

Abstract
Theoretical studies using the state-of-the-art density functional theory and dynamicalmean-field theory (DFT + DMFT) method show that weak electronic correlation effects are crucial for reproducing the experimentally observed pressure-induced phase transitions of calcium from β-tin to Cmmm and then to the simple cubic structure. The formation of an electride state in calcium leads to the emergence of partially filled and localized electronic states under compression. The electride state was described using a basis containing molecular orbitals centered on the interstitial site and Ca-d states. We investigate the influence of Coulomb correlations on the structural properties of elemental Ca, noting that approaches based on the Hartree–Fock method (DFT + U or hybrid functional schemes) are poorly suited for describing correlated metals. We find that only the DFT + DMFT method reproduces the correct sequence of high-pressure phase transitions of Ca at low temperatures.
Export citation and abstract BibTeX RIS
1. Introduction
Metallic Ca attracts considerable interest because of its unexpectedly complex phase diagram, the highest among elements critical temperature of 25 K for superconductivity under high pressure [1], and a loss of metallic properties under high pressure [6].
These unique properties are mostly a result of peculiarities of the electronic structure, which manifest themselves as pressure increases. The phase diagram of Ca was studied both experimentally [2–4] and theoretically [5–10]. The x-ray diffraction experiments showed that at low temperatures Ca undergoes a series of phase transitions: from bcc (stable below 32 GPa) to β-tin (I41/amd) tetragonal structure in the pressure range of 32 to 40 GPa, then to Cmmm structure, which can be regarded as a slightly distorted simple cubic (SC) structure, and, finally, to the SC above 47 GPa [5]. It was shown [2] that the simple cubic phase Ca exhibits an anomalous resistance behavior in a wide pressure range of up to 109 GPa, associated with unusual electronic properties arising from the s–d electron transfer.
Most of the previous attempts to theoretical modeling of the electronic structure of calcium were based on density functional theory (DFT). In particular, the existence of the β-tin (I41/amd) structure was predicted theoretically [5]. Later, this phase was confirmed to be stable below 7 K in an experiment [11] that showed a sequence of pressure-induced phase transitions: fcc → bcc → β-tin → Cmmm → SC. Li et al [11] argued that SC structure at room temperature can be metastable and stability of β-tin and Cmmm phases can be explained as a compromise between density, temperature and enthalpy factors. However, they mentioned that the s–d electron transfer is responsible for complex behavior of Ca. Stabilization of the SC phase above 40 GPa and temperatures above 300 K due to anharmonic effects was proposed earlier [4, 5, 12]. Nevertheless, the nature of stability of the SC structure at low temperatures remains unclear. Since the impact of phonon entropy vanishes with decreasing temperature due to freezing of lattice degrees of freedom and cannot explain the fact that the SC structure is also stable at very low temperatures down to 6 K in a wide pressure range above 55 GPa, another mechanism is needed to explain the stability of all these phases at low temperatures. It is possible that DFT does not model accurately the seemingly simple calcium metal, possibly due to the underestimation of Coulomb correlation effects. It was suggested earlier that s–d electron transfer occurring under pressure [3, 12] and Coulomb correlations play an important role in bonding [13] and intriguing electronic properties of Ca [2]. The transition to the SC phase was first modeled theoretically using a hybrid functional [14]. However, the transition to the intermediate Cmmm phase was not reproduced. Electron–electron interactions are implicitly present in the exchange–correlation part of the hybrid functional. The hybrid functional, however, has a number of hidden parameters and does not fully include the effect of Coulomb repulsion on the electronic structure and physical properties of materials. At the moment, there is no systematic view on the relationship between structural changes under compression and Coulomb correlations in high-pressure phases of Ca. Taking into account the Coulomb interactions between electrons in the mixed atomic states with s- and d-symmetry, we will assess the role of correlation effects in determining both electronic and structural properties of elemental calcium. For this purpose, we employ the method combining DFT and dynamical mean-field theory, DFT + DMFT [15, 16], which can be applied to both strongly and weakly correlated materials since it works perfectly in the whole range of U/W ratio, where U is Coulomb repulsion and W stands for bandwidth. It can describe both uncorrelated metallic and strongly correlated insulating states within the same approach in contrast to simple band-structure methods where different techniques, DFT and DFT + U, are used to obtain metallic or insulating solutions, separately. Also, DFT + DMFT allows to treat paramagnetic state which is expected due to a large overlap between orbitals. It was shown for many strongly correlated compounds that even after the magnetic collapse (i.e. high-spin–low-spin transition) there are still non-zero local fluctuating magnetic moments [17–19]. Here, for the first time we reproduced the phase diagram of elemental calcium under high pressure. This was not possible with either DFT, due to the lack of proper account for the correlation effects, or DFT + U and hybrid functional approaches based on Hartree–Fock method, which are well suited only for insulators. We also show that the weak Coulomb correlations are responsible for stabilizing the simple cubic phase of Ca at low temperatures.
2. Methods
To get the theoretical equation of state, we started with a full relaxation of the crystal structures for a series of cell volumes using the VASP package [20]. When performing DFT calculations, we used the PBE exchange–correlation functional and the projector augmented wave (PAW) [21] potentials from the standard VASP library. Then, for the relaxed structures, we constructed a correlated states Hamiltonian in Wannier function (WF) basis. For every structure, the GGA calculation was performed using the pseudopotential method as implemented in Quantum ESPRESSO [22]. The precision of the total energy calculation in DFT was set to 10−6 eV. Then we used the Wannier90 package [23] to extract the noninteracting GGA Hamiltonian HGGA in real space, which included both Ca 3d and Ca 3s states, the latter was centered in the middle of the unit cell. Then the Hamiltonian was transformed to reciprocal space. The Coulomb correlations were taken into account for the constructed Hamiltonian within the DFT + DMFT approach. Parameter of Coulomb interaction U was calculated within so-called constrained DFT procedure [24] for the same basis which was chosen for non-interacting Hamiltonian construction. In this method, the noninteracting band structure  obtained using DFT takes into account all the peculiarities of
obtained using DFT takes into account all the peculiarities of  for a given material, while DMFT was used to calculate many-body effects such as Coulomb correlations [25, 26]. Recently, this method was successfully used to investigate phase transitions under pressure [27–33]. The DFT + DMFT calculation was carried out for the inverse temperature value β = 1/kBT = 10 eV−1, where kB is the Boltzmann constant and T is the absolute temperature. We used the simplified fully localized limit form for the double counting correction in a self-consistent manner. The continuous time quantum Monte-Carlo hybridization-expansion solver from the AMULET package [34] was employed to solve the effective DMFT quantum impurity problem [35]. The error of the DMFT energy did not exceed 10−4 eV. The details of the total energy calculations in the DFT + DMFT method can be found elsewhere [36].
for a given material, while DMFT was used to calculate many-body effects such as Coulomb correlations [25, 26]. Recently, this method was successfully used to investigate phase transitions under pressure [27–33]. The DFT + DMFT calculation was carried out for the inverse temperature value β = 1/kBT = 10 eV−1, where kB is the Boltzmann constant and T is the absolute temperature. We used the simplified fully localized limit form for the double counting correction in a self-consistent manner. The continuous time quantum Monte-Carlo hybridization-expansion solver from the AMULET package [34] was employed to solve the effective DMFT quantum impurity problem [35]. The error of the DMFT energy did not exceed 10−4 eV. The details of the total energy calculations in the DFT + DMFT method can be found elsewhere [36].
3. Results and discussion
The idea of the electride formation in Ca under pressure [6] was based on the analysis of an unusual distribution of the valence electron localization function (ELF) [37] in simple cubic Ca. There, the ELF maxima are located not only at Ca sites but also in the cubic voids between them, forming a CsCl-type structure. Based on this, we also investigated the lower-symmetry β-tin and slightly distorted Cmmm structures to track the evolution of the electride state during phase transitions.
The valence charge density distribution calculated using the DFT method for SC (a) and β-tin (b) Ca phases is shown in figure 1. We do not show the charge density distribution for the Cmmm structure since it has minor visual differences from SC. The SC structure has a pronounced charge density maximum in the middle of the unit cell. Because of the significant lattice compression in the SC phase, the s-orbitals of the eight nearest Ca atoms overlap in the void of the unit cell and form a bonding orbital. Bader analysis [38] revealed that this area contains 0.67 electrons. These facts are in favor of viewing the SC phase of calcium as a zero-dimensional electride, with a compact region of charge density localization of anionic electrons of the Bohr radius size, which cannot be interpreted as a gas of free electrons. On the other hand, the β-tin structure has narrow 'serpent-like' 1D channels with high charge density extending in c axis direction. This change in the charge distribution between the SC and β-tin phases reflects the distortions of the crystal structure. Note, that shape of electride charge accumulation does not change with pressure for each phase. The charge density and WFs for all phases were constructed for different values of external pressure in the range from 25 to 55 GPa. For all considered pressures β-tin structure has narrow 1D channels with high charge density extending along the c-axis direction and SC and Cmmm phases have almost spherical electride region. These states correspond to rather flat bands which are near the Fermi level and cross it. Since the band width is inversely proportion to the particle lifetime, this indicates the importance of taking into account the Coulomb correlations when describing the properties of electride system.

Figure 1. Valence charge density for SC (a) and β-tin (b) structures as obtained in the GGA calculation at P = 42.6 GPa. Plotted using VESTA [43].
Download figure:
Standard image High-resolution imageStarting from the high-pressure SC phase and following the structure transformation as pressure decreases, the first transition into the Cmmm phase is characterized by an increase in γ, an angle between the crystallographic axes a and b, to 92°, and nonequivalent a and b cell parameters. Bader analysis shows that the number of electrons in the middle of the unit cell goes down slightly to 0.61. The distance between the Ca site and electride site (by the 'electride site' we mean the position of a critical point [38] that corresponds to a maximum of the charge density within the voids) in different structures at the same pressure characterizes the overlap of a bonding s-orbital in the middle of the unit cell and atom-centered Ca d-orbitals. These distances are 2.3031 Å and 2.3679 Å for Cmmm and 2.3086 Å for SC phase, meaning that the hybridization should weaken during the Cmmm → SC transition. With further pressure decrease, the distortions of the crystal structure grow: Ca atoms shift from the cubic structure position to an alternating pattern, forming corrugated planes with different Ca–Ca distances. These distances in the β-tin phase at P = 38 GPa are 2.8962 Å, 2.0523 Å, and 3.5338 Å. Both the increase in distance and lowering of symmetry make an overlap of Ca-s orbitals inside the voids weaker, almost suppressing the electride formation (figure 1(b)). At low-pressure bcc phase, the empty space in the middle of the cell is occupied with Ca atom and no electride bonding orbital can be formed in the cell voids.
To investigate the band structure of Ca during phase transitions, we used the Wannier function (WF) projection procedure. To reflect the electride idea, we started with six WFs: five with d-symmetry, centered on Ca sites, and one with s-symmetry, centered in the middle of the unit cell, in the cubic void. Figure 2 shows the spatial distribution of the constructed WFs for electride state with s-symmetry (left top panel) and two WFs with dxz- and dxy-symmetry (right top panel). Lower panel shows the contribution of s-symmetry (blue) and d-symmetry (yellow) WF to the band structure of SC Ca.
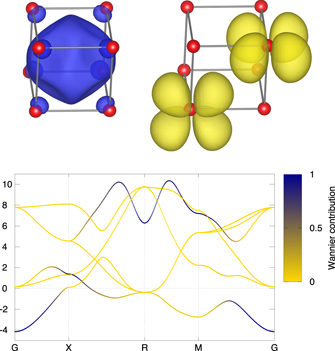
Figure 2. Wannier function with s-symmetry centered in the cubic void within the SC cell (top left) and WFs with dxz, dxy-symmetry (top right). Ca atoms are shown as red spheres. Bottom panel: band structure of SC Ca and contribution of the WF with s-symmetry (showed by the deepness of blue color) centered on the interstice to the Bloch states. d-states are shown with yellow lines.
Download figure:
Standard image High-resolution imageThe lowest almost occupied band originates from two types of WF s- and d-symmetry (t2g) strongly hybridized with each other (figure 2 low panel) and can be described as a molecular orbital formed by Ca t2g and electride states. The expected correlation effects on this partially filled flat molecular orbital band can be essential. To check this guess, we carried out the self-consistent DFT + DMFT calculations using model Hamiltonian in WF basis. The DMFT calculations were performed for all structures, namely SC, β-tin and Cmmm for a series of unit cell volumes, corresponding to pressure range from 25 to 55 GPa. The Coulomb parameter U was calculated in the constrained DFT procedure [24] for the SC phase using the supercell containing 27 Ca atoms to be sure that a charge perturbation on the one (central) atom does not affect the nearest atoms. This approach contains no free parameters since it uses the same WFs basis as the subsequent DFT + DMFT. It was shown [39] that using the same basis for both calculation of Coulomb parameters and DFT + DMFT is essential for obtaining stable result regardless of particular basis. The obtained value of U = 0.47 eV was used then for all phases under investigation. This value is smaller than the typical one for 3d metals varying from 2–3 eV for Ti or V to 8–10 eV for Ni or Cu calculated on atomic local orbitals, which can be explained by the effective screening by WF which are more spread to nearest sites, and by appropriate choice of the basis function [40]. Note also, that a similarly small value of the U parameter was obtained for iron in LaOFeAs using the same method [39]. Since the occupied orbitals are formed by WFs with s- and d-symmetry strongly hybridized with each other, all the six WFs in small Hamiltonian are considered correlated, and the DMFT impurity problem has the dimension of 6 × 6 (12 spin-orbitals).
In addition to the DFT and DFT + DMFT calculations, we check the results using the DFT + U method, which also allows one to take into account the Coulomb correlations. To evaluate the pressure, we fit to our total energies the third-order Birch–Murnaghan equation of state [41], separately for each phase. The enthalpy (H = E + PV) of each phase was then calculated to investigate the phase stability and transition pressures. The enthalpies of β-tin and Cmmm phases compared to that of the SC structure (shown by zero line), calculated using the DFT, DMFT, and DFT + U methods, are presented in figure 3.

Figure 3. Enthalpy of β-tin (red) and Cmmm (black) phases of Ca under pressure obtained using the DFT (dashed lines), DFT + U (dash-dotted lines) and DFT + DMFT (solid lines) methods. Zero line shows the enthalpy of the SC phase.
Download figure:
Standard image High-resolution imageIn agreement with previous calculations, the enthalpy of the SC phase calculated using DFT is always higher than that of the β-tin phase, while the enthalpy of the Cmmm structure lies between them. However, when the correlation effects are taken into account, we suddenly find a sequence of phase transitions that agrees with the experiment. Even a small U value makes the Cmmm phase stable at pressures higher than 45 GPa. As pressure increases, the transition to the SC phase occurs at 52 GPa. Obtained theoretical transition pressures are in reasonable agreement with the experimental critical pressures: approximately ≈40 GPa for the β-tin to Cmmm phase transition and 47 GPa for Cmmm to SC phase transition [11]. This is in contrast to the results obtained by the hybrid functional method [14] according to which the enthalpy of Cmmm phase is always higher than that of the SC and β-tin phases. The DFT + U method also did not reproduce the correct sequence of phase transitions (figure 3).
The total energy corrections due to the correlation effects are almost independent of the applied pressure for all the structures under consideration and their average values for β-tin, SC and Cmmm are equal to 0.37, 0.31 and 0.32 eV/atom, respectively. Unexpectedly, the correction is larger in the β-tin phase, while the electride is more pronounced in the SC phase. Therefore, the states that are considered correlated should be more localized in the cubic phase. The value of the DFT + DMFT total energy correction calculated for the Cmmm phase lies between that of the β-tin and SC phases. We suggest the following explanation: the most important change resulting from the phase transitions from β-tin to Cmmm and then to SC structures is an increase in local symmetry. E.g., the partially filled t2g orbitals of Ca, which are completely nondegenerate in the β-tin phase and nearly degenerate in Cmmm, become triply degenerate in the SC phase. The critical value of the Coulomb interaction parameter Uc needed for the metal–insulator transition in the half-filled degenerate Hubbard model, is  ([42]), where N is degeneracy and
([42]), where N is degeneracy and  is the critical U value for a nondegenerate case. Therefore, the same U value, used in the DMFT calculations, will have a lower impact for degenerate electronic states than for a system with a low symmetry. These phases of Ca are far from the metal–insulator transition, but the strength of the correlation effects should decrease if we go from the β-tin to the SC phase.
is the critical U value for a nondegenerate case. Therefore, the same U value, used in the DMFT calculations, will have a lower impact for degenerate electronic states than for a system with a low symmetry. These phases of Ca are far from the metal–insulator transition, but the strength of the correlation effects should decrease if we go from the β-tin to the SC phase.
However, the correlation effects are strong enough to result in the correct phase sequence. Even though the β-tin, Cmmm and SC phases are weakly correlated, the correlation effects are more pronounced in the β-tin phase due to its lower symmetry.
To evaluate the correlation strength, we calculated the band structure using the DFT + DMFT method (figure 4). No significant difference was observed in comparison with the DFT-calculated band structure, which is expected for such a small U value.

{kind=link}
{kind=link}
{kind=link}
Figure 4. ARPES for the SC structure obtained in the DFT + DMFT calculation for V = 18.23 Å3.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
We showed that weak Coulomb correlations influence the stabilization of the simple cubic (SC) phase of elemental calcium and the experimentally observed sequence of structural transitions from the β-tin to Cmmm and, finally, to the SC phase under increasing pressure. Our calculations show that at low temperatures these phase transitions exist due to a delicate balance between different factors. We found that the difference in total energies is very tiny, hence small distortion of crystal structure, defects, nonstoichiometry, or temperature effects can make one of the structures more stable which can explain the variation of experimental data. Note, that at low temperature when all crystal degrees of freedom are frozen the contribution to the total energy from Coulomb interaction between electrons on electride states, though small, is sufficient to change the picture of phase transitions and bring it to agreement with experimental observations. We found that the DFT + DMFT approach, which works well for any U/W and can be applied to correlated metals, is suitable for reliable electronic structure calculations for high-pressure phases of calcium. We also showed that electron correlations in calcium occur due to the electron transfer from the 4s- to 3d-states under pressure, leading to the existence of partially filled electronic states. The electron transfer also results in the localization of the corresponding electronic density in the cell interstitial area, making this phase of calcium an electride. Our findings for Ca might be relevant for other electride materials, while the idea of electron–electron correlations in high-pressure phases with hollow structures needs a further study.
Acknowledgments
The phase diagram calculated using the DFT and DFT + U methods was obtained within the state assignment of Ministry of Science and Higher Education of the Russian Federation (theme 'Electron' No. AAAA-A18-118020190098-5). The pressure dependence of enthalpy and ARPES evaluated using the DFT + DMFT method were supported by the Russian Science Foundation (Project 19-72-30043).