

Abstract
The nitrogen-rich compounds are promising candidates for high-energy-density applications, owing to the large difference in the bonding energy between triple and single/double nitrogen bonds. The exploration of stable copper–nitrogen (Cu–N) compounds with high-energy-density has been challenging for a long time. Recently, through a combination of high temperatures and pressures, a new copper diazenide compound (P63/mmc-CuN2) has been synthesized (Binns et al 2019 J. Phys. Chem. Lett. 10 1109–1114). But the pressure-composition phase diagram of Cu–N compounds at different temperatures is still highly unclear. Here, by combining first-principles calculations with crystal structure prediction method, the Cu–N compounds with different stoichiometric ratios were searched within the pressure range of 0–150 GPa. Four Cu–N compounds are predicted to be thermodynamically stable at high pressures, Pnnm-CuN2, two CuN3 compounds with the P-1 space group (named as I-CuN3 and II-CuN3) and P21/m-CuN5 containing cyclo-N5−. Finite temperature effects (vibrational energies) play a key role in stabilizing experimentally synthesized P63/mmc-CuN2 at ∼55 GPa, compared to our predicted Pnnm-CuN2. These new Cu–N compounds show great promise for potential applications as high-energy-density materials with the energy densities of 1.57–2.74 kJ g−1.
1. Introduction
At ambient conditions, molecular nitrogen is chemically inert because the N≡N triple bond is one of the strongest chemical bonds. Poly-N materials with single (N–N) and double (N=N) bonds are promising high-energy-density materials owing to the large difference in bonding strength between triple and single/double bonds [1, 2]. The polymeric N has never been formed at ambient pressure, but high-temperature and high-pressure provide an effective way to stabilize them. At high pressures (>110 GPa) and high temperature (>2000 K), the cg-N2 structure has been obtained by Eremets et al in 2004 [3]. More recently, another kind of nitrogen polymerization with the black phosphorus structure, (bp-N2), was also synthesized at 140 GPa and 4000 K [4, 5].
An alternative way to stabilize poly-N phases is constructing N-rich compounds with additional elements [6, 7], such as alkali (Li, Na, K, Cs) [7–13], alkaline earth (Mg, Ca, Be) [14–18], transition metals (Fe, Ru, Ir, Rh) [19–23] and main group elements (Al, B, S, Se, Rb and so on) [12, 17, 24–29]. In addition to the previously known lithium nitride (Li3N) and lithium azide (LiN3) [8, 30], new Li-N compounds like LiN2, LiN and LiN5 have recently been predicted to be stable and synthesizable [9, 10]. Calculations based on the machine-learning-accelerated technique predicted that BeN10 and MgN10 are metastable and can release a large amount of energy when decomposition occurs [1]. For the Fe-N compounds, Wu et al have performed a structure search within the pressure rang of 0–100 GPa through the particle swarm optimization (PSO) technique [23]. The high-energy-density-materials Fe3N8, FeN3, FeN6 and FeN8 were considered stable at high pressure of 28–100 GPa, 30–100 GPa, 20–64 GPa and 24–60 GPa, respectively [23]. In 2018, Bykov et al synthesized three kinds of Fe-N compounds, Fe3N4, FeN2 and FeN4 structures, at 50 GPa, 58 GPa and 106 GPa by heating Fe and N2 in a laser-hearted diamond anvil cell [19]. For the S-N compounds, the tetra-sulfur tetra-nitride (S4N4) can be used as an explosive and was first prepared in 1835 by Gregory [31]. More recently, Li et al have predicted that two new S-N compounds (SN2, SN4) are stable under high pressure and revealed the high-pressure structural phase transitions of S-N compounds [28].
However, the synthesis of stable N-containing compounds with coinage metals (Cu, Ag and Au) has been more challenging [32]. For a long time, only two kinds of metastable Cu–N compounds are known to exist: copper azide (Pm m-CuN3) as an explosive [33], and copper nitride (Cmcm-Cu3N) as a lithium battery electrode material [34]. Till recently, by using a combination of high pressure (>50 GPa) and high temperature (1500 K) conditions, Binns et al reported the observation of direct reaction between copper and nitrogen to forming a previously unknown copper diazenide (P63/mmc-CuN2) [32]. Despite these progresses, the pressure-composition phase diagram of Cu–N compounds at different temperatures is largely unknown. So, a systematic structure search for stable Cu–N compounds with varying Cu:N ratios under high pressure is needed.
In this work, through a combination of first-principles calculation and structure search, we explored the pressure-composition phase diagram of Cu–N compounds. Four Cu–N structures were found stable under high pressures: Pnnm-CuN2, two CuN3 compounds with the P-1 space group (named as I-CuN3 and II-CuN3) and P21/m-CuN5 containing cyclo-N5 - ring. Besides P21/m-CuN5 (previously proposed by Li et al in 2019 [35]), the other three kinds of Cu–N compounds are all newly discovered Cu–N structures. Finite temperature effect (vibrational energy) was found important in stabilizing the experimentally observed P63/mmc-CuN2 at ∼55 GPa [32]. Among all these newly predicted Cu–N structures, only P21/m-CuN5 shows semiconductor characteristic, with the others being metals. Phonon dispersion calculations confirm the dynamical stability of these novel Cu–N compounds. These newly discovered Cu–N compounds show energy densities of 1.57–2.74 kJ g−1, higher than those of previously known Pm m-CuN3 and P63/mmc-CuN2. The study provides a guide for researchers who are exploring stable N-containing compounds with coinage metals.
2. Computational methods
The structural prediction was performed with the PSO algorithm as implemented in the CALYPSO code [36]. Seven kinds of Cu:N ratios (3:1, 2:1, 3:2, 3:4, 1:2, 1:3, 1:5) were considered at five different pressures (0, 55, 70, 100 and 150 GPa). Simulation cells containing one to four formula units (f.u.) were used during the structure search. For each round of search, we considered up to 1000 structures with 60 percent of them produced by PSO and 40 percent generated randomly under symmetry constraints.
The total-energy calculation and structural optimization were performed based on the density functional theory method [37] as implemented in the Vienna ab initio simulation package (VASP) [38]. The projector-augmented wave method within the Perdew–Burke–Ernzerhof (PBE) functional was used. The Γ-centered Monkhorst–Pack method with a k-point density higher than 2π × 0.03 Å−1 and a plan-wave energy cutoff of 600 eV were employed. The convergence criteria of Hellmann–Feynman force on the ion were set to 0.01 eV Å−1. Phonon dispersions were calculated by using the finite displacement method in PHONOPY package [39]. Both PBE and hybrid functional (HSE06) [40, 41] were used to study the electronic band structures. Synchrotron x-ray-diffraction simulations of Cu–N compounds were carried out within the MERCURY 4.2 software package [42]. The crystal orbital Hamilton population (COHP) curves implemented in the LOBSTER program [43], which are presented in the supplemental information (SI) (https://stacks.iop.org/JPCM/34/025401/mmedia).
3. Results and discussion
3.1. Phase stability of Cu–N binary compounds
To investigate the stabilities of various Cux Ny structures, the formation enthalpies of them (per atom) were calculated as , where , HCu and HN are enthalpies of Cux Ny (per f.u.), Cu (per atom) and N (per atom) reference systems at corresponding pressure. Based on calculated enthalpies, the convex hull diagrams for Cu–N compounds were plotted in figures 1(a)–(e). Any structure whose formation enthalpy lies on the convex hull (solid lines) could be considered stable and synthesizable.
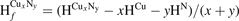
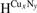

Figure 1. The formation enthalpy-composition curves for Cu–N compounds at (a)–(e) 0 GPa, 55 GPa, 70 GPa, 100 GPa and 150 GPa, with temperature of 0 K. The convex hulls are shown by solid lines. (f) Pressure-composition phase diagram of the Cu–N compounds with Cu:N ratios of 1/5, 1/3 and 1/2, at 0 K. The supper-cell structure of (g) Pnnm-CuN2 (2*2*3) at 55 GPa, (h) P21/m-CuN5 (2*1*1) at 55 GPa, (i) I-CuN3 (3*3*2) at 70 GPa and (j) II-CuN3 (3*3*2) at 150 GPa. The Cu and N atoms are shown by blue (large) and gray (small) spheres, respectively.
Download figure:
Standard image High-resolution imageAs shown in figure 1, four kinds of Cu–N compounds can be deemed stable at high-pressures. They are Pnnm-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5. Among them, the P21/m-CuN5 was previously proposed to be metastable at ambient condition [35]. While the other three are all newly discovered stable Cu–N structures. The crystal structures (supercell models) of them are presented in figures 1(g)–(j). The schematic of the pressure-composition phase diagram for the Cu–N compounds at 0 K are also shown in figure 1(f), with the pressure regions where these four predicted Cux Ny structures are stable marked by different colors. Pnnm-CuN2 is thermodynamically stable within the pressure range between 20 to 150 GPa. For CuN3 compounds, two kinds of structures, I-CuN3 and II-CuN3, are stable in the pressure range from 48–150 GPa. The phase transition between these two phases occurs at ∼84 GPa, as shown in figure 2(b). We also found that the P21/m-CuN5, which was previously reported to be metastable at ambient condition, is stable between 45–75 GPa. But it should be acknowledged that this pressure-composition phase diagram for Cu–N compounds is based on static DFT calculations at temperature of 0 K. The finite temperature effects will be discussed in the following.

Figure 2. (a) The enthalpy difference curves of P63/mmc-CuN2 structure (relative to Pnnm-CuN2) as a function of pressure. (b) The enthalpy difference curves of I-CuN3 (relative to II-CuN3) along with pressure. In (a) and (b), the black, red and blue solid lines represent the enthalpy results based on calculated total-energy (T = 0 K), with ZPE correction and then including vibrational energies (TS term with T = 1500 K), respectively.
Download figure:
Standard image High-resolution imageAs mentioned above, the P63/mmc-CuN2 is not the most favorable CuN2 structure (0 K DFT result) within the pressure range studied, as shown in figure 1. But in experiment [32], P63/mmc-CuN2 has been synthesized at the pressure of 52 GPa and temperature above 1500 K. To understand this discrepancy, we further calculated the phonon spectra and then the lattice vibrational energies at 1500 K. As shown in figure 2(a), the enthalpy of newly discovered Pnnm-CuN2 was set to 0, which acts as the reference enthalpy. The relative enthalpies of P63/mmc-CuN2 are presented. Black, red and blue solid lines represent relative enthalpies based on calculated total-energy (T = 0 K), total-energy plus zero-point energy (ZPE) correction and plus vibrational energies (TS term with T = 1500 K), respectively. ZPE correction does not change the relative enthalpy of P63/mmc-CuN2 much. But the finite temperature effects at 1500 K shift the relative enthalpy curve downwards by 20–35 meV atom−1, making the P63/mmc-CuN2 phase thermodynamically more stable than Pnnm-CuN2 at pressure lower than 60 GPa. This result rationalizes the experimentally observed P63/mmc-CuN2 structure and provides useful guidance for future experimental exploration of Pnnm-CuN2 phase.
For the phase transition between I-CuN3 and II-CuN3, the ZPE correction and finite temperature effects were also studied and then shown in figure 2(b). Both ZPE and vibrational energy corrections increase the phase transition pressure between I-CuN3 and II-CuN3. The vibrational energy (Ev) introduced by the phonon of lattice at finite temperature is expressed as
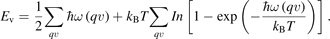
According to the formula of the added free energy of phonon, both the ZPE (T = 0) and the vibrational energy (T > 0) of lattice are positively correlated with the averaged vibrational frequency integrated in the whole q-space and all branches of phonon. By the careful examination of the two CuN3 structures, we found that an adding of nitrogen–nitrogen bond is associated with the structural transformation from I-CuN3 to II-CuN3. In a unit cell, I-CuN3 has five nitrogen–nitrogen bonds while II-CuN3 contains six, both of which correspond to the high frequency peaks of optical modes in the density states of phonon, as shown in supplementary figure S1. Although I-CuN3 has a double bond (N=N, bond length of 1.19 Å) with the highest frequency of 53 THz, the added N–N bond in II-CuN3 helps a larger averaged frequency than that of I-CuN3. Thus, the ZPE and vibrational energy at finite temperature in II-CuN3 are larger than those in I- CuN3, and additional pressure is needed to overcome these energy differences between the two phases. The phase transition from I-CuN3 to II-CuN3 occurs at ∼84 GPa. Notably, this structural transformation is associated with the increasing the N–N bond and the appearance of metal-metal bond. In I-CuN3, metal ions are effectively separated by nitrogen ions with the edge-shared polyhedrons to avoid the formation of Cu–Cu bond. However, strong compression weakens the Cu–N bonds and strengthens the N–N and Cu–Cu bonds for II-CuN3, which is consistent with the claim in a previous review that the pressure will favor the formation of homonuclear bonds instead of heteronuclear bonds [44]. The calculated phonon dispersions of these four Cu–N compounds under different pressures are shown in figure S2. There are no imaginary phonon frequencies in the whole Brillouin zone, indicating the dynamic stabilities of them.
3.2. Crystal structure and stability
Figure 1 gives the atomic structures of the above four Cu–N compounds (supercell models). For CuN2, both of newly discovered Pnnm-CuN2 in figure 1(g) and previously synthesized P63/mmc-CuN2 [32] contain the octahedrally coordinated Cu atoms. Each Cu atom is bonded to six N atoms. N atoms exist in the form of N2 dimer. The N–N distance of N2 dimer is 1.22 Å in Pnnm-CuN2 and 1.2 Å in P63/mmc-CuN2, being typical N=N bond. With the increase of nitrogen content, the N2 dimer in the CuN2 transform to a two-dimensional (2D) nitrogen sheet in I-CuN3/II-CuN3 and then to the cyclo-N5 − ring in P21/m-CuN5. For P21/m-CuN5 and II-CuN3, the distances between N atoms in the cyclo-N5 − ring and N sheet are about 1.3 Å and 1.28 Å, which lies between the N–N single bond (1.45 Å) and N=N double bond lengths (1.25 Å) [26, 45]. The distances between nitrogen atoms in I-CuN3 are 1.19 Å, 1.26 Å and 1.29 Å, and there is an N=N (1.19 Å). Indeed, the criterion depending on bond length to decide N–N or N=N bond is just only an empirical method, though this method has been adopted by many previous works [1, 6, 23, 26]. The bond strength is strongly related to the phonon vibration frequency. Experiments take Raman or infrared spectroscopy to determine single and double bonds. In theoretical calculation, it is reasonable to use phonon vibration frequency to judge single or double bonds. We illustrated the phonon density of states for these four high pressure phases at the same pressure of 55 GPa in figure S3. There are two distinct high frequency peaks with frequencies of about 48 (Pnnm-CuN2) and 50 THz (I-CuN3), which obviously correspond to two double bonds in the two structures. If we treat the N–N bonds of cyclo-N5 ring in P21/m-CuN5 as single bonds with the maximal frequency of 40 THz, and those peaks between the two frequencies may be considered as the mixed bond of N=N and N–N. The bond lengths of N–N are labeled at supplementary figure S4. The crystallographic parameters and unit-cell structures of these Cux Ny compounds are also provided in supplementary table S1 and figure S4.
The electronic band structures of these four Cu–N compounds at different pressures are presented in figure 3. The Pnnm-CuN2, I-CuN3 and II-CuN3 are metals at pressure of 55, 70 and 150 GPa, respectively. The P21/m-CuN5 at 55 GPa is a semiconductor with an indirect band gap energy of 0.073 eV, based on PBE functional calculation. Hybrid HSE06 functional calculation leads to a much larger band-gap value (1.28 eV), but with similar dispersion of energy bands. The band-gap energy decreases with the increased pressure, as shown in figure S5. The special local structure with the formation of the closed aromatic cyclo-N5 ring and the ionic interaction between the cyclo-N5 − and Cu+ in P21/m-CuN5 are the origin of the band gap. The N–N bond lengths in P21/m-CuN5 vary slightly between 1.31 and 1.34 Å, which agree very well with previously reported semiconductive aromatic cyclo-N5 − compounds, such as CuN5 [35], LiN5 [46], NaN5 [7] and MgM10 [1]. We found that the appearance of P21/m-CuN5 phase in 45 to 75 GPa is correlated with the formation of electronic band gap, which lowers the energy of electronic states approached to Fermi level. The increased pressure will gradually close the band gap (figure S5), and lifted the energy of electronic states, which leads to the instability of P21/m-CuN5 at high pressures.

Figure 3. Electronic band structures of (a) Pnnm-CuN2 at 55 GPa, (b) P21/mCuN5 at 55 GPa, (c) I-CuN3 at 70 GPa and (d) II-CuN3 at 150 GPa using PBE (black line) and hybrid functional HSE06 (red line). The point, where the energy value is zero, is the Fermi level position.
Download figure:
Standard image High-resolution image3.3. Chemical bonding and energy density
In order to further analyze the bonding characteristics, we have performed the Bader charge and electron localization function (ELF) calculations and showed the results in figure 4. The covalent bond and lone pair can be represented by a high ELF value (≫0.5); while the ELF value ≪0.5 and close to 0.5 are often indicators of ionic bonds and metal bonds. The calculated ELF suggested the valance bonding between N atoms, the ionic bonding between N and Cu atoms and lone pairs at several N atoms. The Bader charge results also show that copper in these four structures exist as Cu+ ion. Our ELF and Bader charge analysis also show that the bonding in the N5 − anion is covalent and stabilized by the electron transferred from nearby Cu atom, as show in figure S6. The formation of a large sharp peak of Cu–N antibonding approaching to Fermi level in figure S7 also suggested the ionic interaction between Cu+ and N5 −. The effect of pressure on charge transfer has also been investigated. As shown in figure S8, the pressure has little effect on the Bader charge results within the pressure range studied by us. The calculated COHP for these four Cu–N compounds are presented in figure S7. The integrated COHP values of Cu–N bonds and N–N bonds are −2 to −4 eV pair−1 and −14 to −16 eV pair−1, indicating that the N–N bonds are stronger than the Cu–N bonds, which is consistent with our ELF analysis.

Figure 4. The 2D ELF of Cu–N compounds for (a) Pnnm-CuN2 at 55 GPa in (0 0 1) plane, (b) P21/m-CuN5 at 55 GPa in (0 1 0) plane, (c) I-CuN3 at 70 GPa in (0 1 0) plane, (d) II-CuN3 at 150 GPa in (0 1 0) plane; the atomic Bader charge of (e) Pnnm-CuN2 at 55 GPa, (f) P21/m-CuN5 at 55 GPa, (g) I-CuN2 at 70 GPa and (h) II-CuN2 at 150 GPa. The red number represents the amount of charge transferred (eV), which means that each copper atoms transfers about an electron to its neighbor N atoms.
Download figure:
Standard image High-resolution imagePrevious experiment has provided the Rietveld refinement of synchrotron XRD pattern of P63/mmc-CuN2 [32]. To provide additional information for future experiments, the simulated XRD patterns of Pnnm-CuN2, P63/mmc-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5 at corresponding pressures have been obtained with λ = 0.3344 Å which is known as the wavelength of the incident x-ray beam, in order to keep consistent with that of the XRD pattern of experimental P63/mmc-CuN2 [32]. Among them, the data of Pnnm-CuN2 and P63/mmc-CuN2 are presented in figure 5, shown along with the experimental result for P63/mmc-CuN2. The rest of them are plotted in figures 5(b)–(e). The Miller indices (h k l) for each XRD peaks have been marked. By comparing our simulated XRD patterns with the experimental one, the calculated XRD pattern of P63/mmc-CuN2 agrees much better with the experimental obtained XRD data, supporting that the previously synthesized CuN2 is indeed P63/mmc-CuN2 phase. In addition, we hope the simulated XRD patterns of Pnnm-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5 structures can provide insights for further experimental efforts to find them.

Figure 5. Simulated synchrotron XRD patterns (λ = 0.3344 Å) of (a) the Pnnm-CuN2 phase at 52 GPa (blue line), the P63/mmc-CuN2 phase at 52 GPa (red line). Powder XRD pattern of P63/mmc-CuN2 phase at 52 GPa (black line). Simulated synchrotron XRD patterns (λ = 0.3344 Å) of (b) Pnnm-CuN2 at 55 GPa, (c) P21/m CuN5 at 55 GPa, (d) I-CuN3 at 70 GPa and (e) II-CuN3 at 150 GPa. The Miller indices (h k l) for the diffraction peaks of the Pnnm-CuN2 phase and P63/mmc-CuN2 are marked.
Download figure:
Standard image High-resolution imageThe N-rich compounds were considered as promising high-energy-density materials. Especially, the cyclo-N5 − has been extensively studied because of the high chemical energy stored in it. At last, we investigated the explosive performances of these novel stable Cu–N compounds by calculating the energy-densities of them. In the dissociation of , the calculated energy densities for Pnnm-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5 are 1.57, 2.39, 2.62 and 2.74 kJ g−1, respectively. These values are higher than those of the two known structures, P63/mmc-CuN2 (1.57 kJ g−1) and Pm m-CuN3 (1.51 kJ g−1), similar to the CO/N2, TATB and RDX (1–3 kJ g−1) [47], but lower than the TNT (4.3 kJ g−1) and HMX (5.7 kJ g−1) [45].
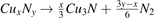

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusions
To summarize, we reported here a theoretical study on new stable Cu–N compounds at high pressure (0–150 GPa), with the combination of DFT calculation and structure search method. By plotting the formation enthalpy-composition curves at 0 K, we proposed that four Cu–N compounds (Pnnm-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5) are thermodynamically stable at high pressure studied. Phonon dispersion calculations also suggest that they are dynamically stable. Among them, only P21/m-CuN5 is a semiconductor, while the others are all metals. By further including the finite temperature effects, the recently synthesized P63/mmc-CuN2 become more stable than Pnnm-CuN2 at pressure <60 GPa, which rationalized the experimental observation of P63/mmc-CuN2 at ∼55 GPa. The calculated pressure-composition phase diagram and XRD patterns for these four Cu–N compounds could provide guidance for future experimental efforts to synthesize these new phases. The energy densities of Pnnm-CuN2, I-CuN3, II-CuN3 and P21/m-CuN5 are 1.57, 2.39, 2.62, and 2.74 kJ g−1,respectively, indicating that they are potential high-energy-density materials.
Acknowledgments
This work was supported by the National Science Foundation of China (Grant Nos. 11974105, 11704111 and 11634001). We would like to acknowledge the computational resources provided by the supercomputer TianHe in Tianjin, China.
Date availability statement
All date that support the findings of this study are included within the article (and any supplementary files)