

Abstract
The ultraviolet (UV)-induced dissociation and photofragmentation of gas-phase CH2BrI molecules induced by intense femtosecond extreme ultraviolet (XUV) pulses at three different photon energies are studied by multi-mass ion imaging. Using a UV-pump–XUV-probe scheme, charge transfer between highly charged iodine ions and neutral CH2Br radicals produced by C–I bond cleavage is investigated. In earlier charge-transfer studies, the center of mass of the molecules was located along the axis of the bond cleaved by the pump pulse. In the present case of CH2BrI, this is not the case, thus inducing a rotation of the fragment. We discuss the influence of the rotation on the charge transfer process using a classical over-the-barrier model. Our modeling suggests that, despite the fact that the dissociation is slower due to the rotational excitation, the critical interatomic distance for charge transfer is reached faster. Furthermore, we suggest that charge transfer during molecular fragmentation may be modulated in a complex way.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Inner-shell ionization by extreme-ultraviolet (XUV) or soft-x-ray pulses and the subsequent Auger relaxation is an efficient way of creating multiply-charged molecular ions, which often fragment extensively via a process known as Coulomb explosion [1–4, 6]. In particular, heavy-atom containing molecules exhibit large inner-shell photoabsorption cross sections in the XUV region. The photon energy can often be chosen such that the ionization is either site-specific or distributed over several atomic constituents, providing the opportunity to tailor the probe pulse to address different parts of the molecule. When performing Coulomb explosion experiments with femtosecond pulses from free-electron lasers (FELs), molecular dynamics and fragmentation processes of gas-phase samples can be studied in a time-resolved fashion [5, 7–11].
Here, we focus on the ultraviolet (UV)-induced photodissociation of bromoiodomethane, CH2BrI, with the intent to investigate charge transfer after XUV inner-shell ionization at different molecular sites. Bromine and iodine have inner-shell binding energies (see table 1) that are conveniently within the range of the FLASH free-electron laser [12], and the molecules' photochemistry in the UV region is well-characterized [13–19]. The wavelength of 271 nm used in the present study lies in the A-band of the CH2BrI absorption spectrum, ranging from approximately 240 to 300 nm. The dissociation energies of the C–I and C–Br bond are 2.39 eV [14] and 3.25 eV [14]. One photon at 271 nm corresponds to 4.56 eV, therefore either carbon-halide bond could potentially cleave upon single photon absorption, but cleavage of both bonds by a single-photon absorption is energetically not possible. Isomerisation of the CH2BrI molecule and elimination of a BrI fragment is not observed following absorption in the A-band [14], but is seen for shorter wavelengths in the B-band at 193 and 210 nm. The elimination is attributed to long-lived molecular Rydberg states [14]. A recent experiment employing 8 eV probe pulses compared time-resolved ion yields of CH2BrI and CH2I2 molecules [20]. In combination with trajectory surface hopping calculations, this showed that all excited neutral states in CH2BrI are directly dissociative. The authors also observed oscillations in the ion yield for delays  300 fs, which were attributed to halogen–carbon vibrational stretching but are not discussed in detail.
300 fs, which were attributed to halogen–carbon vibrational stretching but are not discussed in detail.
Table 1. Relevant inner-shell binding energies and resonant excitations of CH2BrI. Values marked with † correspond to CH3I, values marked with △ correspond to CH3Br, and values marked with ‡ correspond to C2H3Br.
I
| 58.3 eV [38] |
I
| 56.6 eV [38] |
Br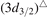
| 77.3 eV [39] |
Br
| 77.3 eV [40] |
Br
| 76.2 eV [39] |
Br
| 76.4 eV [40] |
| Br(3d)→ σ* (C–Br), 3d5/2 | 70.5 eV [16] |
| Br(3d)→ σ* (C–Br), 3d3/2 | 71.5 eV [16] |
| I(4p)† | 129 eV [41, 42] |
A-band photoabsorption is attributed to the transition of a non-bonding electron associated with the iodine atom to a C–I anti-bonding orbital [13, 14]. This excitation primarily results in a dissociation into two neutral partners, I + CH2Br, via direct C–I bond cleavage—as found in many iodine-containing hydrocarbons. The contribution of C–Br bond fission is a minority [14]. The C–I fragmentation occurs on the 5A' potential energy surface and produces ground-state iodine atoms I [13, 14, 18]. Spin-orbit-excited iodine atoms I*(
[13, 14, 18]. Spin-orbit-excited iodine atoms I*( ) are also produced on the 4A' and 4A'' states via an avoided crossing from the 5A' state. Their production ratio was measured to be 4:3 at an excitation wavelength of 248 nm [14], and the I*(
) are also produced on the 4A' and 4A'' states via an avoided crossing from the 5A' state. Their production ratio was measured to be 4:3 at an excitation wavelength of 248 nm [14], and the I*( ) yield was shown to be relatively independent of the excitation wavelength between 222 nm and 280 nm for CH2ICl [21].
) yield was shown to be relatively independent of the excitation wavelength between 222 nm and 280 nm for CH2ICl [21].
In molecules such as CH3I [8, 9, 22] or C6H3F2I [23], in which we have studied ultrafast charge transfer following inner-shell ionization in the past, the center of mass of the molecule lies along the C–I bond, thus almost all available energy is transferred to dissociation along this bond. This allowed studying electron transfer from the neutral co-fragment to the iodine ion in a controlled way. In CH2BrI, the situation is more complex: about 70% of the available energy is transferred into internal energy [17, 19], predominantly into rotation of the CH2Br radical. This rotational excitation is induced by the so-called anchor effect [19] of the second halide atom: the center of mass is shifted away from the C–I bond axis. As a result, the CH2Br radical experiences a torque following excitation via the A-band, subsequently leading to a rotation of the CH2 around the Br. This has implications for the probability of electron transfer to the iodine ion, as we discuss in section 3.2.
2. Methods
The experiment was carried out using the CFEL-ASG Multi-Purpose (CAMP) instrument [24] installed permanently at beamline BL1 of the free-electron laser in Hamburg (FLASH) [12]. Figure 1 shows a schematic overview of the setup. The CH2BrI sample (97% purity, Sigma-Aldrich) was contained in a stainless-steel vessel outside of vacuum at room temperature. The pure target vapor was guided to a 30 μm diameter in-vacuum nozzle, creating a continuous, supersonic expansion into vacuum. The resulting molecular beam was collimated by two skimmers, and was captured in a differentially-pumped beam dump after the interaction region. In the interaction region, the molecular beam was crossed at a 90° angle by the (near-)collinear pump and probe laser beams.
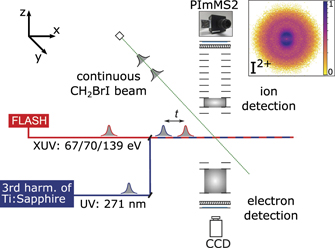
Figure 1. Schematic overview of the experimental setup. A continuous molecular beam of CH2BrI molecules (along y-axis) is intersected by UV and XUV laser pulses (along x-axis) with a controllable delay t. The momentum distributions of ions and electrons are recorded in a double-sided velocity-map imaging (VMI) spectrometer (aligned along the z-axis). An exemplary velocity-map image in the top-right corner shows I2+ ions formed in the UV-pump XUV-probe fragmentation. The spatial overlap of UV and XUV beams was optimized on the ratio of the UV photo-fragments in the image center (indicative of UV dissociation) with respect to the XUV-fragmentation products in the outer feature (created by XUV ionization).
Download figure:
Standard image High-resolution imageThe FEL pulses, with photon energies of 67, 70, or 139 eV, were focused to a nominal focus size of approximately 5 × 7 μm by a pair of Kirkpatrick–Baez mirrors [24]. The shot-to-shot pulse energy fluctuations were monitored by a gas monitor detector upstream of the beamline optics [25]; typical average pulse energies were 60–90 μJ. Thin metallic filters in the beamline were used to attenuate the pulses further. The electron pulse duration was measured to be 63 fs (rms) at 139 eV with the LOLA bunch monitor [26], corresponding to approximately 70 fs (FWHM, full width at half maximum) photon pulse duration [27]. We expect the pulse durations to be roughly comparable at the other photon energies. The FEL was operated in single-bunch mode at 10 Hz to match the repetition rate of the optical laser. The averaged photon energy spectra of the FEL pulses at each of the three photon energies (shown in figure 3) are an accumulation of single-shot spectra that were recorded with a variable-line-space grating spectrometer [28]. The absolute energy was calibrated in advance of the data taking with a high-resolution grating spectrometer in the upstream photon diagnostics section.
UV pulses were generated by frequency-tripling the output of the FLASH pump-probe laser [29]. With a central wavelength of 810 nm (26 nm bandwidth FWHM) and a pulse duration of 55 fs FWHM, this resulted in a central UV wavelength of 271 nm (2.5 nm bandwidth FWHM) with about 22 μJ pulse energy. A prism compressor installed in the 271 nm beam path was used to partially compress the UV pulse to approximately 100 fs FWHM. The laser beam was focused to approximately 50 μm (1/e) diameter by an out-of-vacuum lens and was coupled into the experimental chamber via a drilled in-vacuum mirror, resulting in an intensity of approximately 1013 W/cm2. Downstream of the interaction region, the beam was guided out of the experimental chamber via a window flange in order to reduce stray UV light.
The spatial and temporal overlap of laser and FEL pulses was determined using a fast photodiode, and by optimizing the signal to obtain a maximum yield of low-energy iodine ions, as explained in detail in reference [30]. The delay of the UV pulses with respect to the XUV pulses was adjusted with a mechanical delay stage in steps of 25–50 fs. The stochastic nature of the self-amplified spontaneous emission (SASE) process inherent to FEL radiation typically leads to an arrival-time jitter between the FEL and the optical laser pulses. The arrival time of the FEL pulses was corrected on a shot-to-shot basis using the electron bunch arrival monitors, as described in reference [31].
The molecular beam, the FEL beam, and the UV beam were overlapped in the center of a double-sided VMI spectrometer [24, 32]. Ions and electrons created from photoionization were guided by electric fields towards opposite sides of the spectrometer onto two microchannel-plate (MCP) detectors coupled to phosphor screens and were detected simultaneously.
The momentum distributions of all resulting fragment ion hits on the MCP/phosphor detector (chevron configuration, P47 scintillator) were recorded using the PImMS2 camera [33, 34] outside of vacuum. The data contain flight times and impact positions of each ion, recorded simultaneously in every shot, with a temporal resolution of 40 ns. Multi-mass ion imaging is crucial for being able to obtain the presented results, in particular due to the limited beamtime and the low repetition rate of only 10 Hz which we had to use due to the required optical laser parameters (see above). In the first analysis step, each ion event, which is registered on multiple pixels and time registers, was reduced to a single pixel event in a process called centroiding, as described in reference [35]. Velocity-map images for a given fragment were mirror-symmetrized and Abel-inverted using the pbasex algorithm [36] to retrieve the central slice of the crushed Newton sphere. Kinetic-energy distributions were extracted by angular integration of the inverted images. The kinetic energy was calibrated by simulating the trajectories of the photoions in the spectrometer using the simion software package [37]. Delay-dependent data were not inverted due to insufficient statistics in each delay step. The selection of low-energy ions to generate the data set shown in figure 7 was achieved by applying radial gates to the velocity-map images, which correspond to a projection of the 3D momentum distribution.
Photoelectrons were recorded on a second MCP detector (chevron configuration, P42 scintillator) using a charge-coupled device camera. Electron data are not presented in this manuscript, but the cross-correlation signal of UV laser and FEL in the electron signal was used to determine the temporal overlap of both pulses.
3. Results and discussion
3.1. XUV-induced fragmentation of CH2BrI
Figure 2 shows ion time-of-flight mass spectra of CH2BrI recorded with the PImMS2 camera at photon energies of 67 eV (black), 70 eV (blue), and 139 eV (red). Overall, the spectra look similar and contain the same species for all energies. Dominant fragmentation products are the singly-charged halogens, I+ and Br+, and  with n = 0, 1, 2 bound hydrogens. Moreover, the two radicals CH2Br+ and CH2I+, as well as higher charged atomic halogens up to I5+ and Br3+ are visible.
with n = 0, 1, 2 bound hydrogens. Moreover, the two radicals CH2Br+ and CH2I+, as well as higher charged atomic halogens up to I5+ and Br3+ are visible.

Figure 2. Ion time-of-flight mass spectra of CH2BrI products after irradiation with XUV pulses at 67 eV (black), 70 eV (blue), and 139 eV (red) photon energy. The pulse energies on target were approximately 1 μJ at 67 eV, 3 μJ at 70 eV, and 2 μJ at 139 eV. The spectra are normalized to the integral of the I+ peak. CHn + peaks with n = 0, 1, 2 are overlapping due to their kinetic energy. Water contamination originates from the residual gas in the experimental chamber; it is strongly reduced at 139 eV photon energy due to the ionization cross section being approximately four times lower compared to those at 67 and 70 eV. Peaks labeled with stars are artefacts. Those are created when ions hit the mesh terminating the end of the ion drift tube of the spectrometer, generating secondary electrons. Those electrons are subsequently accelerated towards the ion detector and have shorter flight times compared to the heavier ions, thus they appear earlier in the spectrum than the corresponding ion peak.
Download figure:
Standard image High-resolution imageAt the higher photon energies, the mass spectra reveal a tendency for the formation of higher charge states and dissociation into smaller fragments than at lower energy. In particular, in the (partially overlapping) CHn + peaks one can see a tendency to lose a higher number of hydrogens as the photon energy increases. These trends can be explained by a combination of two effects: (i) the change in photoabsorption cross section (see figure 3), and (ii) the accessibility of the Br(3d), and the I(4p) levels at 77 eV and 129 eV, respectively, (see table 1) at the highest photon energy.

Figure 3. XUV Photoabsorption cross sections for CH3I [43] (dotted line), CH3Br [44] (dashed line), and their sum (dash-dotted line). The features labelled A and B correspond to core excitations of I(4d) and Br(3d) electrons, respectively [16]. Also shown are the measured averaged spectral profiles of the employed FEL pulses at 67 eV (black, 0.8 eV FWHM), 70 eV (blue, 1.0 eV FWHM), and 139 eV (red, 2.2 eV FWHM). The photon energies corresponding to the mean of each distribution are indicated next to the arrows at the top of the figure.
Download figure:
Standard image High-resolution imageFigure 3 depicts literature values for the absolute absorption cross sections as a function of photon energy measured for CH3I [43] (dotted line) and CH3Br [44] (dashed line), as well as their sum (dash-dotted line). The latter provides a good estimate for the photoionization cross section of the CH2BrI molecule. A high-resolution XUV absorption spectrum measured for gaseous CH2BrI between 46 and 73 eV [16] is in good qualitative agreement with the data shown in figure 3. The broad, intense peak centered around 94 eV corresponds to photoionization of the I(4d) shell—the so-called giant resonance. The features around 53 and 71 eV (labelled A and B in figure 3) can be assigned to core excitations of I(4d) and Br(3d) electrons, respectively [16]. The averaged spectral profiles of the FEL pulse used in this work are also indicated in figure 3. We estimate cross sections of 12 Mb at 67 eV, 16 Mb at 70 eV, and 10 Mb at 139 eV for CH2BrI. At 139 eV, the strongest fragmentation is observed, which can be rationalized when considering the average charge state produced by single photoabsorption at each of these photon energies.
To estimate the average charge created by a single photoabsorption at a given photon energy, we consider the relative absorption cross sections of iodine and bromine, as well as charge-state distributions of comparable rare gases at the same photon energy. Ionization of krypton (isoelectronic to Br−) just below the (3d) edge results in about 80% of Kr+ and 20% of Kr2+, while 50 eV above the edge, 60% of Kr2+ and about 30% of Kr3+ are created, see table 2 [45]. For xenon (isoelectronic to I−), the majority of photoionizations between the (4d) and the (4p) shells results in Xe2+ (70%) and Xe3+ (20–30%), with a small contribution of Xe+ from valence ionization. Above the (4p) edge, Xe4+ rises rapidly and Xe+ is virtually absent [45]. Considering that photoabsorption at 67 eV and 70 eV leads predominantly to inner-shell ionization at the iodine site in CH2BrI, we estimate an average molecular charge of about 2.2 per inner-shell photoabsorption (see table 2). At 139 eV, the partial absorption cross sections at the bromine and iodine site are approximately equal (see figure 3). Absorption at iodine leads to an average charge of about 2.65, and absorption at bromine to about 2.2, thus yielding an average charge of approximately 2.4 per photoabsorption at 139 eV, contributing to the increased fragmentation observed at this photon energy. In addition to the single-photon absorption cross sections and average charge states, we also need to consider multi-photon absorption for the intense FEL pulses. In figure 2, the spectra at all photon energies contain Br3+,I4+, and I5+, indicating a significant contribution from multi-photon absorption.
Table 2. Average charge states created by a single photoabsorption in krypton and xenon, at photon energies corresponding to the same energy difference as for bromine and iodine in the present experiment. The relative yields are taken from reference [45]. The bottom row denotes the average charge.
| krypton (bromine−) | xenon (iodine−) | |||
|---|---|---|---|---|
| Below 3d | 50 eV above 3d | 15 eV above 4d | Above 4p | |
| 1+ | 80% | 10% | ≈5% | — |
| 2+ | 20% | 60% | 70% | ≈50% |
| 3+ | — | 30% | ≈25% | ≈35% |
| 4+ | — | — | — | ≈15% |
| Aver. | 1.2 | 2.2 | 2.2 | 2.65 |
The use of the PImMS2 time-stamping camera allows us to obtain the velocity-map images of all fragments simultaneously, in addition to the mass spectrum. Figure 4 shows the kinetic energy distributions (KEDs) of several species. To a large extent, they are independent of the photon energy. The KEDs of the singly-charged halogens, X+, and the corresponding radicals, CH2X+, are dominated by a peak at approximately 1–3 eV. By comparing the KEDs to the results of a simple, classical Coulomb explosion simulation for partners of different charge states n (indicated by the arrows at the top of each panel), this peak can be assigned to a Coulomb explosion with a singly-charged co-fragment. We also confirmed the existence of these fragmentation channels by ion–ion covariance analysis, which is not addressed further in this manuscript. The photoabsorption of CH2BrI at 67 and 70 eV happens predominantly at the iodine site, but the energies simulated by placing the charges at iodine and bromine, respectively, match the data well. This suggests that the second charge (initially created at the iodine site) is rapidly transferred to the bromine atom. We will elaborate further on this in the analysis of the time-resolved data presented in section 3.2. The additional peak at  1 eV visible for some of the fragments is attributed to a dissociation including a neutral fragmentation partner, resulting from valence ionization. The KEDs of halogens with higher charge states show an increasing contribution of co-fragments with two, three or four charges, consistent with an overall stronger charging of the entire molecule.
1 eV visible for some of the fragments is attributed to a dissociation including a neutral fragmentation partner, resulting from valence ionization. The KEDs of halogens with higher charge states show an increasing contribution of co-fragments with two, three or four charges, consistent with an overall stronger charging of the entire molecule.

Figure 4. Kinetic energy distributions (KEDs) of selected ionic fragments after XUV irradiation at three different photon energies, obtained by angular integration of the corresponding VMI images. The spectra are normalized to an integral of 1. Arrows at the top indicate for comparison kinetic energies calculated for a prompt Coulomb explosion of two charged partners, when starting in the equilibrium geometry and assuming a localization of the charges at the iodine and bromine positions, respectively. The measured KEDs result from multiple possible fragmentation channels and charge locations. The peak at  3 eV in the Br3+ spectrum is caused by residual
3 eV in the Br3+ spectrum is caused by residual  ions from background gas. The flight times of Br2+ and I3+ partially overlap due to their large kinetic energies, as visible in figure 2. Therefore, their KEDs are not fully separated.
ions from background gas. The flight times of Br2+ and I3+ partially overlap due to their large kinetic energies, as visible in figure 2. Therefore, their KEDs are not fully separated.
Download figure:
Standard image High-resolution imageWhen comparing the three photon energies, several differences are discernible in the KEDs in figure 4. The Br2+,I3+, and CHn + distributions, although somewhat noisy, appear to show higher-energy shoulders for 139 eV compared to 67 and 70 eV. The corresponding partner of I3+ in a simple Coulomb explosion model would need to have three to five charges to explain an energy of 15–25 eV, implying a total molecular charge of at least +6 to +8. Such charge states can be reached by absorption of only two photons at 139 eV (see table 2), but at least three photons are required to reach charge states higher than +6 for the lower energies. The strongly enhanced yield of CH2I+ with Br+ as a partner at 139 eV is slightly surprising, but can potentially be explained by a higher average molecular charge leading to this channel, in comparison to the other energies. The corresponding CH2I+ peak in the mass spectrum in figure 2 appears slightly shifted to lower masses, suggesting that more hydrogens may be lost (not resolved). We also note that the valence ionization cross section is lower at 139 eV.
The influence of the bromine resonance around 70 eV on the molecular fragmentation has been investigated by Olney et al, who recorded photoion branching ratios for CH3Br up to 80 eV [44]. Although of limited energy resolution, these indicate that the main differences at 70 eV (compared to 67 eV) are: (i) an increase of Br+, (ii) a trend towards overall enhanced fragmentation and hydrogen loss, and (iii) the appearance of a small contribution (≈2%) of Br2+. Our KEDs for 67 and 70 eV in figure 4 look very similar for most fragments. CH2Br+ and CH2I+ appear to occur slightly more often with a charged partner at 70 eV compared to 67 eV—consistent with the findings of Olney et al [44]; Br2+ occurs less often with a singly-charged cofragment, and more often with higher charged partners; and I3+ and Br3+ show an increase in combination with a partner of three or four charges. The peaks of I2+ and Br2+ are slightly enhanced for 70 eV in figure 2. We attribute those changes to an increased probability of multi-photon absorption at 70 eV, due to the higher cross section compared to 67 eV, in combination with a slightly higher pulse energy for the case of 70 eV.
3.2. Intramolecular charge (re-)distribution
In previous work, we have studied ultrafast charge transfer in different dissociating molecules [8, 9, 22, 23]. In those studies, the center of mass of the molecular fragment lay along the axis of the dissociating bond, and thus could be modeled well as two point-like particles undergoing prompt Coulomb explosion. A classical over-the-barrier model [46, 47] was found to describe the critical distance for electron transfer towards the iodine surprisingly well. In ring-like molecules, it was necessary to revisit the effective distance of the repelling charges [23], but the dissociation could still be described by two point-like charges.
In the case of CH2BrI, the initial UV photoexcitation is very similar to the other studied systems containing a C–I bond. However, as described in the introduction, a large fraction of the total available energy is not converted into kinetic energy of the dissociation, but into rotation of the CH2Br radical since the C–I dissociation is not symmetric with respect to the center of mass of the molecule. This influences the transfer of electrons to the iodine atom in different aspects. While it is intuitive that the dissociation effectively becomes slower, it is not a priori clear whether electron transfer to the iodine is best described as occurring from the carbon or the bromine site (or from the bromine via the carbon, or from a (delocalized) molecular orbital). One would expect two aspects to play an important role: (i) the preferential localization of the lowest bound electron in the CH2Br radical, and (ii) the orientation of the radical with respect to the iodine. We will discuss the influence of both in the following.
Figure 5 shows a sketch of the molecular orbital energies of the CH2Br radical. As in other halomethyl radicals, a C–X π-bond is formed upon interaction of the 4py orbital with the 2py orbital on the carbon atom, while the 4px orbital forms a non-bonding lone-pair on the bromine atom [48, 49]. The π* (C–Br) and 4px orbitals, calculated at the density functional level of theory using the B3LYP functional and an aug-cc-pVTZ basis set [50–52], are also shown in figure 5. The unpaired electron in the π* (C–Br) HOMO is localized on the carbon atom, which is in accordance with electronic structure calculations [49] and experimental studies employing hyperfine-resolved microwave spectroscopy [53]. The next occupied orbital is the 4px lone pair [49], which is localized at the bromine site. Both, the unpaired electron in the HOMO and the bromine lone pair, could potentially serve as a source of electrons that may be transferred to the iodine site, as long as the iodine is closer than the critical distance. In order to model the charge-transfer probability as a function of the pump-probe delay, it is therefore necessary to take the rotation of the radical into account.

Figure 5. Schematic of the molecular orbitals in the CH2Br radical (molecular orbitals derived from the combination of C and H atomic orbitals are not shown). The HOMO is the π* anti-bonding orbital formed by the Br(4p) and the C(2p) atomic orbitals.
Download figure:
Standard image High-resolution imageFigure 6 shows the time evolution of the CH2BrI molecular geometry after UV-induced dissociation of the C–I bond, retrieved according to the procedure described in appendix

Figure 6. Calculated time-dependent geometries of the CH2BrI molecule after UV excitation. (a) Sketch of transient geometries at different times; (b) C–I distance and (c) I–Br distance as a function of time after UV excitation. The hollow circles at early times correspond to the mean of the ab initio trajectories from reference [19]; the raw data were provided by the Bañares group. The solid blue line in (b) shows the extrapolated average C–I distance up to 1.5 ps, for the average rotation speed of 1.34°/fs (see appendix
Download figure:
Standard image High-resolution imageTo understand how this geometry evolution would be reflected in the electron transfer probability, we employ the classical over-the-barrier model [46, 47] for both the carbon and the bromine sites. This requires 'site-specific' ionization energies (IEs) as an input. The IE of the CH2Br radical, corresponding to the IE of the HOMO located at the carbon (see figure 5), has been reported as EC = 8.72 eV [54]. We determine the IE for an electron in the 4px bromine lone pair by calculating the zero-point corrected energy of the CH2Br radical in its doublet ground electronic state, as well as the energy of the singly-charged CH2Br+ ion in its triplet configuration (at the geometry of the neutral radical). The calculations have been performed using the Gaussian 09 software suite [55] at the CCSD(T)/aug-cc-pVTZ level of theory [50–52]. The difference between the two zero-point corrected energies is the vertical IE of the CH2Br radical from the 4px − EBr = 10.97 eV.
The critical distances for electron transfer from the bromine or the carbon atom of a neutral CH2Br radical to the iodine can be calculated as [9]:  , with q being the iodine charge state. This distance corresponds to the case in which the potential barrier equals the electron binding energy. The results for different iodine charge states q are indicated with dashed lines in figures 6(b) and (c). The critical distances for electron transfer from carbon to I2+–I5+ (blue filled circles in figure 6(b)) are reached for pump-probe delays between 600 and 900 fs, and for delays between 450 and 750 fs for electron transfer from bromine (black filled circles in figure 6(c)). Also shown for comparison are the hypothetical Rcrit for electron transfer from carbon when ignoring the rotation of the cofragment (red filled circles in figure 6(b)), which lie between 700 and 1100 fs. This illustrates that although a considerable amount of energy is spent on the rotational excitation, somewhat counter-intuitively, the rotation effectively shortens the delay at which the critical distance to the carbon atom is first reached by 100–200 fs, depending on the charge state. For almost all pump-probe delays, the delay-dependent interatomic distance is larger for the case of a rotating cofragment than a model ignoring the rotation would suggest (the blue curve in figure 6(b) lies above the red one for most delays). Rotation of a molecular co-fragment has previously been identified as a reason for unexpectedly fast dissociation of Br
, with q being the iodine charge state. This distance corresponds to the case in which the potential barrier equals the electron binding energy. The results for different iodine charge states q are indicated with dashed lines in figures 6(b) and (c). The critical distances for electron transfer from carbon to I2+–I5+ (blue filled circles in figure 6(b)) are reached for pump-probe delays between 600 and 900 fs, and for delays between 450 and 750 fs for electron transfer from bromine (black filled circles in figure 6(c)). Also shown for comparison are the hypothetical Rcrit for electron transfer from carbon when ignoring the rotation of the cofragment (red filled circles in figure 6(b)), which lie between 700 and 1100 fs. This illustrates that although a considerable amount of energy is spent on the rotational excitation, somewhat counter-intuitively, the rotation effectively shortens the delay at which the critical distance to the carbon atom is first reached by 100–200 fs, depending on the charge state. For almost all pump-probe delays, the delay-dependent interatomic distance is larger for the case of a rotating cofragment than a model ignoring the rotation would suggest (the blue curve in figure 6(b) lies above the red one for most delays). Rotation of a molecular co-fragment has previously been identified as a reason for unexpectedly fast dissociation of Br Cl [56] and CHBr3 molecules [57] after an initial halogen-carbon bond fission. Furthermore, the rotation leads to the fact that the critical distance for C–I can be crossed twice or even three times for certain charge states, which accordingly is expected to lead to a non-trivial oscillation of the charge-transfer probability.
Cl [56] and CHBr3 molecules [57] after an initial halogen-carbon bond fission. Furthermore, the rotation leads to the fact that the critical distance for C–I can be crossed twice or even three times for certain charge states, which accordingly is expected to lead to a non-trivial oscillation of the charge-transfer probability.
The electron transfer towards the iodine ion is reflected in the delay-dependent yield of low-kinetic-energy iodine ions following UV-dissociation of the C–I bond. This channel only occurs for cases where the internuclear distance between iodine and the rest of the molecule is sufficiently large upon XUV-photoabsorption, such that charge transfer between them is inhibited. As observed in earlier experiments [8, 9, 22, 23], the step-wise increase in the yield of these low-energy ions therefore does not occur at time overlap between the pulses, but is shifted towards the UV-early side (i.e. longer delay times). The delay-dependent ion yields for different iodine charge states and photon energies are plotted in figure 7. A step-like increase of the ion yield is visible in the yields of I2+ and I3+ ions for all photon energies, and in addition for I4+ and I5+ ions at 139 eV. At the two lower photon energies, one XUV photoabsorption at iodine results in either two or three charges (see section 3.1). Charges of +4 or +5 would require two-photon absorption, which we consider unlikely, because the I(4d) binding energies of the I2+ and I3+ ions are most likely higher than 70 eV, thus the second photoabsorption can only take place in the valence shell. For the case of 139 eV, the situation is different. One photoabsorption at the I(4p) shell can result in four charges (see table 2), therefore I4+ can be created by a single photon. The I(4p) binding energy of the neutral atom is 137.7 eV [58], therefore a charged iodine can likely not be ionized at the I(4p) level. However, the I(4d) and the Br(3d) levels are both still accessible and have a significant absorption cross section such that a second photoabsorption can take place in an I2+ or I3+ ion, contributing to the I4+ and I5+ ion yield. Moreover, the data at 139 eV were recorded at a higher XUV pulse energy, but for a shorter total acquisition time, leading to lower statistics in this data set. We therefore doubt the significance of the seemingly slower rise time of the step functions at 139 eV, since our charge-transfer model gives no reason to expect a photon-energy dependence for a given charge state.

Figure 7. Delay-dependent yields of iodine ions of different charge states for the different photon energies. Shown is the integrated signal in the channel corresponding to UV-dissociated molecules (kinetic energy of ⩽0.55 eV). The XUV pulse energies on target were approximately 4 μJ at 67 eV, 3 μJ at 71 eV, and 35 μJ at 139 eV. A fit using a Gaussian cumulative distribution function is shown as a red line. Also indicated with triangles at the top are the delays at which the critical distances for electron transfer from carbon or bromine are reached for each charge state (see also figures 6(b) and (c)).
Download figure:
Standard image High-resolution imageThe colored triangles in each panel of figure 7 indicate the delays at which critical internuclear distances for electron transfer are reached in different scenarios (see also figure 6). Blue triangles correspond to electron transfer from the carbon atom of a rotating CH2Br, black triangles to electron transfer from the bromine atom (the rotation is practically irrelevant in this case, as the center of mass lies very close to Br). The red triangle shows the expectation for the hypothetical case ignoring the rotation. The latter case provides the worst fit to the data, as all steps clearly occur at shorter delays in the experiment. This is expected, as the rotation shortens the delay at which the critical distances are reached, as described above. The delay times for electron transfer from carbon or bromine (blue and black triangles) lie relatively close to each other, but the black triangles match the onset of the step better. The calculated delay at which the black triangles are located depends on the precise IE of the 4px bromine lone pair orbital. As shown in figure 5, the HOMO of the CH2Br radical is located at the carbon site. However, the HOMO is only occupied by one electron, while the 4px lone pair, located at the bromine, contains two electrons, which might be reflected in an increased probability for electron transfer from the HOMO-1.
We note that the blue triangles correspond to the first time at which the critical internuclear distance is reached. However, charge transfer can again become possible up to 100–200 fs later due to the rotation (see figure 6). This would lead to a temporary decrease in the low-energy ion yield, and therefore an oscillation in the ion yields as a function of the pump-probe delay might be expected, reflecting the oscillating electron transfer probability due to the rotation of the radical with a period of about 250 fs. However, the overall temporal resolution of the experiment and the limited statistics for each delay point were not sufficient to clearly distinguish this in the present experiment. A further point to consider is the dephasing of the trajectories over time, leading to the fact that an ensemble of molecules with different geometries is probed at a given delay. As is visible in figure 6, significant deviations already start to occur for delays between 500 and 900 fs, which is when the critical distances for electron transfer are reached and the neutral channel starts to be populated.
4. Conclusion and outlook
We have shown the influence of the choice of photon energy on the XUV-induced fragmentation of bromoiodomethane molecules, and have demonstrated the importance of taking into account the center of mass in a two-body dissociation, and discussed in detail possible implications of the rotational excitation on the electron transfer during the fragmentation. While the present data are limited by statistics and temporal resolution, they provide a clear route towards future time-resolved studies of photochemistry. Few-femtosecond laser and FEL pulses are now available, with very promising recent developments towards sub-femtosecond x-ray pulses [59] and few-femtosecond synchronization schemes [60]. In the present case, the repetition rate of the experiment was limited to 10 Hz due to the need for intense optical laser pulses suitable to generate the third harmonic with sufficient intensity, which the burst-mode laser at FLASH1 could not provide. However, existing and upcoming high-repetition-rate FELs such as FLASH2020+, the European XFEL and the future LCLS-II facility also feature high-repetition-rate optical lasers suitable for UV generation. Combined with the high single-pulse energies and freely tunable photon energy due to variable-gap undulators at these FEL facilities, this provides excellent opportunities for future studies of real-time molecular dynamics, in particular in combination with coincident or covariant detection of several ions.
Acknowledgments
We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at FLASH. Beamtime was allocated for proposal F-20140072EC. We gratefully acknowledge the tireless efforts and hospitality of the scientific and technical teams at FLASH, who have made these experiments possible. We acknowledge the Max Planck Society for funding the development and the initial operation of the CAMP end-station within the Max Planck Advanced Study Group at CFEL and for providing this equipment for CAMP@FLASH. The installation of CAMP@FLASH was partially funded by the BMBF Grants 05K10KT2, 05K13KT2, 05K16KT3 and 05K10KTB from FSP-302. We thank Luis Bañares and Jesus Gonzalez-Vazquez for providing the raw data of the calculated trajectories, and Evgeny Savelyev, Jonathan Underwood, Rolf Treusch, Erland Müller, and Pascal Lablanquie for help during the beamtime. The Kansas team acknowledges support from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, Grants No. DE-FG02-86ER13491 (AR, FZ, DR) and DE-SC0019451 (EW). TM acknowledges financial support from the French Agence Nationale de la Recherche (ANR) through the ATTOMEMUCHO Project (ANR-16-CE30-0001). ASM and PKO acknowledge German-Russian Interdisciplinary Science Center (Grants No. G-RISC, No. C-2015a-6, No. C-2015b-6, and No. C-2016b-7) funded by the German Federal Foreign Office via the German Academic Exchange Service (DAAD) and Saint-Petersburg State University for financial support. DMPH is grateful to the Science and Technology Facilities Council (United Kingdom) for financial support. The Sorbonne University team acknowledges support from the CNRS PEPS SASLELX program. This work was supported by the Agence Nationale pour la Recherche (under Contracts No. ANR11-EQPX0005-ATTOLAB and No. ANR14-CE320010-Xstase). MBr, SRM and CV gratefully acknowledge the support of EPSRC Programme Grants EP/L005913/1 and EP/T021675/1. CV and MBr additionally thank the EPSRC for their support through Programme Grant No. EP/V026690/1. MBu (EP/S028617/1 and EP/L005913/1) and JWLL (EP/L005913/1)) are also grateful to the EPSRC. JL, SM and PE-J acknowledge support from the Swedish Research Council and the Swedish Foundation for Strategic Research. We acknowledge support by Deutsches Elektronen-Synchrotron DESY, a member of the Helmholtz Association (HGF). This work was supported by the Deutsche Forschungsgemeinschaft through the Clusters of Excellence 'Center for Ultrafast Imaging' (CUI, EXC 1074, ID 194651731) and 'Advanced Imaging of Matter' (AIM, EXC 2056, ID 390715994). SB acknowledges funding from the Initiative and Networking Fund of the Helmholtz Association through the Young Investigators Group program (VH-NG-1104).
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Appendix A.: Molecular dynamics simulations
The dissociation dynamics were simulated by a classical model, in which the relative velocity between the two dissociating fragments was assumed to reach the asymptotic velocity (vf) with an exponential rise function of lifetime τ, as suggested by Burt et al [17], while the CH2Br was treated as a rigid rotor. This functional form was found to match well with both the experimental data and calculated ab initio trajectories [19] at early dissociation times.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Sampled internuclear distance of (a) C–I, (b) C–Br, and (c) molecular angle I–C–Br. 5000 points are included in the sampling process.
Download figure:
Standard image High-resolution image{kind=link}
The time-dependent relative velocity v(t) and distance R(t) between the iodine atom and the center of mass of CH2Br are thus described by
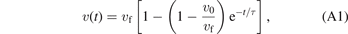
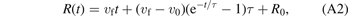
where v0 and R0 are the initial relative velocity and initial center-of-mass distance, respectively. The time constant, τ, is an empirical fit parameter that was set to 50 fs, in agreement with the 'dissociation lifetime' reported for C–I bond dissociation by Attar et al [16]. The initial configuration of v0 and R0 was sampled by a Wigner distribution to account for the zero-point vibration of CH2BrI. In the sampling process, the nuclear wavefunction of CH2BrI is described by a harmonic oscillator, and the Wigner distribution links the wavefunction to a probability distribution in phase space. We use the values for the iodine kinetic energy from Burt et al [17], 0.29 eV and 0.17 eV for I and I
and I , respectively, to determine the asymptotic velocity (vf). The ratio between ground and spin-orbit-excited iodine was chosen as 4:3, in accordance with the findings of reference [17]. The coupling between translational and rotational motions is included in the ab initio simulation [19]. Our model, which ignores this coupling since we focus on describing the dynamics at large delays, agrees well with the average results of these trajectory calculations.
, respectively, to determine the asymptotic velocity (vf). The ratio between ground and spin-orbit-excited iodine was chosen as 4:3, in accordance with the findings of reference [17]. The coupling between translational and rotational motions is included in the ab initio simulation [19]. Our model, which ignores this coupling since we focus on describing the dynamics at large delays, agrees well with the average results of these trajectory calculations.
The rotational velocity of the CH2Br rigid rotor is described by an exponential rise function with the same lifetime as the bond dissociation. Based on the ab initio calculation of Murillo-Sanchez et al [19], the averaged value of the asymptotic rotational velocity is estimated to be 1.34°/fs. The sampled C–I and C–Br internuclear distances and the C–I–Br molecular angle are shown in figure 8. The zero-point vibration results in an uncertainty of approximately ±0.1 Å, and ±6° to the initial bond distances and angle, respectively. Considering the vibrational frequency of the scissors vibration mode of CH2BrI (136 cm−1) [61], the rotational velocity uncertainty from the zero-point vibration is estimated to be ±0.05°/fs.