

Abstract
In order to understand the role of W-site substitution on properties of cubic tungsten carbide ( -WC), we have investigated the structural, mechanical, and electronic properties of WXC2 (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U) using first principles calculations based on density functional theory, within generalized gradient approximation. The structural optimization has carried out for all these compounds using force as well as stress minimization. The optimized structural parameters for experimentally known compounds are in good agreement with the available x-ray diffraction measurements and structural parameters for nineteen WXC2 compounds are newly predicted. The W-site substitution of the above-listed elements into
-WC), we have investigated the structural, mechanical, and electronic properties of WXC2 (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U) using first principles calculations based on density functional theory, within generalized gradient approximation. The structural optimization has carried out for all these compounds using force as well as stress minimization. The optimized structural parameters for experimentally known compounds are in good agreement with the available x-ray diffraction measurements and structural parameters for nineteen WXC2 compounds are newly predicted. The W-site substitution of the above-listed elements into  -WC reduces the symmetry of the primitive lattice to tetragonal structure. The heat of formation (
-WC reduces the symmetry of the primitive lattice to tetragonal structure. The heat of formation ( ) and the mechanical stability studies are carried out to investigate the stability of these systems. The single-crystal elastic constants cij, elastic moduli of the polycrystalline aggregates, anisotropy in elastic constants and related properties of the WXC2 materials have calculated and discussed in detail. The hardness of the above materials is predicted using two different criteria, based on the softest elastic mode as well as the Pugh's modulus ratio. There is a correlation in the hardness predicted from these two approaches except in the case of
) and the mechanical stability studies are carried out to investigate the stability of these systems. The single-crystal elastic constants cij, elastic moduli of the polycrystalline aggregates, anisotropy in elastic constants and related properties of the WXC2 materials have calculated and discussed in detail. The hardness of the above materials is predicted using two different criteria, based on the softest elastic mode as well as the Pugh's modulus ratio. There is a correlation in the hardness predicted from these two approaches except in the case of  -WC. The chemical bonding interaction between the constituents is analysed using the density of states, crystal orbital Hamiltonian population, and charge density for selected systems. All these compounds are predicted to be metal and our calculations suggest that W-site substitutions do not improve the hardness of
-WC. The chemical bonding interaction between the constituents is analysed using the density of states, crystal orbital Hamiltonian population, and charge density for selected systems. All these compounds are predicted to be metal and our calculations suggest that W-site substitutions do not improve the hardness of  -WC. However, from the heat of formation studies, we have identified five new stable compounds such as CrWC2, NbWC2, ScWC2, YWC2, and UWC2 with reasonably good hardness and those need experimental verifications.
-WC. However, from the heat of formation studies, we have identified five new stable compounds such as CrWC2, NbWC2, ScWC2, YWC2, and UWC2 with reasonably good hardness and those need experimental verifications.
Export citation and abstract BibTeX RIS
1. Introduction
In recent decades, tungsten carbide (WC) has attracted much attention from researchers and technologists for its application in the manufacturing of drilling and mining tools, mill products including various end-mills and mill inserts, for the making of jewellery, surgical tools, industrial alloys etc [1–7]. Along with its applications in hard-metal industries, WC is found as a low cost alternative for Pt catalyst [8]. The practical interest on WC is due to its high elastic moduli, melting point, hardness and catalytic property [7, 8]. These are interesting properties useful for various practical applications. The WC belongs to the carbides of group IV–VI transition metal compounds which have the highest melting points and are the hardest among all the compounds known [9]. Even though WC is not the carbide with the highest value of hardness or melting point [9], it is the most useful among others. This is mainly due to its sufficiently good high-temperature stability (up to 1000–1200 K) [10] compared to other carbides. In addition, the WC has a factor of 1.5–2 higher elastic moduli and a factor of 1.5–2 smaller thermal expansion coefficient in comparison with other transition metal carbides [11]. The traditional applications of WC in hard-metal industry is due to its attractive mechanical properties. Where as, the studies of Levy and Boudart [8] on the catalytic properties of WC made this material as an alternative for highly expensive Pt electrode in hydrogen evolution reactions. Recently many people have investigated the catalytic behaviour of WC using experiments and theory [12–16].
The history of tungsten carbide started when it was first synthesized by Moissan in 1893 [17]. Following decades, the commercial-scale production and industrial applications made this material popular among researchers and technologists [18–23]. The experimental and theoretical studies show that the W–C system includes two phases namely, higher carbide (WC) and semi-carbide (W2C) [9] with several of their structural modifications. Different types of carbon atom distribution causes the formation of four polymorphs ( ,
,  ,
,  , and
, and  ) of semi-carbide of tungsten (W2C). Higher tungsten carbide exists in two poly-morphs such as room temperature hexagonal phase (
) of semi-carbide of tungsten (W2C). Higher tungsten carbide exists in two poly-morphs such as room temperature hexagonal phase ( -WC) and high-temperature (stable only above 2525 °C) cubic phase (
-WC) and high-temperature (stable only above 2525 °C) cubic phase ( -WC) [24]. The
-WC) [24]. The  -WC was discovered by Sara in 1964 [24]. There are so many literature available on phase stability, electronic properties, elasticity, hardness etc of higher carbide WC [25–32]. The
-WC was discovered by Sara in 1964 [24]. There are so many literature available on phase stability, electronic properties, elasticity, hardness etc of higher carbide WC [25–32]. The  -WC phase is also extensively investigated [6, 25–28, 33–38]. But there are comparatively less number of studies have done on
-WC phase is also extensively investigated [6, 25–28, 33–38]. But there are comparatively less number of studies have done on  -WC. Even though
-WC. Even though  -WC is a high-temperature phase of WC, through a rapid quenching process, one can stabilize it at room temperature [7]. It was first considered as the high-temperature structural modification of W2C [39–41]. But at later, it is concluded that
-WC is a high-temperature phase of WC, through a rapid quenching process, one can stabilize it at room temperature [7]. It was first considered as the high-temperature structural modification of W2C [39–41]. But at later, it is concluded that  -WC is a high-temperature modification of higher carbide WC due to its ability to exist in stoichiometric form [10]. The studies on the structural stability of all the six W-C systems shows that
-WC is a high-temperature modification of higher carbide WC due to its ability to exist in stoichiometric form [10]. The studies on the structural stability of all the six W-C systems shows that  -WC and
-WC and  -WC are the most and least stable respectively [25, 28, 42]. The hardness value of the
-WC are the most and least stable respectively [25, 28, 42]. The hardness value of the  -WC is slightly higher than that of the
-WC is slightly higher than that of the  -WC [28] and this motivated us to substitute W site in
-WC [28] and this motivated us to substitute W site in  -WC to analyze the elastic properties and hardness. Because, a costless alternative for diamond like superhard material is essential for practical application and it keep the investigations for superhard/hard-metals more vibrant [43–46]. Some of the recent research works pointed out that the carbides are promising class of materiel for hard-metal industries [47–51]. Also, the application of the
-WC to analyze the elastic properties and hardness. Because, a costless alternative for diamond like superhard material is essential for practical application and it keep the investigations for superhard/hard-metals more vibrant [43–46]. Some of the recent research works pointed out that the carbides are promising class of materiel for hard-metal industries [47–51]. Also, the application of the  -WC in the hard-metal industry is rare because of its meta-stable state at room temperature.Hence, it is important to improve its stability by various substitutions without compromising its hardness. There is substantial works are done on improving the stability of transition metal nitrides without compromising their hardness [52–60].It may be noted that the addition of other cubic carbides like VCy can stabilize the cubic structure of WC [11].
-WC in the hard-metal industry is rare because of its meta-stable state at room temperature.Hence, it is important to improve its stability by various substitutions without compromising its hardness. There is substantial works are done on improving the stability of transition metal nitrides without compromising their hardness [52–60].It may be noted that the addition of other cubic carbides like VCy can stabilize the cubic structure of WC [11].
The purpose of the present study is to understand the role of W-site substitution on structural, mechanical, and electronic properties of  -WC. Here, we use the first-principles calculation in the framework of density functional theory (DFT). Since hardness is an important but poorly defined property in microscopic scale; there are several traditional and empirical models which correlate hardness with other material properties especially with the elastic properties of crystals such as bulk modulus, shear modulus, Young's modulus, and Pugh's modulus ratio [61–66]. Recently, Rong Yu's group reported [67] that the softest elastic mode shows a better correlation with hardness compared to other elastic properties. So, here the value of hardness is calculated based on the smallest Eigenvalue [67] as well as Pugh's modulus ratio [66]. The results from present calculations may be useful for the practical applications of W-site substituted systems instead of pure
-WC. Here, we use the first-principles calculation in the framework of density functional theory (DFT). Since hardness is an important but poorly defined property in microscopic scale; there are several traditional and empirical models which correlate hardness with other material properties especially with the elastic properties of crystals such as bulk modulus, shear modulus, Young's modulus, and Pugh's modulus ratio [61–66]. Recently, Rong Yu's group reported [67] that the softest elastic mode shows a better correlation with hardness compared to other elastic properties. So, here the value of hardness is calculated based on the smallest Eigenvalue [67] as well as Pugh's modulus ratio [66]. The results from present calculations may be useful for the practical applications of W-site substituted systems instead of pure  -WC.
-WC.
2. Computational details
The ab initio total energy calculations for all the systems in this work have been performed using the DFT as implemented in the Vienna ab initio simulation package (VASP) [68–70]. As the generalized gradient approximation (GGA) generally improves the prediction of structural and elastic properties [71], the exchange-correlation potential term has been considered within the GGA formulated by Perdew, Burke and Ernzerhof [72] with projector augmented wave (PAW) method. We have used 1  1
1  2 supercell with four atoms for all the WXC2 systems. The valence electrons considered in the calculations for most of the transition metals (Ti, Cr, Ru, Rh, Pd, Ag, Cd, Hf, Ta, Os, Ir, and Pt), except Sc, V, Y, Zr, Nb, Mo, and Re are from the outermost s and d orbitals. But in the remaining cases, we considered the semicore p orbital electrons also apart from the electrons from outermost s and d orbitals. Whereas, s and p electrons are considered for p-block elements, and the electrons from the outermost f and d orbitals are included for the f-block elements. For W, the outermost orbital electrons such as 6 s 5 d are considered and for C the 2 s 2 p electrons are accounted in the calculations. Accurate Brillouin zone integration is carried out using the standard special k-points technique of Monkhorst Pack (MP) [73] with a grid size of 12
2 supercell with four atoms for all the WXC2 systems. The valence electrons considered in the calculations for most of the transition metals (Ti, Cr, Ru, Rh, Pd, Ag, Cd, Hf, Ta, Os, Ir, and Pt), except Sc, V, Y, Zr, Nb, Mo, and Re are from the outermost s and d orbitals. But in the remaining cases, we considered the semicore p orbital electrons also apart from the electrons from outermost s and d orbitals. Whereas, s and p electrons are considered for p-block elements, and the electrons from the outermost f and d orbitals are included for the f-block elements. For W, the outermost orbital electrons such as 6 s 5 d are considered and for C the 2 s 2 p electrons are accounted in the calculations. Accurate Brillouin zone integration is carried out using the standard special k-points technique of Monkhorst Pack (MP) [73] with a grid size of 12  12
12  12 for structural optimizations and elastic constant calculations. We have used a
12 for structural optimizations and elastic constant calculations. We have used a  -centered grid of 12
-centered grid of 12  12
12  12 for the density of states (DOS) calculations and crystal orbital Hamiltonian population (COHP) analysis. The plane-wave energy cut-off which restricts the number of plane waves in the basis set is set to be 600 eV in all the calculations. The structures are relaxed until the difference of total energy within the SCF convergence threshold of 10−6 eV/atom.
12 for the density of states (DOS) calculations and crystal orbital Hamiltonian population (COHP) analysis. The plane-wave energy cut-off which restricts the number of plane waves in the basis set is set to be 600 eV in all the calculations. The structures are relaxed until the difference of total energy within the SCF convergence threshold of 10−6 eV/atom.
3. Results and discussions
3.1. Structural details
At room temperature, WC crystallizes in a hexagonal structure with space group P m2 (
m2 ( -WC) [33]. But, the cubic NaCl-type phase (space group Fm
-WC) [33]. But, the cubic NaCl-type phase (space group Fm m) of WC is known to be a high-temperature phase which can be stabilized at room temperature with a rapid quenching process [7]. In
m) of WC is known to be a high-temperature phase which can be stabilized at room temperature with a rapid quenching process [7]. In  -WC, the C atoms occupy all octahedral interstitials in the fcc sublattice of W and can form a stoichiometric compound. It may be noted that the bcc phase of W forms the carbide with fcc structured metal sublattice i.e. the presence of carbon induce changes in the crystal structure of the metal and leads to a changes in the symmetry of the metal sublattice [10]. Already several WXC2 phases were synthesized and their structures are characterized [74–77]. Using the experimental structural information as input, we have obtained the substituted systems WXC2 by replacing 50 percentage of W atoms in
-WC, the C atoms occupy all octahedral interstitials in the fcc sublattice of W and can form a stoichiometric compound. It may be noted that the bcc phase of W forms the carbide with fcc structured metal sublattice i.e. the presence of carbon induce changes in the crystal structure of the metal and leads to a changes in the symmetry of the metal sublattice [10]. Already several WXC2 phases were synthesized and their structures are characterized [74–77]. Using the experimental structural information as input, we have obtained the substituted systems WXC2 by replacing 50 percentage of W atoms in  -WC by other elements (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U). We introduced the new elements in the Wykoff 1c site (0.5, 0.5, 0). Whereas, the remaining W atoms were placed in the Wykoff site of 1b (0, 0, 0.5) and carbon atoms at the 1a (0, 0, 0) and 1d (0.5, 0.5, 0.5) sites. The analysis of structural parameters from structural optimization for the new systems shows a tetragonal P4mmm structure as primitive due to reduction in the symmetry elements by W-site substitution. The estimated equilibrium structural parameters for the ground state geometry for all the considered compounds are listed in table 1, where the comparison is made with available experimental data and the corresponding crystal structure is displayed in figure 1.
-WC by other elements (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U). We introduced the new elements in the Wykoff 1c site (0.5, 0.5, 0). Whereas, the remaining W atoms were placed in the Wykoff site of 1b (0, 0, 0.5) and carbon atoms at the 1a (0, 0, 0) and 1d (0.5, 0.5, 0.5) sites. The analysis of structural parameters from structural optimization for the new systems shows a tetragonal P4mmm structure as primitive due to reduction in the symmetry elements by W-site substitution. The estimated equilibrium structural parameters for the ground state geometry for all the considered compounds are listed in table 1, where the comparison is made with available experimental data and the corresponding crystal structure is displayed in figure 1.
Table 1. The optimized structural parameters for WXC2 systems.
| Compound | a (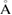 ) ) |
c (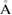 ) ) |
V (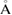 3) 3) |
|---|---|---|---|
| WSiC2 | 2.975 | 4.509 | 39.911 |
| WScC2 | 3.166 | 4.387 | 43.98 |
| WTiC2 | 3.079 | 4.325 | 41.00 |
| 3.045 |
4.306 |
39.92 |
|
| WVC2 | 3.033 | 4.268 | 39.27 |
| 2.988 |
4.226 |
37.74 |
|
| WCrC2 | 3.037 | 4.248 | 39.19 |
| WGeC2 | 3.038 | 4.673 | 43.14 |
| WYC2 | 3.307 | 4.546 | 49.72 |
| WZrC2 | 3.204 | 4.490 | 46.09 |
| 3.182 |
4.500 |
45.56 |
|
| WNbC2 | 3.128 | 4.423 | 43.28 |
| WMoC2 | 3.092 | 4.384 | 41.92 |
| WRuC2 | 3.084 | 4.328 | 41.17 |
| WRhC2 | 3.106 | 4.318 | 41.66 |
| WPdC2 | 3.145 | 4.355 | 43.75 |
| WAgC2 | 3.194 | 4.420 | 45.10 |
| WCdC2 | 3.227 | 4.539 | 47.28 |
| WSnC2 | 3.152 | 4.847 | 48.16 |
| WHfC2 | 3.185 | 4.248 | 45.36 |
| 3.148 |
4.452 |
44.12 |
|
| WTaC2 | 3.127 | 4.429 | 43.31 |
| 3.083 |
4.360 |
41.44 |
|
| WReC2 | 3.089 | 4.370 | 41.69 |
| 2.899 |
4.101 |
34.49 |
|
| WOsC2 | 3.103 | 4.344 | 41.83 |
| WIrC2 | 3.130 | 4.329 | 42.41 |
| WPtC2 | 3.152 | 4.403 | 43.75 |
| WThC2 | 3.463 | 4.780 | 57.34 |
| WUC2 | 3.312 | 4.568 | 50.11 |
aDenbnovetskaya [74] (ICSD 618 962). bRogl et al [75] (ICSD 619 083). cEremenko et al [76] (ICSD 619 104). dEremenko et al [76] (ICSD 618 065). eDenbnovetskaya [74] (ICSD 618 866). fLawson [77] (ICSD 618 713).
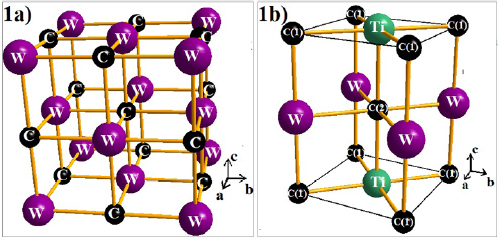
Figure 1. Optimized crystal structures of (a)  -WC and (b) TiWC2.
-WC and (b) TiWC2.
Download figure:
Standard image High-resolution imageFrom the comparative analysis of predicted equilibrium structural parameters with corresponding experimental data, it can be seen that most of the systems show good agreement. Even though they are very close, one can notice that the computed lattice constants are slightly overestimated since we have used GGA potential for all our calculations. As all the considered compounds are having transition metals, the Coulomb correlation effect may be important to consider to predict the equilibrium lattice parameters accurately. So, we have made additional test calculations for ZrWC2 including Coulomb correlation effect using GGA+ U method with U value of 4 eV for both Zr and W [78, 79]. The comparative analysis of structural parameters from both the calculations show that, GGA calculations give better agreement with the experimental lattice parameters than the GGA+U results (GGA gives 1.16 %, deviation in equilibrium volume with experimental value, whereas, GGA+U shows 2.48 % deviation). So, we have proceeded with GGA calculations for remaining systems since it is more reliable and computationally less expensive.
Further, the experimental lattice parameters for WReC2 reported in the literature [77] it is stated that the strong x-rays scattering from the transition metal compared with C atom, they could observe the position of transition metal alone reliably. For the position of C atom they have assumed that it will occupy the octahedral voids. Interestingly, the equilibrium lattice parameter reported in this literature for WReC2 (4.101  ) is found to be in excellent agreement with the lattice parameter of 4.100
) is found to be in excellent agreement with the lattice parameter of 4.100  , reported for WReC0.8 recently [80]. So, one can conclude that the equilibrium lattice parameter for WReC2 reported in the literature is in fact that of WReC0.8. This observation could explain why the calculated equilibrium volume for WReC2 is deviating very much from the corresponding experimentally reported value than that for all the other compounds considered in the present study.
, reported for WReC0.8 recently [80]. So, one can conclude that the equilibrium lattice parameter for WReC2 reported in the literature is in fact that of WReC0.8. This observation could explain why the calculated equilibrium volume for WReC2 is deviating very much from the corresponding experimentally reported value than that for all the other compounds considered in the present study.
Apart from various approximations involved in our calculations the temperature effect also will cause some differences since the results from the present study is applicable for 0K whereas, the experimental results correspond to finite temperature.
3.2. Enthalpy of formation
Enthalpy of formation is an important parameter to understand the thermodynamic stability of solids. At ambient conditions the volume change in a solid is quite small and hence the PV terms can be neglected in the free energy of the system. Therefore, the enthalpy of formation can be compared with internal energy change. By excluding the effect of entropy contribution, the enthalpy of formation for  -WC can be calculated as,
-WC can be calculated as,
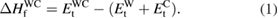
Where E , E
, E , and E
, and E are the total energy for
are the total energy for  -WC, bcc-W and C(graphite), respectively obtained from structural optimization. Similarly, enthalpy of formation for WXC2 can be calculated as,
-WC, bcc-W and C(graphite), respectively obtained from structural optimization. Similarly, enthalpy of formation for WXC2 can be calculated as,
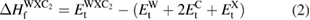
where E and E
and E are the ground state total energy of the WXC2(tetragonal P4mmm) and X systems, respectively obtained from our structural optimizations. By substituting the total energy values from first principles calculations in equations (1) and (2), we have obtained the enthalpy of formation of
are the ground state total energy of the WXC2(tetragonal P4mmm) and X systems, respectively obtained from our structural optimizations. By substituting the total energy values from first principles calculations in equations (1) and (2), we have obtained the enthalpy of formation of  -WC and WXC2 compounds and are given in table 2. From these values, it is clear that Sc, Ti, V, Cr, Y, Zr, Nb, Hf, Ta and U substituted systems are thermodynamically stable. It may be noted that WTiC2, WVC2, WZrC2, WHfC2 and WTaC2 are experimentally found to be stable compounds and our theoretical predictions are in consistent with these experimental observations. Among the considered systems WScC2, WCrC2, WYC2 and WNbC2 also found to be thermodynamically stable according to our total energy calculations. We hope that the present study will motivate the experimentalists to synthesis these newly predicted systems.
-WC and WXC2 compounds and are given in table 2. From these values, it is clear that Sc, Ti, V, Cr, Y, Zr, Nb, Hf, Ta and U substituted systems are thermodynamically stable. It may be noted that WTiC2, WVC2, WZrC2, WHfC2 and WTaC2 are experimentally found to be stable compounds and our theoretical predictions are in consistent with these experimental observations. Among the considered systems WScC2, WCrC2, WYC2 and WNbC2 also found to be thermodynamically stable according to our total energy calculations. We hope that the present study will motivate the experimentalists to synthesis these newly predicted systems.
Table 2. Enthalpy of formation ( ) in kJ mol−1 and eV/atom of WXC2 compounds.
) in kJ mol−1 and eV/atom of WXC2 compounds.
| Compound | 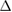 H H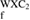 (kJ/mol) (kJ/mol) |
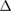 H H (eV/atom) (eV/atom) |
Experimentally Available |
|---|---|---|---|
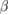 -WC -WC |
9.47 | 0.05 | |
| WSiC2 | 175.6 | 0.46 | No |
| WScC2 | −164.48 | −0.43 | No |
| WTiC2 |
−195.66 | −0.51 | Yes |
| WVC2 |
−109.55 | −0.28 | Yes |
| WCrC2 | −9.21 | −0.02 | No |
| WGeC2 | 280.49 | 0.73 | No |
| WYC2 | −10.41 | −0.03 | No |
| WZrC2 |
−160.82 | −0.42 | Yes |
| WNbC2 | −134.06 | −0.35 | No |
| WMoC2 | 5.73 | 0.01 | No |
| WRuC2 | 179.11 | 0.46 | No |
| WRhC2 | 191.29 | 0.50 | No |
| WPdC2 | 276.43 | 0.72 | No |
| WAgC2 | 438.45 | 1.14 | No |
| WCdC2 | 453.55 | 1.18 | No |
| WSnC2 | 361.10 | 0.94 | No |
| WHfC2 |
−185.41 | −0.48 | yes |
| WTaC2 |
−128.99 | −0.33 | Yes |
| WReC2 |
192.51 | 0.50 | Yes |
| WOsC2 | 279.70 | 0.72 | No |
| WIrC2 | 282.46 | 0.73 | No |
| WPtC2 | 329.79 | 0.85 | No |
| WThC2 | 127.42 | 0.33 | No |
| WUC2 | −57.90 | −0.15 | No |
aDenbnovetskaya [74] (ICSD 618 962). bRogl et al [75] (ICSD 619 083). cEremenko et al [76] (ICSD 619 104). dEremenko et al [76] (ICSD 618 065). eDenbnovetskaya [74] (ICSD 618 866). fLawson [77] (ICSD 618 713).
From the calculated  values, it is clear that Sc, Ti, V, Cr, Y, Zr, Nb, Mo, Hf, Ta and U substitutions in the W-site form stable phase and among them Ti is the most favourable substituent to have stable phase.
values, it is clear that Sc, Ti, V, Cr, Y, Zr, Nb, Mo, Hf, Ta and U substitutions in the W-site form stable phase and among them Ti is the most favourable substituent to have stable phase.
3.3. Role of W-site substitution on elastic properties of  -WC
-WC
The single crystal elastic constants are significant parameters that characterize the deformation capacity of materials under an externally applied stress. Determination of single crystal elastic constants are essential because they are closely related to various fundamental physical properties of the system. It gives information about the chemical bonding, mechanical stability, stiffness, and sound velocity. Moreover, it is used as a tool to predict the material's hardness. Investigation of the elastic properties also provides valuable information about thermodynamic properties such as specific heat, thermal expansion, and Debye temperature. It implies that the elastic constants are important parameters while selecting suitable materials for many practical applications. There are several experimental methods to find out the single crystal elastic constants of materials. It needs a perfect single crystal of the materials with sufficient dimension which in most of the cases difficult to synthesis. So, it signifies the importance of the theoretical calculation of elastic constants. It is well known that the elastic properties of a solid belong to the most fundamental ground-state properties that can be predicted from the first-principles total-energy calculations. [71]
The single crystal elastic constant values measure the response of the material to the external stress. The first three diagonal elements c11, c22, c33 are the coefficient of uniaxial stress along the principal directions  1 0 0
1 0 0 ,
,  0 1 0
0 1 0 , and
, and  0 0 1
0 0 1 , respectively [81]. Similarly, the remaining diagonal elements c44, c55, c66 are the coefficients corresponds to the shear stress between planes (1 0 0) and (0 1 0), (0 1 0) and (0 0 1), (0 0 1) and (1 0 0), respectively. Whereas, the off-diagonal elements i.e. cij with
, respectively [81]. Similarly, the remaining diagonal elements c44, c55, c66 are the coefficients corresponds to the shear stress between planes (1 0 0) and (0 1 0), (0 1 0) and (0 0 1), (0 0 1) and (1 0 0), respectively. Whereas, the off-diagonal elements i.e. cij with  are called biaxial elements represents the shear and normal strains.
are called biaxial elements represents the shear and normal strains.
In this work the single crystal elastic constants of WXC2 (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U) are calculated at their equilibrium volume obtained from strctural optimization using the change in total energy under various deformation as implemented in VASP. The elastic constants of a material can be tuned and modified by substitutions. The corresponding modified values of elastic constants depend on the properties of the substituents, and the substituent-host matrix interactions. As discussed earlier, due to the W-site substitution in  -WC, the system loses some of its symmetry elements and hence the primitive cell of all the WXC2 compounds can be described in simple tetragonal structure as shown in figure 1. The elastic properties of cubic crystals can be fully described through three independent elastic constants, i.e c11, c12, and c44. On the other hand, the tetragonal structure has six independent elastic constants such as c11, c12, c13, c33, c44, and c66 [82]. The knowledge of single crystal elastic constants is important because the whole variety of polycrystalline elasticity can be described by means of it [83]. The calculated values of elastic constants (cij), bulk modulus B, shear modulus G, Young's modulus E, Poisson's ratio
-WC, the system loses some of its symmetry elements and hence the primitive cell of all the WXC2 compounds can be described in simple tetragonal structure as shown in figure 1. The elastic properties of cubic crystals can be fully described through three independent elastic constants, i.e c11, c12, and c44. On the other hand, the tetragonal structure has six independent elastic constants such as c11, c12, c13, c33, c44, and c66 [82]. The knowledge of single crystal elastic constants is important because the whole variety of polycrystalline elasticity can be described by means of it [83]. The calculated values of elastic constants (cij), bulk modulus B, shear modulus G, Young's modulus E, Poisson's ratio  and inverse of Pugh's modulus ratio or Pugh's index (B/G) for all the systems considered in the present study are listed in table 3. Also, the table 3 contains calculated.
and inverse of Pugh's modulus ratio or Pugh's index (B/G) for all the systems considered in the present study are listed in table 3. Also, the table 3 contains calculated.
Table 3. Calculated values of the independent elastic constants (cij, in GPa), bulk moduli (B, in GPa), shear moduli (G, in GPa), Young modulus (E, in GPa), Poisson's ratio ( ), and inverse of Pugh's index (B/G) for
), and inverse of Pugh's index (B/G) for  -WC and WXC2 systems.
-WC and WXC2 systems.
| Compound | c11 | c12 | c13 | c33 | c44 | c66 | B | G | E | B/G |  |
|---|---|---|---|---|---|---|---|---|---|---|---|
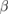 -WC -WC |
846.2 | 192.4 | 85.7 | 410.3 | 151.9 | 405.6 | 2.70 | 0.307 | |||
| 696.5 |
215.5 |
122 |
375.9 |
160.7 |
422.7 |
2.34 |
0.313 |
||||
| 706.5 |
191.4 |
151.6 |
363.1 |
187.7 |
480.4 |
1.93 |
0.279 |
||||
| WSiC2 | 756.97 | 162.64 | 132.84 | 438.69 | 119.58 | 265.89 | 302 | 187.7 | 466 | 1.6 | 0.242 |
| WScC2 | 618.82 | 140.90 | 119.31 | 739.42 | 178.73 | 146.48 | 303 | 201 | 494 | 1.5 | 0.228 |
| WTiC2 | 756.91 | 153.63 | 145.69 | 800.80 | 178.41 | 157.51 | 356 | 218 | 543 | 1.6 | 0.246 |
| WVC2 | 793.73 | 168.24 | 158.40 | 825.46 | 151.95 | 150.55 | 376 | 206 | 522 | 1.8 | 0.268 |
| WCrC2 | 784.50 | 183.71 | 171.43 | 797.42 | 100.26 | 115.88 | 380 | 164 | 430 | 2.3 | 0.311 |
| WGeC2 | 219.13 | 72.67 | 132.29 | 215.47 | 87.76 | 111.94 | 146 | 72 | 185 | 2.0 | 0.289 |
| WYC2 | 453.51 | 125.67 | 104.99 | 600.60 | 158.31 | 106.15 | 241 | 158 | 389 | 1.5 | 0.231 |
| WZrC2 | 516.43 | 248.08 | 123.40 | 702.10 | 167.01 | 233.38 | 302 | 190 | 471 | 1.6 | 0.241 |
| WNbC2 | 779.59 | 160.50 | 151.70 | 788.32 | 149.71 | 137.04 | 364 | 199 | 505 | 1.8 | 0.269 |
| WMoC2 | 808.15 | 186.64 | 187.95 | 791.56 | 81.70 | 89.84 | 392 | 146 | 390 | 2.7 | 0.334 |
| WRuC2 | 729.15 | 216.20 | 188.16 | 778.16 | 32.41 | 67.28 | 380 | 98 | 270 | 3.9 | 0.382 |
| WRhC2 | 810.68 | 224.51 | 188.83 | 827.58 | 25.32 | 104.68 | 406 | 103 | 286 | 3.9 | 0.383 |
| WPdC2 | 562.54 | 184.03 | 184.63 | 687.19 | 32.99 | 90.39 | 323 | 88 | 243 | 3.7 | 0.375 |
| WAgC2 | 484.43 | 163.53 | 163.76 | 593.09 | 43.25 | 87.69 | 282 | 89 | 242 | 3.2 | 0.356 |
| WCdC2 | 455.42 | 148.34 | 156.16 | 474.09 | 67.62 | 95.40 | 256 | 101 | 268 | 2.5 | 0.326 |
| WSnC2 | 596.59 | 172.24 | 115.17 | 342.71 | 95.21 | 191.34 | 249 | 143 | 359 | 1.7 | 0.260 |
| WHfC2 | 711.31 | 151.08 | 133.69 | 752.08 | 170.04 | 144.15 | 335 | 205 | 510 | 1.6 | 0.246 |
| WTaC2 | 827.00 | 169.70 | 161.75 | 824.44 | 148.86 | 140.06 | 385 | 204 | 520 | 1.9 | 0.275 |
| WReC2 | 787.85 | 225.68 | 219.28 | 777.16 | 13.96 | 14.70 | 409 | 72 | 204 | 5.7 | 0.417 |
| WOsC2 | 724.12 | 236.54 | 222.51 | 774.88 | 7.28 | 18.62 | 398 | 62 | 177 | 6.4 | 0.426 |
| WIrC2 | 662.10 | 239.71 | 195.43 | 819.84 | −1.73 | 74.72 | 378 | 55 | 158 | 6.9 | 0.430 |
| WPtC2 | 561.59 | 222.15 | 215.26 | 642.35 | −10.45 | 86.61 | 341 | 29 | 84 | 11.8 | 0.459 |
| WThC2 | 325.75 | 116.20 | 127.87 | 423.30 | 94.34 | 57.81 | 200 | 93 | 241 | 2.2 | 0.299 |
| WUC2 | 470.05 | 172.41 | 148.52 | 657.29 | 90.69 | 40.92 | 280 | 103 | 276 | 2.7 | 0.336 |
aCalculated by CASTEP with GGA scheme in [82]. bCalculated by FLAPW with GGA scheme in [83].
For the requirement of mechanical stability of a tetragonal (I) class (4/mmm) crystal structure, the values of independent elastic constants should satisfy the conditions  ,
,  , c44 > 0, c66 > 0 [84]. From the table 3, the estimated values of elastic constants reveal that Ir and Pt substituted
, c44 > 0, c66 > 0 [84]. From the table 3, the estimated values of elastic constants reveal that Ir and Pt substituted  -WC systems are mechanically unstable; whereas, all the other systems under investigation satisfy the above Cauchy's mechanical stability criteria. The bulk moduli(B) and shear moduli(G) for polycrystalline materials are usually obtained from single crystal elastic constants based on two averaging methods namely Voigt and Reuss methods. The former method assumes uniform strain throughout a polycrystal and defines Bv and Gv as a function of elastic stiffness constants cij [84]. But the later method assumes uniform stress and gives BR and GR as a function of elastic compliance constants sij, which are the inverse matrix element of cij [85]. According to these methods, B and G for the tetragonal system are given as [25],
-WC systems are mechanically unstable; whereas, all the other systems under investigation satisfy the above Cauchy's mechanical stability criteria. The bulk moduli(B) and shear moduli(G) for polycrystalline materials are usually obtained from single crystal elastic constants based on two averaging methods namely Voigt and Reuss methods. The former method assumes uniform strain throughout a polycrystal and defines Bv and Gv as a function of elastic stiffness constants cij [84]. But the later method assumes uniform stress and gives BR and GR as a function of elastic compliance constants sij, which are the inverse matrix element of cij [85]. According to these methods, B and G for the tetragonal system are given as [25],
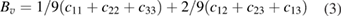
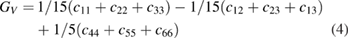
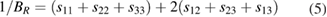
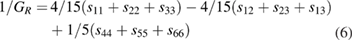
where the subscript V denotes the Voigt value (upper bound for all lattices), R denotes the Reuss value (lower bound for all lattices). According to Voigt–Reuss–Hill (VRH) approximation, the arithmetic average of Voigt and Reuss values give the best estimation for the B and G of a polycrystalline material [86]. In this work, the B and G values are calculated based on the VRH approximation.
The bulk modulus is a measure of the substance's resistance to uniform compression. The low values of B for X substituted  -WC systems reveal that these systems are more vulnerable to hydrostatic pressure compared to
-WC systems reveal that these systems are more vulnerable to hydrostatic pressure compared to  -WC. There are some systems such as WReC2 and WRuC2 have bulk moduli comparable to that of
-WC. There are some systems such as WReC2 and WRuC2 have bulk moduli comparable to that of  -WC. The comparative analysis of shear moduli G shows that Ti, Hf or Y substituted systems have considerably higher values than undoped
-WC. The comparative analysis of shear moduli G shows that Ti, Hf or Y substituted systems have considerably higher values than undoped  -WC; whereas, Ta, Pd, Ag, Cd, or Cr substituted
-WC; whereas, Ta, Pd, Ag, Cd, or Cr substituted  -WC have relatively smaller shear moduli values. This means that the substituted systems show a wide range of values for resistance to shear stress, depending on the substituents.
-WC have relatively smaller shear moduli values. This means that the substituted systems show a wide range of values for resistance to shear stress, depending on the substituents.
The Young's modulus and Poisson's ratio are measured frequently for polycrystalline materials and these values can also be calculated using the bulk modulus and shear modulus as follows:
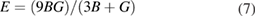
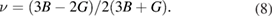
The Young's modulus E is also a proportionality constant between stress and strain. It is a measure of the stiffness of solids to change in the length. Among all the considered systems, WYC2 has the highest stiffness which is noticeably higher than that of pure  -WC. The Poisson's ratio
-WC. The Poisson's ratio  is a measure of relative lateral expansion (compression) of a material due to the relative longitudinal compression (expansion). The Poisson's ratio,
is a measure of relative lateral expansion (compression) of a material due to the relative longitudinal compression (expansion). The Poisson's ratio,  corresponds to, no volume change during elastic deformation [71]. It characterizes the volume change associated with their deformations. Poisson's ratio also provides information about the directionality of the bonding. The value of
corresponds to, no volume change during elastic deformation [71]. It characterizes the volume change associated with their deformations. Poisson's ratio also provides information about the directionality of the bonding. The value of  is much less than 0.25 (around 0.1) for a typical covalent compound, while it is nearly 0.25 or more for a typical ionic compound [87]. In the present study, all the compounds show positive value of
is much less than 0.25 (around 0.1) for a typical covalent compound, while it is nearly 0.25 or more for a typical ionic compound [87]. In the present study, all the compounds show positive value of  and none of them has
and none of them has  . Based on the Poisson's ratio one can interpret that most of the WXC2 compounds show ionic nature. The central-force solids are limited within the range of Poisson's ratio of 0.25 to 0.5 [71]. Even though many of the WXC2 systems are central-force solids, the WAgC2, WHfC2, WNbC2, and WZrC2 are non-central. The table 3 contains the B/G ratio called inverse of Pugh's index which roughly distinguish the brittle or ductile behaviour of materials. According to Pugh's criteria [88], the material is brittle if B/G < 1.75 otherwise, the material behaves in a ductile manner. So we can conclude that the WXC2 with X = Si, Sc, Ti, Y, Zr, Sn or Hf are brittle and others are ductile as
. Based on the Poisson's ratio one can interpret that most of the WXC2 compounds show ionic nature. The central-force solids are limited within the range of Poisson's ratio of 0.25 to 0.5 [71]. Even though many of the WXC2 systems are central-force solids, the WAgC2, WHfC2, WNbC2, and WZrC2 are non-central. The table 3 contains the B/G ratio called inverse of Pugh's index which roughly distinguish the brittle or ductile behaviour of materials. According to Pugh's criteria [88], the material is brittle if B/G < 1.75 otherwise, the material behaves in a ductile manner. So we can conclude that the WXC2 with X = Si, Sc, Ti, Y, Zr, Sn or Hf are brittle and others are ductile as  -WC.
-WC.
3.4. Directional dependence of elastic properties
The investigation of directional dependence of the elastic properties is important for the practical application of a material. The anisotropy in the elastic properties vary with symmetry in solids. If one knows the cij values, one can easily estimate the directional dependency of elastic properties such as bulk modulus, shear modulus, Young's modulus and Poisson's ratio. [71, 89] We have already done such calculations for orthorhombic [71] and hexagonal [90] crystals and given the methodology elaborately. In the same way, the directional dependence of the elastic properties of selected materials are investigated and depicted in figure 2 using MATLAB software [91] from the elastic stiffness constants predicted from our DFT calculations.
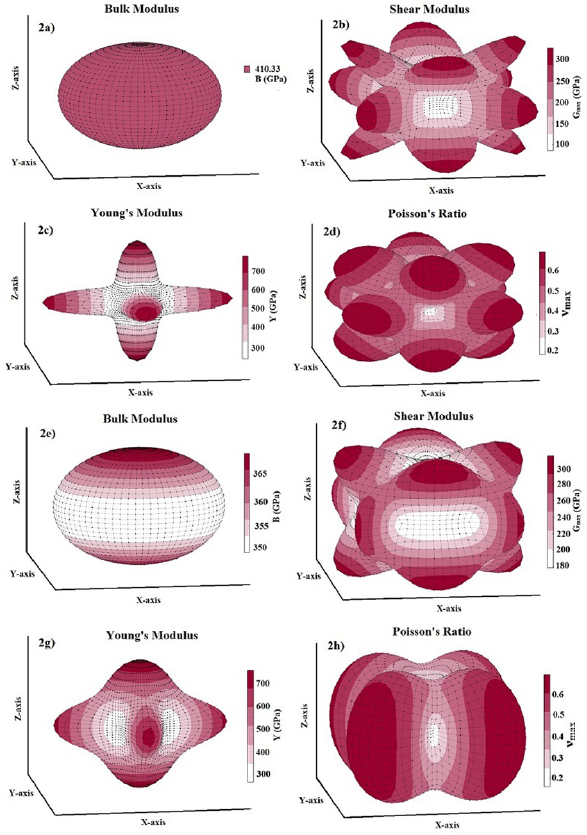
Figure 2. Directional dependence of elastic constants of  -WC and WTiC2 systems. Figures (a)–(d) represent the anisotropy in bulk modulus, shear modulus, Young's modulus and Poisson's ratio respectively of
-WC and WTiC2 systems. Figures (a)–(d) represent the anisotropy in bulk modulus, shear modulus, Young's modulus and Poisson's ratio respectively of  -WC and figures (e)–(h) are the anisotropy in bulk modulus, shear modulus, Young's modulus and Poisson's ratio respectively of WTiC2.
-WC and figures (e)–(h) are the anisotropy in bulk modulus, shear modulus, Young's modulus and Poisson's ratio respectively of WTiC2.
Download figure:
Standard image High-resolution imageThe figures 2(a)–(d) show the directional dependent elastic properties of  -WC, whereas figures 2(e)–(h) shows the same for WTiC2 system. The partial substitution of W by Ti atom makes considerable changes in the directional dependency of elastic properties. If we compare the variation of bulk modulus with direction for Ti substituted
-WC, whereas figures 2(e)–(h) shows the same for WTiC2 system. The partial substitution of W by Ti atom makes considerable changes in the directional dependency of elastic properties. If we compare the variation of bulk modulus with direction for Ti substituted  -WC, the B is higher along c direction [0 0 1] of the crystal compared to other principle axes. But, the degree of variation is not very large. Whereas, it is isotropic in all directions in the case of
-WC, the B is higher along c direction [0 0 1] of the crystal compared to other principle axes. But, the degree of variation is not very large. Whereas, it is isotropic in all directions in the case of  -WC. From the figures 2(a) and (e), one can also note that the value of B for Ti-substituted system is lower than that of
-WC. From the figures 2(a) and (e), one can also note that the value of B for Ti-substituted system is lower than that of  -WC. Compared with the directional dependent variation of bulk modulus, the shear moduli shows considerable directional dependent behaviour for both
-WC. Compared with the directional dependent variation of bulk modulus, the shear moduli shows considerable directional dependent behaviour for both  -WC and WTiC2. It is interesting to note that, though the bulk modulus of the pure system is isotropic and the Ti-substituted system is anisotropic, the anisotropy in the shear moduli for the pure system is higher than that of the Ti-substituted system as evident from figures 2(b) and (f). The common feature in the shear moduli of both the materials is that the value of shear moduli is minimum along the principal axis whereas, maximum along the angular bisectors of the principal axis. I.e. the shear modulus is small along the principal axis of the crystal implies it is easy to shear along the principal direction compare to all the other direction.
-WC and WTiC2. It is interesting to note that, though the bulk modulus of the pure system is isotropic and the Ti-substituted system is anisotropic, the anisotropy in the shear moduli for the pure system is higher than that of the Ti-substituted system as evident from figures 2(b) and (f). The common feature in the shear moduli of both the materials is that the value of shear moduli is minimum along the principal axis whereas, maximum along the angular bisectors of the principal axis. I.e. the shear modulus is small along the principal axis of the crystal implies it is easy to shear along the principal direction compare to all the other direction.
The comparative analysis of the directional dependent Young's modulus of pure and Ti-substituted system show that Young's modulus of Ti-substituted system has small anisotropic behaviour along (1 0 0) and (0 1 0) planes compared to the (0 0 1) plane. But the parent system shows anisotropy in the calculated Young's modulus and almost the same in all the principal planes. The Poisson's ratio analysis brings out completely different anisotropic behaviour for both, the parent and the Ti substituted systems. The Poisson's ratio reaches the minimum value along the principal axis in both the systems and maximum only along the angular bisectors of the principal axis in the parent system. However, in the case of Ti-substituted system, the Poisson's ratio show highly anisotropic behaviour and hence along [0 0 1] direction the Poisson's ratio is much higher than that in other principal directions. It may be noted from figure 2(d) that for pure  -WC the 2-dimensional projection of Poisson's ratio on all the principal planes are identical. From the above observations, we conclude that Young's modulus and Poisson's ratio of Ti-substituted systems have different behaviour along (0 0 1) plane compared to other principal planes. However, the Poisson's ratio is highly anisotropic along (0 0 1) where Young's modulus show more isotropic behaviour.
-WC the 2-dimensional projection of Poisson's ratio on all the principal planes are identical. From the above observations, we conclude that Young's modulus and Poisson's ratio of Ti-substituted systems have different behaviour along (0 0 1) plane compared to other principal planes. However, the Poisson's ratio is highly anisotropic along (0 0 1) where Young's modulus show more isotropic behaviour.
3.5. Analysis of Debye temperature obtained from elastic constants
Debye temperature,  is defined as the temperature of a crystal's corresponds to the highest normal mode of vibration. It is one of the fundamental parameters of a solid and correlated with physical properties such as specific heat, melting temperature, elastic constant etc. It determines thermal properties of the crystal such as electron-phonon interaction in the bulk etc. So, the estimation of Debay temperature from single crystal elastic constants is of current interest and it can be calculated from single crystal elastic constants as follows [92],
is defined as the temperature of a crystal's corresponds to the highest normal mode of vibration. It is one of the fundamental parameters of a solid and correlated with physical properties such as specific heat, melting temperature, elastic constant etc. It determines thermal properties of the crystal such as electron-phonon interaction in the bulk etc. So, the estimation of Debay temperature from single crystal elastic constants is of current interest and it can be calculated from single crystal elastic constants as follows [92],
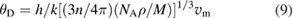
where h is Planck's constant, k is Boltzmann constant, NA is Avogadro's number,  is the density, M is the molecular weight, n is the number of atoms in the molecule and vm is the average wave velocity. vm can be calculated as a function of vt and vl which represents the transverse and longitudinal wave velocities of the material as given in equations (10)–(12) [92].
is the density, M is the molecular weight, n is the number of atoms in the molecule and vm is the average wave velocity. vm can be calculated as a function of vt and vl which represents the transverse and longitudinal wave velocities of the material as given in equations (10)–(12) [92].
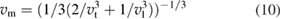
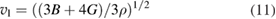

where the values of vl and vt directly depend on shear modulus and bulk modulus. i.e. the Debye temperature of a material can be easily calculated from the elastic constants. In this work, we have calculated the Debye temperature for all the WXC2 systems using this method and the values are given in table 4. As the calculated density get changed, depend upon the substituents the sound velocity change accordingly and hence the Debye temperature. Among the considered compounds in the present study WTiC2 show the highest Debye temperature and hence the constituents are tightly bonded to each other resulting higher phonon frequency. For WPtC2 the calculated Debye temperature is very small compared to that of all the considered systems. The smaller value of Debye temperature in WPtC2 could explain its unstable behaviour (positive heat of formation).
Table 4. The density  in g cm−3, longitudinal, transverse, average elastic wave velocity (
in g cm−3, longitudinal, transverse, average elastic wave velocity ( ,
,  ,
,  in m s−1), and the Debye temperature (
in m s−1), and the Debye temperature ( D) of
D) of  -WC and WXC2 systems obtained from single crystal elastic constants.
-WC and WXC2 systems obtained from single crystal elastic constants.
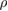 |
 |
 t t |
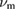 |
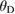 |
|
|---|---|---|---|---|---|
 -WC -WC |
15.292 | 6310 | 3120 | 3502 | 474 |
| WSiC2 | 9.817 | 7497 | 4372 | 4850 | 670 |
| WScC2 | 9.546 | 7738 | 4591 | 5084 | 680 |
| WTiC2 | 10.357 | 7901 | 4587 | 5090 | 697 |
| WVC2 | 10.944 | 7707 | 4335 | 4824 | 670 |
| WCrC2 | 11.258 | 7293 | 3819 | 4271 | 598 |
| WGeC2 | 9.126 | 5143 | 2803 | 3126 | 398 |
| WYC2 | 9.911 | 6749 | 3992 | 4422 | 568 |
| WZrC2 | 10.776 | 7179 | 4195 | 4652 | 613 |
| WNbC2 | 11.540 | 7383 | 4151 | 4619 | 621 |
| WMoC2 | 12.035 | 6986 | 3485 | 3910 | 531 |
| WRuC2 | 12.460 | 6401 | 2801 | 3163 | 432 |
| WRhC2 | 12.828 | 6510 | 2839 | 3206 | 442 |
| WPdC2 | 12.117 | 6031 | 2700 | 3045 | 410 |
| WAgC2 | 11.625 | 5870 | 2772 | 3119 | 414 |
| WCdC2 | 11.248 | 5892 | 2995 | 3356 | 438 |
| WSnC2 | 11.260 | 6246 | 3558 | 3954 | 513 |
| WHfC2 | 14.144 | 6554 | 3805 | 4222 | 559 |
| WTaC2 | 14.907 | 6637 | 3698 | 4117 | 554 |
| WReC2 | 15.696 | 5672 | 2141 | 2429 | 331 |
| WOsC2 | 11.206 | 6553 | 2354 | 2675 | 324 |
| WIrC2 | 15.665 | 5367 | 1879 | 2136 | 289 |
| WPtC2 | 15.294 | 4979 | 1373 | 1567 | 210 |
| WThC2 | 12.739 | 5043 | 2699 | 3014 | 369 |
| WUC2 | 14.776 | 5313 | 2642 | 2965 | 380 |
3.6. Estimation of hardness from the calculated elastic constants
The term hardness describes the ability of a material to resist the deformation under a mechanical load [93]. It is also defined as the resistance to plastic deformation. Extensive research on correlating hardness with physical properties in solids continues over the past several decades to identify superhard materials for various applications. Although hardness is a fundamental mechanical property of materials, it is not as well defined as other physical properties at the microscopic level. It remains a poorly understood property. So, the main focus of this field of research is to understand the hardness microscopically and identify new superhard materials. Superhard materials are defined as the materials having a hardness value above 40 GPa [94]. There are few conventional superhard materials such as diamond, cubic boron nitride etc. The WC also has a high value of hardness i.e. 30 GPa [95]. Even though the superhard materials have technological importance, they are not abundant. Also, they have limitations in certain applications. This makes the field of research on identifying new superhard materials more attractive. In several studies, hardness numbers have been compared with other properties of materials. It is widely believed that correlating elastic properties with hardness is the best tool for the search of superhard materials. Because, factors governing both of these properties are almost the same namely bond length, nature of the bond, bond strength, valence electron density etc. There are many works in which correlation between average elastic constants and hardness are reported. For example, Sung et al [66]and Liu et al used bulk modulus for predicting the hardness, Teter [68] and Brazhkin et al [69] proposed that shear modulus can also be used as an indicator. Chen et al suggested an equation based on Pugh's modulus ratio to predict the value of hardness, which correlates the B and G simultaneously with the hardness value of materials [66]. Recently, Rong Yu et al [67] predicted that the softest elastic mode of the elastic constants matrix show far good correlation with hardness. According to them, the eigenvalues of an elastic constants matrix describe the ability of a material to resist the deformation in independent elastic modes and therefore the softest elastic mode sets the limit of the hardness.
In the present work, we have predicted hardness value using two different methods based on lowest eigenvalue as discussed by Rong Yu et al [67] and Pugh's criteria as discussed by Chen et al [66]. In the first method, we have reproduced the relation between hardness and the lowest eigenvalue using the data given in [67]. The graphical representation of this result is given in figure 3. We have used the linear fit of the source data as a tool to predict the hardness of WXC2 materials by calculating the lowest eigenvalue from single crystal elastic constants. In the second method, we have calculated the hardness value using equation (13). Where k is the G/B value. The calculated values of softest eigenvalue and hardness from both the methods are listed in table 5

Figure 3. Correlation between the hardness values and softest elastic mode reported in [67].
Download figure:
Standard image High-resolution imageTable 5. The estimated values of hardness (in GPa) based on the correlation between hardness versus minimum eigenvalues/Pugh's modulus ratio for WXC2 systems.
| Compound | Minimum eigenvalue (GPa) | Hardness (GPa) (based on minimum eigenvalue) | Hardness (GPa) (Using Pugh's modulus ratio) |
|---|---|---|---|
 -WC -WC |
86 | 10.9 | 34.8 |
| WSiC2 | 120 | 15.9 | 18.2 |
| WScC2 | 146 | 19.7 | 24.5 |
| WTiC2 | 158 | 21.4 | 23.3 |
| WVC2 | 150 | 20.2 | 19.3 |
| WCrC2 | 122 | 16.2 | 11.8 |
| WGeC2 | 63 | 7.5 | 7.6 |
| WYC2 | 106 | 13.8 | 20.6 |
| WZrC2 | 167 | 22.7 | 21.9 |
| WNbC2 | 137 | 18.4 | 18.8 |
| WMoC2 | 82 | 10.3 | 8.6 |
| WRuC2 | 32 | 3.0 | 3.0 |
| WRhC2 | 25 | 2.0 | 3.1 |
| WPdC2 | 33 | 3.2 | 3.0 |
| WAgC2 | 43 | 4.6 | 4.1 |
| WCdC2 | 68 | 8.3 | 7.0 |
| WSnC2 | 95 | 12.2 | 15.9 |
| WHfC2 | 144 | 19.4 | 22.3 |
| WTaC2 | 140 | 18.8 | 18.3 |
| WReC2 | 14 | 0.4 | 0.2 |
| WOsC2 | 7 | −0.64 | −0.4 |
| WIrC2 | −2 | −2.0 | −0.8 |
| WPtC2 | −10 | −3.1 | −2.2 |
| WThC2 | 59 | 7.0 | 8.5 |
| WUC2 | 41 | 4.3 | 6.4 |
The calculated hardness for  -WC based on Pugh's criteria and softest eigenvalue are 34.8 and 10.7 GPa, respectively. The hardness value of the
-WC based on Pugh's criteria and softest eigenvalue are 34.8 and 10.7 GPa, respectively. The hardness value of the  -WC was previously predicted as 32 GPa. This value is in good agreement with our results estimated based on Pugh's criteria. It implies that the Pugh's criteria is more accurate than the softest eigenvalue method in predicting the hardness of
-WC was previously predicted as 32 GPa. This value is in good agreement with our results estimated based on Pugh's criteria. It implies that the Pugh's criteria is more accurate than the softest eigenvalue method in predicting the hardness of  -WC. But most of the other cases, the hardness estimated from both these approach are close enough. In the case of WRuC2, we could get exactly the same results from both the approach, i.e. the method based on softest elastic mode to estimate hardness is successful in predicting the hardness of some materials. Further studies are essential to understand the predicting capability of these two methods. From the results, it is clear that the substitution in
-WC. But most of the other cases, the hardness estimated from both these approach are close enough. In the case of WRuC2, we could get exactly the same results from both the approach, i.e. the method based on softest elastic mode to estimate hardness is successful in predicting the hardness of some materials. Further studies are essential to understand the predicting capability of these two methods. From the results, it is clear that the substitution in  -WC makes considerable changes in its hardness. According to the Pugh's criteria, none of the substituted systems show the hardness greater than that of
-WC makes considerable changes in its hardness. According to the Pugh's criteria, none of the substituted systems show the hardness greater than that of  -WC and obviously, none of the substituted systems is superhard. However, some of the stable systems (mechanically as well as thermally) such as the WHfC2, WNbC2, WTaC2, WTiC2, WVC2, WYC2 and WZrC2 have a relatively higher value of hardness. But they still posses hardness much lower than that of
-WC and obviously, none of the substituted systems is superhard. However, some of the stable systems (mechanically as well as thermally) such as the WHfC2, WNbC2, WTaC2, WTiC2, WVC2, WYC2 and WZrC2 have a relatively higher value of hardness. But they still posses hardness much lower than that of  -WC. The mechanically unstable compounds such as WIrC2 and WPtC2 show negative eigenvalue and hardness. Whereas, the WOsC2 is an exception which is mechanically stable but it still shows a negative value of hardness.
-WC. The mechanically unstable compounds such as WIrC2 and WPtC2 show negative eigenvalue and hardness. Whereas, the WOsC2 is an exception which is mechanically stable but it still shows a negative value of hardness.
3.7. Electronic structure and chemical bonding from density of states analysis
In order to understand the basic features of chemical bonding and structural phase stability, we have calculated the total density of states (TDOS) and partial density of states (PDOS) of some of the systems considered for the present study at their equilibrium geometries and the results are presented in figure 4. Here, we discuss the compounds which exhibit different behaviour in their mechanical stability, thermodynamic stability and hardness values. For example, other than WPtC2, all the WXC2 systems in the figure 4 are mechanically stable even though only WTiC2 is thermodynamically stable. While considering the hardness, one can notice from table 5 that WOsC2 and WPtC2 show negative hardness values; whereas, WReC2 shows a very small positive hardness value and WTiC2 exhibits comparatively high hardness value.
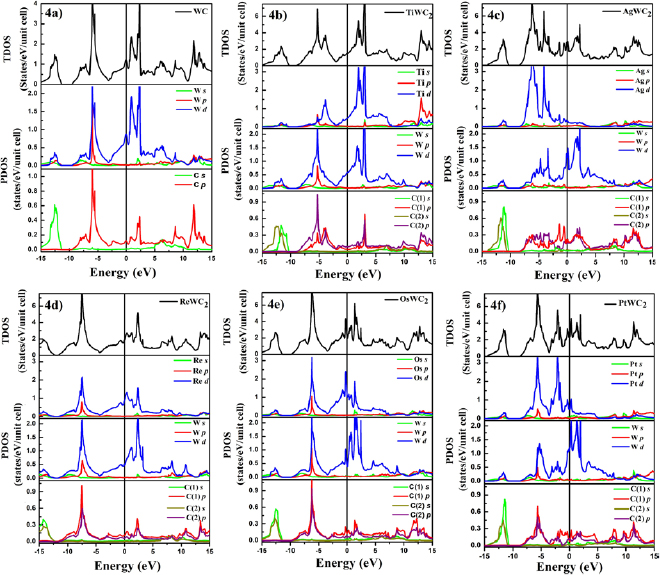
Figure 4. Total density of states (TDOS) and partial density of states (PDOS) of  -WC (a) and the selected WXC2 systems (b-WAgC2, c-WOsC2, d-WPtC2, e-WReC2, f-WTiC2).
-WC (a) and the selected WXC2 systems (b-WAgC2, c-WOsC2, d-WPtC2, e-WReC2, f-WTiC2).
Download figure:
Standard image High-resolution imageIn order to have covalent bonding between constituents they should spatially adjacent to each other and energetically their outermost energy levels should have degenerate behaviour. So, from the partial DOS analysis we can identify the position of electron energy states of valance electrons and from that we can identify the bonding and anti-bonding hybrids and does covalent bonding. If the material has strong covalency, their energy states are usually broad due to strong overlap interactions. In the case of ionic solids, due to large electron negativity difference between cations and anions their valance electrons energy states are well separated with each other. Further, the number of electrons in the valance band obtained from integrated site projected partial DOS for the cation will be smaller than the valance electrons of the corresponding neutral atom. Moreover, due to negligibly small overlap interactions between cations and anions, the DOS will have narrow band features.
In most of the ordered intermetallic compounds a deep valley in the density of states (DOS) curve in the vicinity of Fermi level is observed and this deep valley is called pseudogap. The origin of the pseudogap in ordered intermetallic compounds is traced to (i) charge transfer or ionicity, (ii) hybridization or covalence and (iii) d-resonance [96, 97]. If the electronegativity difference between the constituents is large, due to screening or charge neutrality consideration, a drastic alteration in band shape takes place due to the redistribution of electrons in energy. As a consequence, the screening electrons arc assigned mostly to low-lying states in the band leading to the creation of a minimum in the DOS curve. If the constituents in the intermetallic compounds offer electrons in the same energy range their wave functions are strongly mixed with each other and this covalent hybridization increases the bond strength implying a transfer of electrons to lower energy range, thereby producing a pseudogap. In transition metals and their compounds, the presence of narrow d-states near the Fermi level pulled towards lower energy range from Fermi level due to resonance effect and, hence, deep valley closer to Ef appears. As the  -WC and the transition metals substituted substituted WXC2 compounds considered in this study have mixed bonding nature, the combined effects of the above mechanisms are responsible for the changes in the DOS at the Fermi level.
-WC and the transition metals substituted substituted WXC2 compounds considered in this study have mixed bonding nature, the combined effects of the above mechanisms are responsible for the changes in the DOS at the Fermi level.
A detailed discussion on structural phase stability and position of Fermi level in the DOS curve is given in [98–102]. The comparative analysis of TDOS for the  -WC obtained from our calculations is in good agreement with that from the previous theoretical study [6]. The non-vanishing DOS at the Fermi level indicates the metallic nature of all these systems in figure 4. For Ag, Os, and Pt substituted
-WC obtained from our calculations is in good agreement with that from the previous theoretical study [6]. The non-vanishing DOS at the Fermi level indicates the metallic nature of all these systems in figure 4. For Ag, Os, and Pt substituted  -WC system, one can notice that there is sharp peak like features in the DOS curve in the vicinity of Fermi level (see figures 4(b)–(d)). This indicates that the small external perturbation by temperature, pressure, magnetic field, electric field etc there will be large changes in the DOS at the Fermi level and this explains the unstable nature of these Ag, Os, and Pt substituted WXC2 compounds. It may be noted that in
-WC system, one can notice that there is sharp peak like features in the DOS curve in the vicinity of Fermi level (see figures 4(b)–(d)). This indicates that the small external perturbation by temperature, pressure, magnetic field, electric field etc there will be large changes in the DOS at the Fermi level and this explains the unstable nature of these Ag, Os, and Pt substituted WXC2 compounds. It may be noted that in  -WC also the Fermi level falls on a small peak and this may be the reason why this compound is not stable at ambient conditions and stable only at high temperatures. In WReC2, the TDOS at the Fermi level is having reasonably high value (2.36 states (eV f.u)−1) and however, the DOS profile in the vicinity of Fermi level is not having any sharp peaks. In the case of WTiC2 the DOS at the Fermi level is very small and the Fermi level falls just above a pseudogap feature and hence, one can expect relatively more stability in this system [103] (see the enthalpy of formation given in table 2). It may be noted that the DOS at the Fermi level is dominantly contributed by the transition metal d- states in all the systems considered in the present calculations.
-WC also the Fermi level falls on a small peak and this may be the reason why this compound is not stable at ambient conditions and stable only at high temperatures. In WReC2, the TDOS at the Fermi level is having reasonably high value (2.36 states (eV f.u)−1) and however, the DOS profile in the vicinity of Fermi level is not having any sharp peaks. In the case of WTiC2 the DOS at the Fermi level is very small and the Fermi level falls just above a pseudogap feature and hence, one can expect relatively more stability in this system [103] (see the enthalpy of formation given in table 2). It may be noted that the DOS at the Fermi level is dominantly contributed by the transition metal d- states in all the systems considered in the present calculations.
The partial density of states analysis shows that the occupied band (i.e. electronic states below Fermi level) are dominantly contributed by the transition metal d- and the carbon p -sates. In the occupied band, both transition metal d-states and carbon states are energetically degenerate and this is the indication for covalent interaction between transition metal and carbon atoms. i.e. from the partial DOS plot one can notice that the transition metal d-states and carbon p -states are distributed the same energy window and have similar electronic states distribution in the occupied band. Therefore the electrons at C atom and transition metals leads to covalent bond interactions. In the  -WC, WAgC2, and WPtC2 systems, W- d and C- p orbitals form the electronic states just below the Fermi level. However, in all the other considered cases the d-states of substituted atoms also highly contribute to the topmost region of the occupied band. The TDOS of the
-WC, WAgC2, and WPtC2 systems, W- d and C- p orbitals form the electronic states just below the Fermi level. However, in all the other considered cases the d-states of substituted atoms also highly contribute to the topmost region of the occupied band. The TDOS of the  -WC shows peaks in the energy range between −10 to −2.5 eV originating from the W- d and C- p orbitals and leads to a strong covalent bonding between them. There is a significant electron transfer from the W- s to the C- p orbitals and gives ionic characteristic also. Because, in the elemental state, the the outer most s-orbital of W atom is fully filled, whereas, C atoms have only 2 electrons in its p orbital. But, from the figure 4(a) it can be noted that the outer most s orbital of W is almost empty while the p orbital of C is nearly half filled. Similar kind of bonding interaction is present in transition metal substituted
-WC shows peaks in the energy range between −10 to −2.5 eV originating from the W- d and C- p orbitals and leads to a strong covalent bonding between them. There is a significant electron transfer from the W- s to the C- p orbitals and gives ionic characteristic also. Because, in the elemental state, the the outer most s-orbital of W atom is fully filled, whereas, C atoms have only 2 electrons in its p orbital. But, from the figure 4(a) it can be noted that the outer most s orbital of W is almost empty while the p orbital of C is nearly half filled. Similar kind of bonding interaction is present in transition metal substituted  -WC also. However, in those cases the covalent interaction is weaker than that in
-WC also. However, in those cases the covalent interaction is weaker than that in  -WC. For example, in the case of Ag substituted system, due to weak covalent interaction between Ag with its neighbours the electronic state distribution of W- d states at the occupied band does not have same feature as other substituted systems.
-WC. For example, in the case of Ag substituted system, due to weak covalent interaction between Ag with its neighbours the electronic state distribution of W- d states at the occupied band does not have same feature as other substituted systems.
The weakening of these covalent bonds could explain why the hardness of transition metal substituted systems are smaller than that of  -WC. The difference in electronegativity between the transition metals and carbon may also attribute to the strengthening of bonds by adding some ionic character. Bond strength is one of the important characteristics behind the hardness; therefore, comparatively weak bonds present in substituted systems may be the main reason behind the decrease in hardness value of such compounds. To explain the bonding nature more systematically, we have analysed the COHP and charge density in the following sections.
-WC. The difference in electronegativity between the transition metals and carbon may also attribute to the strengthening of bonds by adding some ionic character. Bond strength is one of the important characteristics behind the hardness; therefore, comparatively weak bonds present in substituted systems may be the main reason behind the decrease in hardness value of such compounds. To explain the bonding nature more systematically, we have analysed the COHP and charge density in the following sections.
3.7.1. The chemical bonding analysis from crystal orbital Hamiltonian population (COHP).
The chemical bonding in  -WC and WXC2 compounds were investigated using the Crystal Orbital Hamilton Populations (COHP) between constituents obtained from Lobster package [104] based on the VASP output. A quantitative bonding analysis is made possible on the basis of the shape and energy integrals (ICOHP—integrated value of COHP) of the COHP given in figure 5 and table 6. The calculated ICOHP of all phases indicates that the W-C1 bond has a higher value than the X-C2 bond. The W-C bond in
-WC and WXC2 compounds were investigated using the Crystal Orbital Hamilton Populations (COHP) between constituents obtained from Lobster package [104] based on the VASP output. A quantitative bonding analysis is made possible on the basis of the shape and energy integrals (ICOHP—integrated value of COHP) of the COHP given in figure 5 and table 6. The calculated ICOHP of all phases indicates that the W-C1 bond has a higher value than the X-C2 bond. The W-C bond in  -WC is stronger than the W-C bond in other substituted carbides due to strong covalent bonding between W-C in
-WC is stronger than the W-C bond in other substituted carbides due to strong covalent bonding between W-C in  -WC. The weakening of the covalent bonding by transition metals substitution in
-WC. The weakening of the covalent bonding by transition metals substitution in  -WC will be the reason for the smaller hardness value in substituted systems.
-WC will be the reason for the smaller hardness value in substituted systems.

Figure 5. Crystal orbital Hamiltonian population (COHP) between constituents in  -WC (a) and that for selected WXC2 systems such as (b) WAgC2, (c) WOsC2, (d) WPtC2, (e) WReC2, and (f) WTiC2.
-WC (a) and that for selected WXC2 systems such as (b) WAgC2, (c) WOsC2, (d) WPtC2, (e) WReC2, and (f) WTiC2.
Download figure:
Standard image High-resolution imageTable 6. The calculated integrated crystal orbital Hamiltonian population (ICOHP) (eV/bond) up to Fermi level between constituents in  -WC and that for selected WXC2(X = Ti, Ag, Re, Os, or Pt) systems.
-WC and that for selected WXC2(X = Ti, Ag, Re, Os, or Pt) systems.
| Compound | X-C1 | X-C2 | W-C1 | W-C2 |
|---|---|---|---|---|
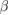 -WC -WC |
−17.427 | |||
| WTiC2 | −11.144 | −5.385 | −7.113 | −13.546 |
| WAgC2 | −3.907 | −1.846 | −6.620 | −12.596 |
| WReC2 | −14.126 | −7.322 | −7.968 | −15.299 |
| WOsC2 | −9.219 | −4.550 | −6.467 | −12.596 |
| WPtC2 | −5.951 | −2.900 | −6.080 | −10.906 |
In figure 5 all the bonding states for W-C bonding pair in  -WC located in the valance band while the anti-bonding states solely appear in the unoccupied region i.e. above the Fermi level. According to band filling of the bonding states analysis, the system will have maximum stability when all the bonding states are filled and all the antibonding states are empty. The COHP between W-C in
-WC located in the valance band while the anti-bonding states solely appear in the unoccupied region i.e. above the Fermi level. According to band filling of the bonding states analysis, the system will have maximum stability when all the bonding states are filled and all the antibonding states are empty. The COHP between W-C in  -WC show such behaviour (see figure 5(a) and hence one could expect relatively high stability for this system. In contrast, in the case of WAgC2, WOsC2, WPtC2, and WReC2 the COHP between X-C in figure 5 show that there is noticeable antibonding states are get occupied. As the filling of antibonding states weakens the structural phase stability as well as bond strength, this could explain why these compounds are not having high stability according to our heat of formation study (see table 2). However, the Ti substituted system also exhibits similar bonding interaction as
-WC show such behaviour (see figure 5(a) and hence one could expect relatively high stability for this system. In contrast, in the case of WAgC2, WOsC2, WPtC2, and WReC2 the COHP between X-C in figure 5 show that there is noticeable antibonding states are get occupied. As the filling of antibonding states weakens the structural phase stability as well as bond strength, this could explain why these compounds are not having high stability according to our heat of formation study (see table 2). However, the Ti substituted system also exhibits similar bonding interaction as  -WC that the COHP between constituents given in figure 5(b) show that all the bonding states are filled and all the antibonding states are empty. This could explain the experimental observation of stable WTiC2 compound. The present COHP analysis suggests that the formation of C vacancy at the C-site closer to X will stabilize most of the compounds mentioned above those have poor structural phase stability. This may be the reason why it is experimentally reported that transition metal substituted WC systems having carbon vacancy such as OsWC0.6, MoWC1.5, RuWC0.6, RhWC0.5 etc.
-WC that the COHP between constituents given in figure 5(b) show that all the bonding states are filled and all the antibonding states are empty. This could explain the experimental observation of stable WTiC2 compound. The present COHP analysis suggests that the formation of C vacancy at the C-site closer to X will stabilize most of the compounds mentioned above those have poor structural phase stability. This may be the reason why it is experimentally reported that transition metal substituted WC systems having carbon vacancy such as OsWC0.6, MoWC1.5, RuWC0.6, RhWC0.5 etc.
3.7.2. The chemical bonding analysis from charge density.
Finally, in order to gain more insights into the bonding interactions between constituents in WXC2 crystals, we have analysed the charge density plot of selected systems. The charge density for a plane where one could see the bonding interaction between all the constituents is depicted in figure 6. The homogeneous charge density distribution at the interstitial region in all the systems considered in the present study indicates the presence of metallic bonding. However, the directional nature of the charge density distribution between W and C indicate the presence of covalent interaction between W-C in all these systems. The charge density plot for  -WC shows that there is a depletion in charge density around the W atom; whereas, the population of electrons in the carbon sites is increased compared with the neutral carbon atom. This indicates that there is noticeable ionic bonding also present between tungsten and carbon. So one can characterise that the bonding interaction between W-C is metallic with iono-covalent nature. Similarly, the chemical bonding between the W and C(1) shows more ionic character than that between W and C(2) and hence more charge is accumulated in the C(2) site as evident from figure 6 for all the WXC2 compounds. In addition, the analysis of charge density between W-site substituted atoms and C atoms shows the directionality in charge density distributions for Re, Os, and Pt substituted systems; which is the fingerprint for covalent bonding interaction. But, the Ag substituted system does not show such covalent interaction between Ag and C atoms. This may be the reason behind its unstable nature. Even though Ti atom in WTiC2 does not make any covalent interaction with the C atoms there is an ionic bonding by the charge transfer from the Ti site to C site. From the charge density analysis, one can understand that the bonding mechanisms in all the above-mentioned systems are combinations of ionic, covalent and metallic nature with varying degrees from one system to another. Also, the noticeable difference in bonding interaction between transition metal with two different carbon atoms indicates the anisotropy in the bonding interactions and this can lead to anisotropy in the elastic properties.
-WC shows that there is a depletion in charge density around the W atom; whereas, the population of electrons in the carbon sites is increased compared with the neutral carbon atom. This indicates that there is noticeable ionic bonding also present between tungsten and carbon. So one can characterise that the bonding interaction between W-C is metallic with iono-covalent nature. Similarly, the chemical bonding between the W and C(1) shows more ionic character than that between W and C(2) and hence more charge is accumulated in the C(2) site as evident from figure 6 for all the WXC2 compounds. In addition, the analysis of charge density between W-site substituted atoms and C atoms shows the directionality in charge density distributions for Re, Os, and Pt substituted systems; which is the fingerprint for covalent bonding interaction. But, the Ag substituted system does not show such covalent interaction between Ag and C atoms. This may be the reason behind its unstable nature. Even though Ti atom in WTiC2 does not make any covalent interaction with the C atoms there is an ionic bonding by the charge transfer from the Ti site to C site. From the charge density analysis, one can understand that the bonding mechanisms in all the above-mentioned systems are combinations of ionic, covalent and metallic nature with varying degrees from one system to another. Also, the noticeable difference in bonding interaction between transition metal with two different carbon atoms indicates the anisotropy in the bonding interactions and this can lead to anisotropy in the elastic properties.

Figure 6. Charge density of (a)  -WC along (0 0 1) plane and selected WXC2 systems such as (b) WTiC2, (c) WAgC2, (d) WReC2, WOsC2, and WPtC2 along (1 1 0) plane.
-WC along (0 0 1) plane and selected WXC2 systems such as (b) WTiC2, (c) WAgC2, (d) WReC2, WOsC2, and WPtC2 along (1 1 0) plane.
Download figure:
Standard image High-resolution image4. Conclusion
We have used the DFT method to perform a set of first-principles total energy calculations to determine the equilibrium structural parameters of WXC2 (X = Si, Sc, Ti, V, Cr, Ge, Y, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Sn, Hf, Ta, Re, Os, Ir, Pt, Th, U) and investigated the effect of W site substitution on the mechanical and electronic properties of  -WC. From these calculations, we have reached the following conclusions.
-WC. From these calculations, we have reached the following conclusions.
- The equilibrium structural parameters of the WXC2 systems obtained by the present work is in good agreement with the available experimental values and we have predicted the equilibrium structural parameters for other systems.
- Based on the calculated enthalpy of formation, we have found that eleven different substitutions (X = Sc, Ti, Cr, V, Y, Zr, Nb, Mo, Hf, Ta, U) to the W site of -WC make stable WXC2 compounds where five among them (X = Sc, Cr, Y, Nb, Mo, U) are not yet synthesized.
- The calculated values of single crystal elastic constants cij, polycrystalline aggregate elastic moduli and Debye temperature of the WXC2 show that all these properties get change by W-site substitution.
- The mechanical stability analysis using Cauchy's relation shows that other than the Pt and Ir substituted -WC systems all the WXC2 systems are mechanically stable.
- The hardness value of these materials are calculated using two different approaches one is based on the Pugh's modulus ratio and the other is based on softest elastic mode. Even though in certain cases we get different hardness values from these two different methods, in most of the cases the variation is small.
- The W-site substitution in several cases leads to more stable systems whereas, none of these stable compounds gives higher hardness value than -WC. However, the stable compound like the WTiC2, WHfC2, and WSnC2 are promising due to their comparatively higher hardness value and high thermodynamic stability.
- We have also discussed the chemical bonding in selected WXC2 systems using the DOS, COHP and charge density analyses and found that these compounds are metallic with various degree of iono-covalent character. In addition, from the DOS and COHP analysis for the selected compounds, we have explained the reason behind the varying thermodynamic stability of these compounds.
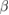
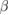
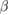
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors are thankful for the financial support from Depart of Science and Technology, India (Grant No. SR/NM/NS-1123/2013), the Indo-Norwegian Cooperation Program (INCP) via Grant No. F. No. 58-12/2014(IC) and the authors are grateful to the Research Council of Norway for providing computer time (under the project number NN2875k) at the Norwegian supercomputer facility. The authors wish to thank Pavel Korzhavy from KTH Royal Institute of Technology, Sweden for the help to do a FORTRAN program to find the eigenvalues of the elastic constant matrix. Anu Maria Augustine also thanks Mr. A Krishnamoorthy for critical reading of the manuscript.
Conflicts of interest
There are no conflicts to declare.