accurate Wang-Govind-Carter (WGC) KEDF [39–41], which yields Ef of the correct sign (2.8 and 0.9 meV/atom for D019 and L12 Mg3Al, respectively). The WGC KEDF also improves the Ef for D03 Mg3Al to 22.7 meV/atom. To test the sensitivity of Ef to the choice of XC functional, we also use KSDFT with the local density approximation (LDA) XC, which produces Ef of −4.2, 34.9, and −2.7 meV/atom, for D019, D03, and L12 Mg3Al, respectively. This is consistent with the general over-binding and under-binding shortcomings of the LDA and PBE XC functionals [42], respectively. Both OFDFT and KSDFT formation energies, however, consistently show that both D019 and L12 Mg3Al are on the verge of being stable compounds. By contrast, the D03 structure consistently exhibits relatively large positive formation energies. We thus rule out D03 as a competitive phase.
Because of the above discrepancies between the OFDFT and KSDFT formation energies, we hereafter use the more accurate KSDFT to study various properties of Mg3Al. The KSDFT-PBE formation energies above suggest that the new D019 Mg3Al structure is more stable than D03 and L12 Mg3Al by 39.1 and 1.5 meV/atom, respectively. Because the L12 phase is so close in energy to the predicted ground-state D019 phase and because its fcc structure could enable it to form by means of a stacking fault within the hcp Mg host lattice, we continue to investigate its properties in what follows.
The formation energy defined in equation (3) contains no contributions from the zero-point energy (ZPE), which could alter the relative stability of D019 and L12 Mg3Al, given their small Ef. We thus determine their ZPEs from KSDFT phonon spectra (figure 3). We then apply the quasi-harmonic approximation with volume-dependent phonon dispersions to compute the temperature-dependent free energies of D019 and L12 Mg3Al, hcp Mg, and fcc Al. Figure 4(a) displays the variation of Ef with temperature. The ZPE contributions to the Ef of D019 and L12 Mg3Al are −37.6 and −38.0 meV/atom, respectively. Such large ZPEs alter the sign of Ef from positive to negative. Increasing the temperature from 0 to 700 K further decreases Ef due to increasing entropy. Ef remains negative within a wide temperature range, confirming that decomposition of D019 or L12 Mg3Al into fcc Al and hcp Mg is endoergic. The variation of the free energy difference per atom between D019 and L12 Mg3Al with temperature (figure 4(b)) shows D019 Mg3Al consistently exhibiting slightly free lower energies than L12 Mg3Al, confirming D019 Mg3Al's slightly greater stability.
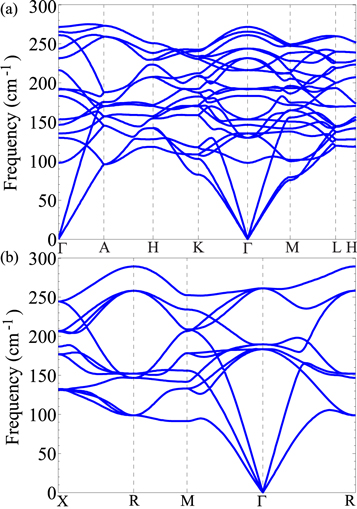
Figure 3. Phonon spectra of (a) D019 and (b) L12 Mg3Al calculated with KSDFT. The coordinates of the high-symmetry q points of D019 Mg3Al are Г(0, 0, 0), A(0, 0, 1/2), H(−1/3, 2/3, 1/2), K(−1/3, 2/3, 0), M(0, 1/2, 0), and L(0, 1/2, 1/2). The coordinates of the high-symmetry q points of L12 Mg3Al are X(1/2, 0, 0), R(1/2, 1/2, 1/2), M(1/2, 1/2, 0), and Г(0, 0, 0).
Download figure:
Standard image High-resolution image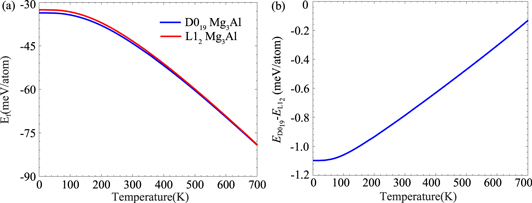
Figure 4. Temperature-dependent (a) formation free energy of D019 and L12 Mg3Al and (b) free energy difference between D019 and L12 Mg3Al calculated with KSDFT.
Download figure:
Standard image High-resolution imageMg3Al is unlikely to decompose directly into fcc Al and hcp Mg. We therefore should examine other possible decomposition reactions before concluding that it is thermodynamically stable. The most likely reaction to consider is

We select Mg17Al12 and Mg as possible products because the composition of Mg in Mg3Al is between that in Mg17Al12 and hcp Mg. Mg17Al12 is additionally one of three intermetallic compounds (Mg17Al12, Mg23Al30, and Mg2Al3) that exist in the experimental phase diagram of Mg-Al alloys [43]. Figure S3 in the supplemental material shows the phonon spectrum of Mg17Al12. Using free energies calculated as discussed earlier, figure S4 in the supplemental material displays the temperature-dependent free energies for Mg3Al decomposing to Mg and Mg17Al12. We find that the above reaction (equation (4)) at 298 K is exoergic by −17.9 and −18.7 meV/atom for D019 and L12 Mg3Al, respectively, i.e., D019 or L12 Mg3Al is susceptible to decomposing into Mg17Al12 and hcp Mg. This decomposition, however, requires an unknown reaction barrier, which we speculate could be large since the structure of Mg17Al12 (see figure S1 in the supplemental material) differs drastically from D019 and L12 Mg3Al. However, currently we have no way to theoretically estimate the barrier to decomposition, so experiments will be needed to confirm or disprove our speculation.
It is of course possible that some other phase of different crystal morphology (non-hcp, non-cubic) not considered here might exist that has a lower formation free energy than that of D019 Mg3Al. Again, we emphasize that our work focuses on Mg-rich Mg-Al alloys with the realistic physical constraints imposed by the Mg lattice. As such, it is very unlikely that adding a small amount of Al to hexagonal Mg at ambient conditions would transform the structure to another morphology incommensurate with hcp, due to the large strain it would engender. A non-equilibrium synthesis process such as the rapid quenching method [44] could be used to precipitate D019 or L12 Mg3Al in hcp Mg, as was used to prepare another metastable intermetallic compound, MgAl2 [45].
We further explore Mg3Al's metastability by evaluating its dynamical and mechanical stability. Beyond enabling calculation of the ZPE, the calculated phonon spectra in figure 3 serve another important purpose: verifying a material's dynamical stability [46], which requires the presence of only real phonon modes in the first Brillouin zone. Indeed, D019 and L12 Mg3Al's phonon modes are all real, confirming both phases' dynamical stability. Figure S5 in the supplemental material shows the phonon spectrum of D03 Mg3Al. D03 Mg3Al is expected to be dynamically unstable, consistent with its positive formation energy Ef of 43.0 meV/atom.
Mechanical stability of D019 and L12 Mg3Al is assessed via calculation of their five (C11, C12, C13, C33, and C44) and three (C11, C12, and C44) independent elastic constants, respectively. The elastic constants for hcp D019 Mg3Al must satisfy the following four constraints [47]:


and

For fcc L12 Mg3Al, three criteria need to be met:


and

The calculated elastic constants of Mg3Al in table 1 clearly satisfy the above four criteria, showing that both D019 and L12 Mg3Al are mechanically stable.
To explore the thermal stability of D019 and L12 Mg3Al, we also simulated their order-disorder phase transition via MC simulations using the OFDFT-derived CE energy expression. Figure 5(a) displays the temperature-dependent total energy and heat capacity of D019 Mg3Al. The total energy rises monotonically with increasing temperature until an abrupt increase occurs at about 460 K. The discontinuous temperature dependence correspondingly appears in the temperature-dependent heat capacity shown in the inset, revealing that an order-disorder phase transition in D019 Mg3Al should occur at ∼460 K. This temperature is sufficiently high to conclude once again that D019 Mg3Al should be at least thermally metastable under ambient conditions. By contrast, the order-disorder transition temperature of L12 Mg3Al is ∼320 K (figure 5(b)). This lower temperature is consistent with the calculated formation energies showing that D019 Mg3Al is slightly more stable.

Figure 5. Temperature-dependent energy and heat capacity (inset) of (a) D019 and (b) L12 Mg3Al.
Download figure:
Standard image High-resolution imageGiven the encouraging stability metrics above, we proceed to study the mechanical properties of D019 and L12 Mg3Al and compare them to those of hcp Mg. We use Pugh's ratio B/G (B: bulk modulus; G: shear modulus) to first assess the expected average ductilities of D019 and L12 Mg3Al and then compare them with that of hcp Mg. This ratio is commonly utilized as an indicator of a material's ductility [48]. According to Pugh, larger B/G implies better ductility. Table 1 lists the bulk and shear moduli converted from the calculated elastic constants according to the Voigt (BV and GV) and Reuss (BR and GR) approximations [35]. These two approximations give rise to upper and lower bounds on the moduli, respectively. Table 1 also reports the bulk and shear moduli based on the Hill approximation, which are the averaged values of the two bounds. The latter approximation is often called the Voigt-Reuss-Hill approximation [35]. For Mg and Mg3Al, the differences between the Voigt and Reuss moduli are negligible. The predicted bulk moduli of D019 and L12 Mg3Al are identical and considerably larger than Mg, whereas the two shear moduli deviate differently: D019 is smaller while L12 is larger than hcp Mg. As a result, the predicted Pugh's ratios B/G of D019 and L12 Mg3Al are 2.65 and 1.96, larger than that of hcp Mg with its calculated B/G of 1.76. The theoretical B/G of hcp Mg is in reasonable agreement with the experimental value of 1.93, derived from the measured elastic constants [49], validating our modeling approach. Given the significantly larger B/G for D019 Mg3Al, we can state with confidence that D019 Mg3Al should be more ductile than hcp Mg, whereas the Pugh's ratio of L12 Mg3Al implies that its ductility should be more similar to that of Mg.
Another indicator of ductility is provided by the GSFE, γGSF. We first consider the calculated γGSF curves of D019 Mg3Al for two common slip systems on the basal plane,  and
and  (figures 6(a) and (b), respectively). Here, the energy scales on the two slip systems are quite different: the γGSF on the
(figures 6(a) and (b), respectively). Here, the energy scales on the two slip systems are quite different: the γGSF on the  system is much smaller than on the
system is much smaller than on the  system. It therefore is much more likely that D019 Mg3Al will deform along the former slip system, similar to Mg: by slipping in this direction, atoms in different planes do not pass directly above each other and can thus minimize repulsions. Table 2 summarizes the four critical γGSF of D019 Mg3Al and Mg on the
system. It therefore is much more likely that D019 Mg3Al will deform along the former slip system, similar to Mg: by slipping in this direction, atoms in different planes do not pass directly above each other and can thus minimize repulsions. Table 2 summarizes the four critical γGSF of D019 Mg3Al and Mg on the  slip system, including the unstable stacking fault (γUSF), stacking fault (γSF), unstable twinning (γUT), and twinning (γTE) energies. A stacking fault corresponds to the stacking sequence ABABABABCACACACA of the close-packed planes in a 16-layer slab model, whereas a twinning fault has the ABABABABCBABABAB stacking pattern. The unstable stacking fault γUSF and unstable twinning γUT refer to the energy barriers to form the stacking and twinning faults, respectively. Table 2 shows that our calculated critical energies of Mg are consistent with previous KSDFT values [50, 51]. Importantly, the slightly reduced γUSF and γUT for D019 Mg3Al will ease formation of stacking and twinning faults, contributing to its ease of plastic deformation. The differences in stacking fault energies (SFEs) between D019 Mg3Al and hcp Mg are small but should be reliable because our calculations are well converged numerically. However, the SFEs of hcp Mg are small in absolute terms, so it is better to compare the SFEs of hcp Mg and Mg3Al in terms of percentage. For example, the reduction in the unstable SFE from hcp Mg to D019 Mg3Al is about 10%. Pugh's ratio (table 1) is a stronger indicator, increasing from 1.76 for hcp Mg to 2.65 for D019 Mg3Al, an increase of 51%. Combining the evaluations of both ductility metrics, D019 Mg3Al should exhibit better ductility than Mg.
slip system, including the unstable stacking fault (γUSF), stacking fault (γSF), unstable twinning (γUT), and twinning (γTE) energies. A stacking fault corresponds to the stacking sequence ABABABABCACACACA of the close-packed planes in a 16-layer slab model, whereas a twinning fault has the ABABABABCBABABAB stacking pattern. The unstable stacking fault γUSF and unstable twinning γUT refer to the energy barriers to form the stacking and twinning faults, respectively. Table 2 shows that our calculated critical energies of Mg are consistent with previous KSDFT values [50, 51]. Importantly, the slightly reduced γUSF and γUT for D019 Mg3Al will ease formation of stacking and twinning faults, contributing to its ease of plastic deformation. The differences in stacking fault energies (SFEs) between D019 Mg3Al and hcp Mg are small but should be reliable because our calculations are well converged numerically. However, the SFEs of hcp Mg are small in absolute terms, so it is better to compare the SFEs of hcp Mg and Mg3Al in terms of percentage. For example, the reduction in the unstable SFE from hcp Mg to D019 Mg3Al is about 10%. Pugh's ratio (table 1) is a stronger indicator, increasing from 1.76 for hcp Mg to 2.65 for D019 Mg3Al, an increase of 51%. Combining the evaluations of both ductility metrics, D019 Mg3Al should exhibit better ductility than Mg.

Figure 6. GSFE curves of D019 Mg3Al and hcp Mg on (a)  (b)
(b)  and (c)
and (c)  slip systems calculated with KSDFT. Note changes in vertical scales for each panel. The inset in (b) illustrates a basal plane and the two slip directions considered. The GSFE curve of L12 Mg3Al on the
slip systems calculated with KSDFT. Note changes in vertical scales for each panel. The inset in (b) illustrates a basal plane and the two slip directions considered. The GSFE curve of L12 Mg3Al on the  slip system is shown in panel (d). a0 is defined as the lattice constant of the unit cell of hcp Mg, D019 Mg3Al, or L12 Mg3Al. Note that a0 of D019 Mg3Al is nearly twice the size of hcp Mg.
slip system is shown in panel (d). a0 is defined as the lattice constant of the unit cell of hcp Mg, D019 Mg3Al, or L12 Mg3Al. Note that a0 of D019 Mg3Al is nearly twice the size of hcp Mg.
Download figure:
Standard image High-resolution imageTable 2.
Unstable stacking fault  stacking fault
stacking fault  unstable twinning
unstable twinning  and twinning
and twinning  energies (mJ/m2) of hcp D019 Mg3Al and hcp Mg on the
energies (mJ/m2) of hcp D019 Mg3Al and hcp Mg on the  slip system and fcc L12 Mg3Al on the
slip system and fcc L12 Mg3Al on the  slip system. The results calculated in this work are in bold. Other KSDFT results from the literature are shown for comparison.
slip system. The results calculated in this work are in bold. Other KSDFT results from the literature are shown for comparison.

|
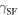
|
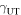
|
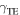
|
|
|---|---|---|---|---|
| D019 Mg3Al | 84 | 29 | 107 | 27 |
| L12 Mg3Al | 91 | 19 | 123 | 90 |
| hcp Mg | 93 | 35 | 113 | 46 |
| 92a | 36a | 111a | 39a | |
| 94b | 28b | 113b | 39b | |
For hexagonal D019 Mg3Al and hcp Mg, we also computed the GSFE for the prismatic  slip system, which is the second lowest energy slip system. Figure 6(c) shows the corresponding GSFE curves, and the energy barriers are determined as 162 and 203 mJ m−2 for D019 Mg3Al and hcp Mg, respectively. We define an anisotropy ratio ʌ as the ratio between the energy barriers for the prismatic and basal slip systems. The ʌ values for D019 Mg3Al and hcp Mg are predicted to be 1.93 and 2.18, respectively. A larger ʌ corresponds to a stronger anisotropy, which results in worse ductility. D019 Mg3Al therefore also exhibits better ductility than hcp Mg by this metric, which is consistent with the assessment using Pugh's ratio.
slip system, which is the second lowest energy slip system. Figure 6(c) shows the corresponding GSFE curves, and the energy barriers are determined as 162 and 203 mJ m−2 for D019 Mg3Al and hcp Mg, respectively. We define an anisotropy ratio ʌ as the ratio between the energy barriers for the prismatic and basal slip systems. The ʌ values for D019 Mg3Al and hcp Mg are predicted to be 1.93 and 2.18, respectively. A larger ʌ corresponds to a stronger anisotropy, which results in worse ductility. D019 Mg3Al therefore also exhibits better ductility than hcp Mg by this metric, which is consistent with the assessment using Pugh's ratio.
The competitive stability of D019 and L12 Mg3Al motivates investigation of the GSFE of L12 Mg3Al on the favored fcc  slip system to assess the energy barrier to form hcp-stacked layers as in D019 Mg3Al. We again use a 16-layer slab model with the initial stacking sequence of ABCABCABCABCABCA. As a result of the stacking fault, the stacking sequence becomes ABCABCABCBCABCAB, i.e., four hexagonally stacked layers (BCBC) occur in the fcc structure. Figure 6(d) shows the resulting GSFE curve of L12 Mg3Al with the four critical energies given in table 2. We predict a barrier to form the stacking fault of 91 mJ m−2, comparable to the barrier (84 mJ m−2) to form three fcc stacking layers (ABC) starting from the D019 structure. Both energy barriers are sufficiently small and consistent with the nearly degenerate energies of the two structures, suggesting that both structures could be present in such precipitates.
slip system to assess the energy barrier to form hcp-stacked layers as in D019 Mg3Al. We again use a 16-layer slab model with the initial stacking sequence of ABCABCABCABCABCA. As a result of the stacking fault, the stacking sequence becomes ABCABCABCBCABCAB, i.e., four hexagonally stacked layers (BCBC) occur in the fcc structure. Figure 6(d) shows the resulting GSFE curve of L12 Mg3Al with the four critical energies given in table 2. We predict a barrier to form the stacking fault of 91 mJ m−2, comparable to the barrier (84 mJ m−2) to form three fcc stacking layers (ABC) starting from the D019 structure. Both energy barriers are sufficiently small and consistent with the nearly degenerate energies of the two structures, suggesting that both structures could be present in such precipitates.
Beyond evaluating the GSFE, we also considered another important planar defect in ordered intermetallic phases that help determine ductility trends, namely, anti-phase boundaries (APBs) [52]. An APB would form in an ordered Mg3Al precipitate by the passing of (partial) dislocations. The formation of an APB in an intermetallic compound introduces a compositional rearrangement, e.g., switched positions of Mg and Al atoms, which could be energetically costly and decrease ductility via setting up an additional effective barrier to dislocation motion. We therefore calculated the APB energies of L12 and D019 Mg3Al with their  and
and  planes shifted along the
planes shifted along the  and
and  vectors, respectively. The atomic coordinates of the surface slabs with and without an APB are given in the supplemental material. We found that the APB energies of L12 and D019 Mg3Al are extremely small, 14 and 27 mJ m−2, respectively. For comparison, we computed the APB energy of L12 Ni3Al with our setup and found it to be an order of magnitude larger, 196 mJ m−2, consistent with previous theoretical and experimental values (e.g., [53, 54]). The significantly smaller APB energies of L12 and D019 Mg3Al imply that such precipitates will not inhibit dislocation motion beyond the barriers predicted by the GSFEs shown in figure 6, which already imply an improved ductility. This conclusion can be drawn because the APB energy is inversely related to the distance between two partial dislocations [52]; the expected large distance between partials in this case then suggests that the ductility of Mg with such Mg3Al precipitates therefore will be determined by the passage of a single partial dislocation, whose ease of movement depends on the GSFEs already discussed.
vectors, respectively. The atomic coordinates of the surface slabs with and without an APB are given in the supplemental material. We found that the APB energies of L12 and D019 Mg3Al are extremely small, 14 and 27 mJ m−2, respectively. For comparison, we computed the APB energy of L12 Ni3Al with our setup and found it to be an order of magnitude larger, 196 mJ m−2, consistent with previous theoretical and experimental values (e.g., [53, 54]). The significantly smaller APB energies of L12 and D019 Mg3Al imply that such precipitates will not inhibit dislocation motion beyond the barriers predicted by the GSFEs shown in figure 6, which already imply an improved ductility. This conclusion can be drawn because the APB energy is inversely related to the distance between two partial dislocations [52]; the expected large distance between partials in this case then suggests that the ductility of Mg with such Mg3Al precipitates therefore will be determined by the passage of a single partial dislocation, whose ease of movement depends on the GSFEs already discussed.
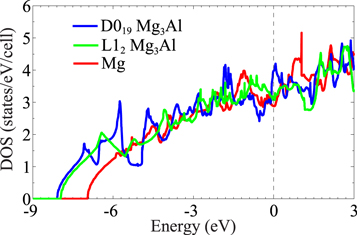
To elucidate the electronic origin of the predicted enhanced ductility of D019 Mg3Al over L12 Mg3Al and hcp Mg, figure 7 compares the density of states (DOS) of a D019 Mg3Al primitive cell, an fcc L12 Mg3Al 2 × 1 × 1 supercell, and an hcp 2 × 2 × 1 Mg supercell (these supercells are used so as to have the same number of atoms in each material's unit cells). The number of states per simulation cell at the Fermi level is 3.98 for D019 Mg3Al, 3.09 for L12 Mg3Al, and 2.88 for hcp Mg. The larger the DOS at the Fermi level, the higher the metallicity [55], again indicating better ductility for D019 Mg3Al over L12 Mg3Al and Mg, consistent with its much higher Pugh's ratio and somewhat lower unstable SFEs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. DOS of an hcp D019 Mg3Al unit cell, an fcc L12 Mg3Al 2 × 1 × 1 supercell, and an hcp Mg 2 × 2 × 1 supercell calculated with KSDFT. The Fermi energy EF is set to zero.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
Using a CE method fit to energies from OFDFT calculations, we discovered a potentially useful new Mg-Al intermetallic compound, Mg3Al, exhibiting a simple D019 hcp structure. This new intermetallic phase satisfies mechanical and dynamical stability criteria, indicating that it is at least metastable. D019 Mg3Al should have better ductility than Mg and a hypothetical L12 Mg3Al phase, according to its calculated Pugh's ratios and unstable SFEs. We attribute the improved ductility to enhanced metallicity, as evidenced by the increased (Al) DOS at the Fermi level. This trend is consistent with the slight polarization of electrons towards Al and away from Mg expected from their relative electronegativity. Bader charge analysis [56, 57] confirms the differentially increased electron density around each Al atom in D019 Mg3Al (2.96 e/atom) versus in L12 Mg3Al (2.82 e/atom).
The potential D019 precipitate structure has two engineering-related important implications. First, its superior Pugh's ratio and unstable SFE imply that D019 Mg3Al precipitates in Mg-rich Mg-Al alloys should improve Mg-rich Mg-Al alloy ductility more than L12 precipitates would. Second, because the in-plane lattice constant (table 1) of D019 Mg3Al is nearly twice of that of hcp Mg, only a small misfit strain δ (∼3%) would be induced between the host Mg matrix and the precipitate. A dislocation-based interface model can be used to estimate the interface energy E caused by this misfit strain [58]: E = Gbδ/2. Here, b is the Burgers vector of Mg, ∼2.5 Å [59]. The resulting E (78 mJ m−2) is sufficiently small to suggest formation of an almost coherent interface that should reduce the risk of fracture failure.
More generally, our work demonstrates the efficacy of coupling OFDFT to the CE method, while searching for new Mg-Al intermetallic compounds. Although the superior efficiency of OFDFT was not fully exploited here due to the simplicity of the binary Mg-Al system, requiring only 101 structures to obtain the CE coefficients, the structure search and characterization strategy demonstrated here is quite general and can be applied to design numerous other complicated lightweight alloys such as Mg-Li-Ca ternary alloys that are of great interest for engineering applications.
Other structure prediction methods [60], such as genetic algorithms, also could be coupled straightforwardly to OFDFT to predict other new complex alloys. The CE method implemented in ATAT has the disadvantage that the predicted structures are constrained to the parent structure symmetry. In contrast, genetic algorithms do not have such a constraint [61]. Developments in OFDFT and enhanced computational power have enabled efficient first-principles molecular dynamics simulations of phase transitions in samples containing more than 1000 metal atoms [62]. Peta-scale OFDFT computations recently demonstrated that the electronic ground state for more than one million metal atoms can be obtained in around one minute [63]. We therefore believe using OFDFT as the energy calculator for genetic-algorithm-based alloy optimization will soon be possible, even for large numbers of atoms.
Acknowledgments
We are grateful to the Office of Naval Research for funding (Grant No. N00014-15-1-2218). We acknowledge use of the TIGRESS high performance computer center at Princeton University. We thank Ms Nari Baughman and Dr Johannes M Dieterich for critical reading of this manuscript. We also thank Dr Qijun Hong for helping with the calculations of order-disorder transition temperatures.