
Abstract
The dissociation dynamics of CH3I at three UV pump wavelengths (279 nm, 254 nm, 243 nm) are measured using an extreme ultraviolet probe in a time-resolved photoelectron spectroscopy experiment. The results are compared with previously published data at a pump wavelength of 269 nm, [2020, Phys. Chem. Chem. Phys., 22, 25695], with complementary photoelectron spectroscopy experiments performed using a multiphoton ionization (MPI) probe [2019, Phys. Chem. Chem. Phys., 21, 11142] and with the recent action spectroscopy measurements of Murillo-Sánchez et al [2020, J. Chem. Phys., 152, 014304]. The measurements at 279 nm and 243 nm show signals that are consistent with rapid dissociation along the C–I bond occurring on timescales that are consistent with previous measurements. The measurements at 254 nm show a significantly longer excited state lifetime with a secondary feature appearing after 100 fs which is indicative of more complex dynamics in the excited state. The time-dependence of the changes are consistent with the previously measured MPI photoelectron spectroscopy measurements of Warne et al, [2019, Phys. Chem. Chem. Phys., 21, 11142]. The consistency of the signal appearance across ionization processes suggests that the extended observation time at 254 nm is not an artefact of the previously used MPI process but is caused by more complex dynamics on the excited state potential. Whether this is caused by complex vibrational dynamics on the dominant 3Q0 state or is due to enhanced population and dynamics on the 1Q1 state remains an open question.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The A-band absorption profile of CH3I covers a broad wavelength range from approximately 220 nm to 300 nm with a maximum absorption at 258 nm [1, 2]. Excitation into the A-band leads to rapid dissociation that has been extensively studied in both the frequency [2–9] and time [10–19] domains. The accessibility of the A-band, and the structural simplicity of the CH3I molecule, combined with the ensuing dissociation dynamics that occur on coupled potential energy surfaces, has resulted in CH3I being used as a prototype system for the study of non-adiabatic dynamics and as a test sample for the development of new experimental techniques. The experiments to date however tend to focus on wavelengths to the red of the absorption maximum with far fewer studies to the blue side of 260 nm. Here we present the results of new time-resolved photoelectron spectroscopy experiments of CH3I using high energy probes that can drive one-photon ionization across a wide range of geometries. The measurements are performed at three new pump wavelengths (243, 254, and 279 nm) which are combined with the previously published data at 269 nm and analysed in conjunction with recent time-resolved product state measurements.
Excitation of the A-band in CH3I results in the promotion of a non-bonding electron localized on the iodine atom into the lowest available anti-bonding molecular orbital (n → σ*) [20], this weakens the C–I bond and results in strongly dissociative character along this coordinate for all of the spin–orbit states within this excited state band [21]. The strong spin–orbit coupling means the excited electronic configuration gives rise to 5 states which, in Mulliken notation, are labelled  ,
,  ,
,  ,
,  and
and  . Of these 5 states, only the 3
Q1,1
Q1 and
. Of these 5 states, only the 3
Q1,1
Q1 and  (the + will be dropped from now on) are optically allowed transitions [20]. For the majority of the A-band absorption profile the strong parallel transition to the 3
Q0 state dominates [2, 3]. The 3
Q1 and 1
Q1 states are coupled via weak perpendicular transitions from the ground state. Most experimental studies indicate that direct transitions into the 3
Q1 and 1
Q1 states only form a significant contribution to the overall absorption cross-section in the wings of the absorption band [3, 5, 6]. Theoretical simulations suggest these states become significant closer to the centre, however these calculations struggle to accurately simulate the absorption spectrum at the wings [5, 22].
(the + will be dropped from now on) are optically allowed transitions [20]. For the majority of the A-band absorption profile the strong parallel transition to the 3
Q0 state dominates [2, 3]. The 3
Q1 and 1
Q1 states are coupled via weak perpendicular transitions from the ground state. Most experimental studies indicate that direct transitions into the 3
Q1 and 1
Q1 states only form a significant contribution to the overall absorption cross-section in the wings of the absorption band [3, 5, 6]. Theoretical simulations suggest these states become significant closer to the centre, however these calculations struggle to accurately simulate the absorption spectrum at the wings [5, 22].
1D cuts of the potential energy surfaces for the three optically allowed states along the C–I coordinate are shown in figure 1. To a first approximation the dynamics following absorption can be understood through consideration of these 1D cuts. If we consider excitation into the dominant 3
Q0 state, the steep gradient along the C–I dissociation coordinate leads to a rapid extension of the C–I bond and dissociation into neutral ground state methyl fragments in conjunction with either a ground, I , or spin–orbit excited,
, or spin–orbit excited,  , iodine atom. The branching between the two fragmentation channels is controlled by the crossing between the 3
Q0 and 1
Q1 electronic states at intermediate C–I bond lengths which allows for population transfer between the two states. Population that remains in the 3
Q0 state dissociates to form the spin–orbit excited iodine atom,
, iodine atom. The branching between the two fragmentation channels is controlled by the crossing between the 3
Q0 and 1
Q1 electronic states at intermediate C–I bond lengths which allows for population transfer between the two states. Population that remains in the 3
Q0 state dissociates to form the spin–orbit excited iodine atom,  , which is seen to dominate the dissociation yield across the vast majority of the absorption profile. Population that transfers onto the 1
Q1 electronic state leads to formation of the ground state atomic fragment. Across the majority of the absorption peak the product distribution is dominated by the
, which is seen to dominate the dissociation yield across the vast majority of the absorption profile. Population that transfers onto the 1
Q1 electronic state leads to formation of the ground state atomic fragment. Across the majority of the absorption peak the product distribution is dominated by the  product with a quantum yield of between 0.6–0.8 [3, 23–27], with the ground state product thought to be almost exclusively formed via the non-radiative transition onto the 1
Q1 state [3]. The quantum yield of I* is defined as
product with a quantum yield of between 0.6–0.8 [3, 23–27], with the ground state product thought to be almost exclusively formed via the non-radiative transition onto the 1
Q1 state [3]. The quantum yield of I* is defined as ![${{\Phi}}_{{\mathrm{I}}^{{\ast}}}=\left[{\mathrm{I}}^{{\ast}}\right]/\left(\left[{\mathrm{I}}^{{\ast}}\right]+\left[\mathrm{I}\right]\right)$](https://content.cld.iop.org/journals/0953-4075/54/13/134003/revision2/bac08f3ieqn11.gif) .
.

Figure 1. 1D cuts along the C–I dissociation coordinate of the diabatic potential energy surfaces of CH3I relevant to the UV photodissociation and photoelectron detection scheme used, adapted from references [21, 28]. Inset: expanded figure of the excited state potentials and the total absorption spectrum of methyl iodide with the location of each pump wavelength marked.
Download figure:
Standard image High-resolution imagePrevious time domain measurements have probed both the excited state population and the appearance time of the products [11, 19, 29–31]. Recent measurements of the excited state population have used both multiphoton and one-photon ionization processes to study the dynamics in time-resolved photoelectron spectroscopy experiments. The multiphoton ionization (MPI) measurements were performed at pump wavelengths, 269 nm and 255 nm [29], while the one-photon XUV measurements were performed at 269 nm [30]. The results from both of these experiments at 269 nm show rapid decay out of the initially populated region of the excited state. The XUV probe could follow the extension of the C–I bond length and provided a time-dependent mapping of the C–I bond. The MPI experiments at 255 nm showed a surprisingly different time-dependence of the initially excited state population. At this pump wavelength the excited state population was observed for an extended time, with a secondary feature appearing after about 100 fs, well after the signal from the other pump wavelength had disappeared. The cause of the extended excited state observation time is not clear and cannot be easily explained by the potential energy curve or by previously completed dynamics calculations. It was suggested that this secondary feature may be a result of dynamics on the 1 Q1 state.
Previous experiments have also measured the appearance time of vibronic state-resolved products at a few pump wavelengths. At wavelengths around 266/268 nm the fragments associated with the dominant spin–orbit excited state products are observed with a formation time of approximately 100 fs while the formation time of the ground state I products has been shown to be more rapid with an appearance time of around 80 fs [11, 19, 31]. The appearance time in this case is defined as the time taken for the C–I bond length to reach approximately 13 Bohr. The 13 Bohr value is derived from calculations of when the experimental resonance conditions are met for the REMPI detection scheme [19]. Increasing the pump energy to 243 nm, Murillo-Sánchez et al [19] observed a decrease in the appearance time for comparable fragments. Formation of the vibrational ground state of CH3 and the ground state iodine,  , had a decreased appearance time of 53 fs and for the equivalent channel when forming the spin–orbit excited iodine, I*
, had a decreased appearance time of 53 fs and for the equivalent channel when forming the spin–orbit excited iodine, I*  , had a similar decrease in appearance time with a value of approximately 80 fs. The measurements show that with increasing vibrational excitation in the methyl fragment the appearance time at the pump wavelength of 243 nm tended to increase while at 268 nm they observed a decrease for ν2 = 1 (umbrella mode) when produced in conjunction with I*
, had a similar decrease in appearance time with a value of approximately 80 fs. The measurements show that with increasing vibrational excitation in the methyl fragment the appearance time at the pump wavelength of 243 nm tended to increase while at 268 nm they observed a decrease for ν2 = 1 (umbrella mode) when produced in conjunction with I*  . This curious result proved difficult to explain based on trajectory calculations on multidimensional potential energy surface with both the magnitude of the change at 243 nm and the decrease in appearance time at 268 nm not showing in the calculations. The observation of rapid dissociation at 243 nm in this work led the authors to suggest that the extended lifetime observed at 255 nm in the MPI measurements may be due to accidental resonances associated with the MPI process.
. This curious result proved difficult to explain based on trajectory calculations on multidimensional potential energy surface with both the magnitude of the change at 243 nm and the decrease in appearance time at 268 nm not showing in the calculations. The observation of rapid dissociation at 243 nm in this work led the authors to suggest that the extended lifetime observed at 255 nm in the MPI measurements may be due to accidental resonances associated with the MPI process.
In the remainder of the article we present XUV TRPES results obtained following excitation of CH3I at 243, 254, and 279 nm and combine these with the previously presented data at 269 nm [30]. We initially compare the XUV TRPES results to the MPI TRPES measurements at the lowest binding energies measured and relevant to the early time dynamics, before considering the extended observation window offered by the high energy probe. We then finish our discussion of the data through comparison with recent product state-distribution measurements of Murillo-Sánchez et al [19]. Our one-photon measurements confirm that the extended lifetime observed at 254 nm is not an artefact of the MPI probing used previously but is due to the underlying dynamics of the molecule.
2. Experimental details
The experimental methods used have been described previously in references [30, 32, 33] such that we provide a brief outline here. A Ti:Sapphire amplified laser system (Red Dragon, KMLabs) pumps an optical parametric amplifier (OPA, HE-TOPAS, Light Conversion). Through second harmonic generation of the sum frequency generated output of an OPA we produce pulses at 279 nm (4.45 eV), 269 nm (4.61 eV), 254 nm (4.87 eV) and 243 nm (5.10 eV) with pulse energies of 2.8 μJ (279 nm), 3.7 μJ (269 nm), 2.2 μJ (254 nm) and 1.5 μJ (243 nm). The wavelengths chosen cover both the red and blue sides of the A-band absorption maximum (258 nm) of methyl iodide. The probe is generated via high harmonic generation using approximately 450 μJ of the second harmonic of the Red Dragon output at 395 nm. The 7th harmonic (54.9 nm, 22.6 eV) is isolated using a time preserving monochromator [34] and reflection focused off a toroidal mirror into the interaction chamber. Overlap between the two laser pulses is ensured with the use of a scintillator crystal and optimised on the electron signal. The pump and probe beams cross the molecular beam about 3 mm from the entrance of the electron time of flight spectrometer (Kaesdorf ETF11). The electron spectrometer has a resolution of approximately 0.1 eV in the 13.5–18.0 eV electron kinetic energy region considered in the manuscript. Both pump and probe beams are linearly polarized along the time of flight axis and cross at an angle of approximately 3°. The CH3I beam is created via an effusive expansion of the room temperature vapour of CH3I through a 200 μm diameter nozzle which is located a few centimetres away from the interaction region where the molecular and laser beams cross. The low pressure effusive expansion minimises any dimer formation and we see no evidence for dimers in the photoelectron spectra obtained. Any signals associated with background residual gas are small and observed at a much higher binding energy than the signals associated with the excited state dynamics presented. Each scan covered pump–probe delays from −200 fs to 300 fs in 25 fs steps. To avoid any issues with long term drifts in power and/or gas density we cycle through each delay position with the data from each cycle being recorded. At each delay the signal is the result of approximately 2 M, 2 M, 2.7 M and 5.2 M laser shots for the 279 nm, 269 nm, 254 nm and 243 nm data respectively. The maximum overlap between the pump and probe pulses, defined as time zero, is taken from a fit to the sum of the time profiles across the region of the strong initial  and
and  features in the spectrum.
features in the spectrum.
3. Results
In figure 2 we plot the excited state photoelectron spectrum at t = 10 fs (the closest spectrum to time-zero) following excitation by 279 nm (4.45 eV) and ionization by a 54.9 nm (22.6 eV) photon. The spectrum is plotted against the one-photon binding energy where eBE = hνprobe − eKE, hνprobe is the probe photon energy and eKE is the kinetic energy of the electron. The electronic configuration of the neutral excited state is  . At >9 eV we observe the onset of the first ground state peak of CH3I. We also observe three excited state peaks. The two lowest binding energy peaks at 5.3 eV and 5.9 eV correspond to the removal of the (σ⋆) electron and ioniziation of the molecule in its Franck–Condon (FC) geometry into the two spin orbit states of the ground electronic state of the cation,
. At >9 eV we observe the onset of the first ground state peak of CH3I. We also observe three excited state peaks. The two lowest binding energy peaks at 5.3 eV and 5.9 eV correspond to the removal of the (σ⋆) electron and ioniziation of the molecule in its Franck–Condon (FC) geometry into the two spin orbit states of the ground electronic state of the cation,  and
and  respectively. The third peak at 8.1 eV corresponds to the removal of an electron from the non-bonding orbital localized on the I atom, leading to the population of a quartet cation state upon ionization. For a more detailed analysis of these assignments we refer the reader to reference [30] which also contains a detailed analysis of the time-dependent changes observed following excitation at 269 nm which we only briefly discuss here.
respectively. The third peak at 8.1 eV corresponds to the removal of an electron from the non-bonding orbital localized on the I atom, leading to the population of a quartet cation state upon ionization. For a more detailed analysis of these assignments we refer the reader to reference [30] which also contains a detailed analysis of the time-dependent changes observed following excitation at 269 nm which we only briefly discuss here.

Figure 2. The excited state photoelectron spectrum following excitation by a 279 nm pulse and ionization by 22.6 eV. The spectrum is the closest result to time zero (t = 10 fs). The intensity has been normalized so the most intense excited state peak is 1. At >9 eV there is the onset of the first ground state peak of CH3I. States are labelled with final ion state symmetry and asymptotic product where known.
Download figure:
Standard image High-resolution imageIn figure 3 we plot the time-dependent photoelectron spectra of CH3I following excitation into the A-band at 279 nm (4.45 eV, (a)), 269 nm (4.61 eV, (b)), 254 nm (4.87 eV, (c)) and 243 nm (5.10 eV, (d)), and subsequent ionization with a 54.9 nm (22.6 eV) probe. Each spectrum is normalized to the highest intensity feature in each measurement. Around time zero each spectrum shows the same three features shown in figure 2. Two intense peaks below 6 eV that are separated by approximately 0.5 eV, correspond to the two spin orbit states of the ground electronic state of the cation,  and
and  . The position of these peaks shifts to lower binding energy with increasing pump energy as expected, with the positions of these peaks marked by dashed vertical lines in each spectrum. A third, broader, peak is also observed at binding energies of ∼7–8 eV, which is the result of ionization to a quartet cation state. Again, we mark the centre of this peak with a dashed vertical line in each spectrum.
. The position of these peaks shifts to lower binding energy with increasing pump energy as expected, with the positions of these peaks marked by dashed vertical lines in each spectrum. A third, broader, peak is also observed at binding energies of ∼7–8 eV, which is the result of ionization to a quartet cation state. Again, we mark the centre of this peak with a dashed vertical line in each spectrum.

Figure 3. Time-dependent photoelectron spectrum of CH3I following excitation at 279 nm (a), 269 nm (b), 254 nm (c) and 243 nm (d). The dashed white lines indicate the position of the initial peaks associated with ionization into the ground and quartet cation states. Each spectrum is normalized to the highest intensity feature in each measurement.
Download figure:
Standard image High-resolution imageAt 269 nm we observe a rapid decay in intensity at the initially observed energies which is accompanied by a shift to higher binding energies as a function of pump–probe delay. This shift to higher binding energies corresponds to the increase in electron binding energy as the C–I bond stretches as the molecule dissociates. Analysis of the time-dependent shift in binding energy provides approximate time-dependent maps of the C–I bond length during the dissociation process as detailed in Warne et al [30]. The measurements at 279 nm and 243 nm, figures 3(a) and (d), show a qualitatively similar structure to that seen at 269 nm, with a rapid decay in the photoelectron intensity combined with a time-dependent shift to higher binding energies at later times. The similar appearance and time scale upon which the changes occur suggests that the dynamics at these three pump energies are very similar, and are indicative of rapid C–I dissociation. In contrast, the 254 nm pump data, figure 3(c), has a quite different structure with an extended excited state lifetime and more complex changes in the time-dependent intensity profile across the spectrum. The time-dependent changes observed between 4 and 6 eV in the 254 nm data, while very different to that observed at the other pump wavelengths, show a similar structure to that observed in recent MPI photoelectron spectroscopy experiments at a similar pump energy. We continue our discussion of the analysis by first comparing the time-dependence of the low binding energy regions that could be measured by both the previously reported MPI probe photoelectron spectroscopy experiments and the new one-photon ionization data presented here. We then extend that discussion to the longer term dynamics and provide a comparison to the results of product state measurements performed at similar pump wavelengths.
3.1. Comparison to MPI measurements
In order to aid comparison with previously reported MPI measurements performed at pump wavelength of 269 nm and 255 nm, we initially consider the signal obtained at binding energies related to ionization of the FC geometry into the  states of the cation as measured by both probes. The intensity profiles over these features for each pump wavelength are plotted in figure 4 and cover a binding energy range of 4.91–6.14 eV, 4.96–5.95 eV, 4.70–5.70 eV and 4.55–5.38 eV for the 279 nm (a), 269 nm (b), 254 nm (c) and 243 nm (d) data respectively. The error bars are derived from a bootstrapping analysis of the data and mark the 95% confidence intervals. The time-dependence of the integrated photoelectron intensity in these regions were fitted to a sum of exponentially decaying functions convolved with a Gaussian instrument response function appropriate for the pump wavelength used. The equation used had the form:
states of the cation as measured by both probes. The intensity profiles over these features for each pump wavelength are plotted in figure 4 and cover a binding energy range of 4.91–6.14 eV, 4.96–5.95 eV, 4.70–5.70 eV and 4.55–5.38 eV for the 279 nm (a), 269 nm (b), 254 nm (c) and 243 nm (d) data respectively. The error bars are derived from a bootstrapping analysis of the data and mark the 95% confidence intervals. The time-dependence of the integrated photoelectron intensity in these regions were fitted to a sum of exponentially decaying functions convolved with a Gaussian instrument response function appropriate for the pump wavelength used. The equation used had the form:
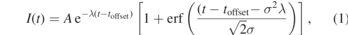
where I(t) is the time-dependent intensity, λ is the decay rate constant and σ is the pump–probe cross-correlation width equivalent to FWHM . toffset is a temporal offset that defines the delay in reaching a maximum intensity. This is measured relative to the experimental time-zero which is defined as the time at which the pump and probe are temporally overlapped. The results of the fits are given in table 1 with the decay represented by the lifetime (τ = 1/λ) rather than the associated decay constant (λ). For the 279 nm, 269 nm and 243 nm pump wavelengths this only required a single exponential term. However, the 254 nm data required two exponentially decaying terms to obtain a good fit with the second term having a variable time offset, with Δt being the separation between the two terms. This behaviour was previously seen at a similar pump wavelength (255 nm) when measured using an MPI probe. The fits to equation (1) are presented as solid lines in figure 4, with the two components of the fit to the 254 nm data presented along with the total.
. toffset is a temporal offset that defines the delay in reaching a maximum intensity. This is measured relative to the experimental time-zero which is defined as the time at which the pump and probe are temporally overlapped. The results of the fits are given in table 1 with the decay represented by the lifetime (τ = 1/λ) rather than the associated decay constant (λ). For the 279 nm, 269 nm and 243 nm pump wavelengths this only required a single exponential term. However, the 254 nm data required two exponentially decaying terms to obtain a good fit with the second term having a variable time offset, with Δt being the separation between the two terms. This behaviour was previously seen at a similar pump wavelength (255 nm) when measured using an MPI probe. The fits to equation (1) are presented as solid lines in figure 4, with the two components of the fit to the 254 nm data presented along with the total.

Figure 4. The photoelectron intensity profile (black data points) over the  state for 279 nm (a), 269 nm (b), 254 nm (c), 243 nm (d), covering binding energy regions 4.91–6.14 eV, 4.96–5.95 eV, 4.70–5.70 eV and 4.55–5.38 eV respectively. Data error bars mark the 95% confidence interval and are obtained from bootstrapping analysis. The yellow line is the fit to the data. For (a), (b), (d) this is an exponential decay, while for (c) this is two exponential decays with variable time zero. The two decays are also plotted with the blue and red curves. The insets in (b) and (c) are the fits and data for the same binding energy region from the MPI results and are reproduced from reference [29] with permission from the PCCP Owner Societies. The fits use the same model as the one-photon ionization results [29]. The values for the fits are presented in table 1. Reproduced from [29] with permission of The Royal Society of Chemistry.
state for 279 nm (a), 269 nm (b), 254 nm (c), 243 nm (d), covering binding energy regions 4.91–6.14 eV, 4.96–5.95 eV, 4.70–5.70 eV and 4.55–5.38 eV respectively. Data error bars mark the 95% confidence interval and are obtained from bootstrapping analysis. The yellow line is the fit to the data. For (a), (b), (d) this is an exponential decay, while for (c) this is two exponential decays with variable time zero. The two decays are also plotted with the blue and red curves. The insets in (b) and (c) are the fits and data for the same binding energy region from the MPI results and are reproduced from reference [29] with permission from the PCCP Owner Societies. The fits use the same model as the one-photon ionization results [29]. The values for the fits are presented in table 1. Reproduced from [29] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageTable 1. Fit parameters from equation (1) for the observation time constants and delays for the Franck–Condon regions obtained in the XUV and MPI [29] experiments. Fits were over the binding regions 4.91–6.14 eV, 4.96–5.95 eV, 4.70–5.70 eV and 4.55–5.38 eV for the 279 nm (a), 269 nm (b), 254 nm (c) and 243 nm (d) data respectively. The errors are 1 standard deviation and are calculated using bootstrapping analysis.
| Probe-XUV | Probe-MPI [29] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pump wavelength (nm) | FWHM (fs) | τ1 (fs) | τ2 (fs) | Δt (fs) | Pump wave-length (nm) | FWHM (fs) | τ1 (fs) | τ2 (fs) | Δt (fs) |
| 279 | 87 ± 9 | <25 | — | — | — | — | — | — | — |
| 269 | 60 ± 8 | 23 ± 6 | — | — | 269 | 64 | 32 | — | — |
| 254 | 77 ± 27 | 52 ± 23 | <20 | 114 ± 38 | 255 | 94 | 40 | 12 | 97 |
| 243 | 57 ± 20 | 46 ± 14 | — | — | — | — | — | — | — |
The intensity profiles for the 279 nm, 269 nm and 243 nm data sets all show a rapid decay in intensity. Fitting the data from each pump wavelength to equation (1) then provides a lifetime equivalent to the time taken for the excited state wavepacket to leave the observation window associated with the FC region of the excited state potential energy surface. From an initial inspection of both figures 3 and 4 the 279 nm data appears longer lived. This appearance is however misleading, the fits to the data show that the apparent extended observation time is due to the much longer pulse duration of the 279 nm pump relative to that of the 269 nm and 243 nm pump as shown in table 1. The relatively long instrument response at 279 nm means we cannot reliably extract a time-constant associated with population leaving the FC region. We obtain our most satisfactory fit with a simple Gaussian envelope which places an upper limit on the lifetime of approximately 25 fs, suggesting very rapid motion out of the FC region. This picture is consistent with that observed at a pump wavelength of 269 nm. The shorter pulse duration at 269 nm means we can extract a lifetime of 23 ± 6 fs utilising the XUV probe which is in good agreement with that previously obtained with a MPI probe (shown as an inset) of 32 fs.
The equivalent 243 nm pump data is plotted in figure 4(d). Fitting to equation (1) provides a characteristic observation lifetime of 46 ± 14 fs which, while slightly longer than those obtained for the 279 nm and 269 nm pump wavelengths, suggests the excited state population rapidly dissociates at this higher pump wavelength as well. While each of these three traces show slight differences in their observation lifetime, they are all consistent with a rapid reduction in measured population that can be accurately described by a simple exponential decay of population out of the FC region. This suggests that the dynamics are broadly similar at each pump wavelength.
The 254 nm data shown in figure 3(c) has a strikingly different appearance. As with the other pump wavelengths, we plot the integrated intensity over the initial excited state features associated with ionization from the FC geometry covering the binding energy range 4.7–5.7 eV, in figure 4(c). This data shows a significant increase in the observation time of the initial excited state features, with significant intensity observed out to over 150 fs. In this case we cannot obtain a satisfactory fit to the data using a single exponentially decaying function. In the previous MPI work at 255 nm, a satisfactory fit was obtained using an empirical fitting function consisting of a sum of two exponentially decaying functions, where one of the two components is delayed relative to time zero. It should be stressed that this provides a purely empirical measure of the intensity profile and care should be taken in trying to extract mechanistic information from the numbers alone. The numbers simply serve to highlight the extended period over which significant signal is maintained and to highlight the difference between this energy region and the others. We use a similar empirical approach here and use the same function to fit the measured data as in the MPI experiments to aid comparison. The results from the MPI experiment are also plotted as an inset in figure 4(c), with the fits in both plotted as solid lines with the associated parameters given in table 1. Comparing the two data sets we see that both show a similar temporal structure but with a much higher intensity delayed component in the MPI measurements than observed in the XUV experiments. The fits to the two data sets provide very similar results for the delay between the two components and their associated lifetimes but with quite different relative amplitudes.
To further compare the MPI and XUV ionization photoelectron spectroscopy measurements around 255 nm, we plot the time-dependent data over the FC energy region that both probes measure in figures 5(a) and (b) respectively. In both data sets the time step used is the same but the MPI data has a significantly higher ultimate energy resolution as defined by the detector pixel spacing for the MPI data while the XUV data resolution is defined by the TOF binning size. The MPI resolution is 0.005 eV as opposed to 0.1 eV for the XUV measurements predominately due to the much lower electron kinetic energies measured, ∼1 eV and ∼17 eV in the MPI and XUV measurements respectively. Despite the difference in energy resolution there are a number of key similarities. In both cases, significant intensity is observed out to almost 200 fs, significantly later than any of the other pump wavelengths. The MPI data shows an initial energy shift to a slightly lower binding energy. The shift of approximately 0.04 eV is smaller than the energy resolution of the XUV data; however, as the pulse has a finite energy width we do see a shift in the centre of the photoelectron peaks in the XUV data to lower binding energies that matches the observations in the MPI data. In the MPI measurement we also observe a long lived feature at 5.5 eV which is highlighted by the white dashed boxes in figure 5. This feature was assigned to  with ionization through a resonant Rydberg state. Although the XUV data does not have the energy resolution to separate this feature from the
with ionization through a resonant Rydberg state. Although the XUV data does not have the energy resolution to separate this feature from the  peak we do not observe any unexpected enhancement of the peak, supporting our original assignment of the feature at 5.5 eV. The resonant enhancement of this ionization process is most likely why the delayed feature appears much stronger in the MPI data than in the XUV data. The similarity in appearance of the photoelectron spectrum and obtained lifetimes from both the MPI and XUV measurements suggest that the extended excited state lifetime observed here is not due to any accidental resonance as previously suggested [19], but is a consequence of the excited states dynamics of methyl iodide in the A-band. The accidental resonance appears to have enhanced the relative intensity of the long lived component in the MPI measurement but its origin is due to some extended excited state dynamics of the system.
peak we do not observe any unexpected enhancement of the peak, supporting our original assignment of the feature at 5.5 eV. The resonant enhancement of this ionization process is most likely why the delayed feature appears much stronger in the MPI data than in the XUV data. The similarity in appearance of the photoelectron spectrum and obtained lifetimes from both the MPI and XUV measurements suggest that the extended excited state lifetime observed here is not due to any accidental resonance as previously suggested [19], but is a consequence of the excited states dynamics of methyl iodide in the A-band. The accidental resonance appears to have enhanced the relative intensity of the long lived component in the MPI measurement but its origin is due to some extended excited state dynamics of the system.

Figure 5. Time-dependent photoelectron spectrum of CH3I following excitation at 255 nm and probed using a multiphoton ionization (a), and at 254 nm and probed using one-photon XUV ionization (b). The dashed white boxed indicates the peak that has previously been assigned to ionization through a Rydberg state. Each spectrum is normalized to the highest intensity feature in each measurement. The data in (a) has previously been published. Reproduced from [29] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution image3.2. Longer term dynamics
To analyse the longer term dynamics of the 279 nm, 254 nm and 243 nm data we follow a similar method to that outlined in Warne et al [30] and plot the integrated photoelectron intensity across a number of other energy regions as a function of pump probe delay, figure 6. The energy regions are selected to provide a series of discrete energy bands below the onset of the ground state spectrum (approx. 9.54 eV) where we can observe the changing intensity profile with time. From the bottom to top of figure 6 the binding energy of the corresponding spectral region plotted increases. The bottom trace in each plot, labelled with an  , is equivalent to that plotted in figure 4 and corresponds to ionization into the
, is equivalent to that plotted in figure 4 and corresponds to ionization into the  state of the methyl iodide cation from the FC region. The second row corresponds to the binding energy region between that covered in
state of the methyl iodide cation from the FC region. The second row corresponds to the binding energy region between that covered in  and the initial energy associated with ionization into the quartet state. The third trace, labelled with a Q, covers the energies associated with ionization into the quartet state from FC geometries, while the fourth plot in the 254 nm and 243 nm data covers the energy region between that associated with the quartet state and the onset of the ground state features. This is absent from the 279 nm data due to a lack of separation between the initial quartet and ground state peaks. The solid lines plotted for the
and the initial energy associated with ionization into the quartet state. The third trace, labelled with a Q, covers the energies associated with ionization into the quartet state from FC geometries, while the fourth plot in the 254 nm and 243 nm data covers the energy region between that associated with the quartet state and the onset of the ground state features. This is absent from the 279 nm data due to a lack of separation between the initial quartet and ground state peaks. The solid lines plotted for the  state region in the 243 nm, 254 nm and 279 nm are the same as those described above for figure 4. For all of the other energy regions the solid lines are fits of the experimental data to equation (1). The scatter of the data at these higher binding energies means that the fits are used more as a qualitative guide to the eye rather than for the extraction of quantitative information from the absolute values obtained. This means that we cannot extract accurate time-dependent shifts for the new data, as we have previously demonstrated on the 269 nm pump data, [29] in order to extract approximate C–I bond lengths. The data at 279 nm and 243 nm however show the same time-dependent shift towards higher binding energy as a function of pump probe delay as seen at 269 nm. The intensity at binding energies between those associated with ionization into the
state region in the 243 nm, 254 nm and 279 nm are the same as those described above for figure 4. For all of the other energy regions the solid lines are fits of the experimental data to equation (1). The scatter of the data at these higher binding energies means that the fits are used more as a qualitative guide to the eye rather than for the extraction of quantitative information from the absolute values obtained. This means that we cannot extract accurate time-dependent shifts for the new data, as we have previously demonstrated on the 269 nm pump data, [29] in order to extract approximate C–I bond lengths. The data at 279 nm and 243 nm however show the same time-dependent shift towards higher binding energy as a function of pump probe delay as seen at 269 nm. The intensity at binding energies between those associated with ionization into the  and quartet states peak at a later time which is indicative of the increasing C–I bond length as the molecule dissociates. The intensity of the quartet state signal masks the continued shift to some extent but in the 243 nm data the continued shift can be seen at the highest binding energy region plotted. The data at 243 nm and 279 nm therefore provides a consistent picture of the rapid dissociation along the C–I with what appears to be a slightly longer lifetime observed at 243 nm.
and quartet states peak at a later time which is indicative of the increasing C–I bond length as the molecule dissociates. The intensity of the quartet state signal masks the continued shift to some extent but in the 243 nm data the continued shift can be seen at the highest binding energy region plotted. The data at 243 nm and 279 nm therefore provides a consistent picture of the rapid dissociation along the C–I with what appears to be a slightly longer lifetime observed at 243 nm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Integrated photoelectron intensity over a number of binding energy regions as labelled for pump wavelengths of 279 nm (a), 254 nm (b) and 243 nm (c). The error bars mark the 95% confidence interval. Each region has been normalized to the highest intensity feature. The bottom traces are over the FC region and are the same as those in figure 4. All other regions have been fitted to a single exponential decay. The second row is over the binding energy just below the appearance of the quartet state. The third row is over the quartet state and the fourth row is just below the ground state features.
Download figure:
Standard image High-resolution image{kind=link}
The data measured at a pump wavelength of 254 nm shows a much longer excited state observation lifetime across each energy region considered. This is most clearly shown in the comparison of the highest energy region covered in the 243 nm and 254 nm data. The 243 nm data at 8–8.6 eV binding energy shows a very sharp peak that reaches a maximum around 50 fs after time zero and decays back to the baseline level within a further 50–60 fs. The equivalent region in the 254 nm data shows a similar delayed increase reaching a maximum intensity at a similar delay to that seen at 243 nm, but in this case it does not return to the baseline level for approximately another 150 fs. Similar increases in the observation time at 254 nm, relative to the other wavelengths measured, are observed in the other binding energy regions plotted suggesting more complex dynamics in the excited state before dissociation. We stress again that the fits here are empirical and, while there are hints of signal related to peaks from multiple components in the time-dependent traces these cannot be asserted with any confidence within the current data set. Current signal levels are limited by a combination of the high-harmonic flux and the gas density at the interaction region.
4. Discussion
Having characterized the excited state dynamics across a range of pump wavelengths, we now compare these to the recent time-resolved measurements of product formation with a particular focus on the recent work of Murillo-Sánchez et al who measured quantum state-resolved appearance times at pump wavelengths of 269 nm and 243 nm.
4.1. 269 nm and 279 nm
Numerous measurements at wavelengths close to 266 nm have shown that the dominant dissociation product channel forms I*  in conjunction with the CH3 co-fragment in its vibrational ground state. Previous product formation experiments measured the appearance time (as defined by the time taken for the C–I bond to reach a length of 13 Bohr) of this dominant fragment was recently measured to be around 114 fs. A shorter appearance time, of approximately 75 fs, was also observed for the next most abundant peak which corresponds to formation of the ground state
in conjunction with the CH3 co-fragment in its vibrational ground state. Previous product formation experiments measured the appearance time (as defined by the time taken for the C–I bond to reach a length of 13 Bohr) of this dominant fragment was recently measured to be around 114 fs. A shorter appearance time, of approximately 75 fs, was also observed for the next most abundant peak which corresponds to formation of the ground state  in conjunction with the CH3 co-fragment in its vibrational ground state. Interestingly, the measurements showed that the product channels associated with the vibrationally excited umbrella mode in CH3 fragments have shorter appearance time than their vibrationless equivalent when produced in conjunction with the excited I*
in conjunction with the CH3 co-fragment in its vibrational ground state. Interestingly, the measurements showed that the product channels associated with the vibrationally excited umbrella mode in CH3 fragments have shorter appearance time than their vibrationless equivalent when produced in conjunction with the excited I*  co-fragment, and longer appearance times when associated with the ground
co-fragment, and longer appearance times when associated with the ground  co-fragment. The shorter appearance times (84 fs for the ν2 = 1 channel) observed for the vibrationally excited fragments in the I*
co-fragment. The shorter appearance times (84 fs for the ν2 = 1 channel) observed for the vibrationally excited fragments in the I*  channel is difficult to explain and is at odds with the modelling performed with both 4D and 9D potential surfaces which consistently show very similar or slightly longer lifetimes for the various vibrational states covered [19]. Our experiments show it takes ∼25 fs for the wavepacket to leave the initial FC region. This gives the wavepacket 50–90 fs to travel about 4.4 Å to be consistent with the product formation experiments. Although timescales measured are not directly comparable, they are, however, consistent, with both showing rapid dissociation. Due to the fact we cannot quantum state-resolve the individual product channels or the vibrational distributions within them, our measurements will be a weighted average of the individual contributions to the lifetime convolved with the laser cross-correlation duration.
channel is difficult to explain and is at odds with the modelling performed with both 4D and 9D potential surfaces which consistently show very similar or slightly longer lifetimes for the various vibrational states covered [19]. Our experiments show it takes ∼25 fs for the wavepacket to leave the initial FC region. This gives the wavepacket 50–90 fs to travel about 4.4 Å to be consistent with the product formation experiments. Although timescales measured are not directly comparable, they are, however, consistent, with both showing rapid dissociation. Due to the fact we cannot quantum state-resolve the individual product channels or the vibrational distributions within them, our measurements will be a weighted average of the individual contributions to the lifetime convolved with the laser cross-correlation duration.
4.2. 243 nm
Other experiments have shown that at the shorter wavelength of 243 nm the I*  in conjunction with the CH3 co-fragment in its vibrational ground state continues to be the dominant product channel. The appearance time was recently measured to be shorter than that observed at 266 nm standing at approximately 83 fs and there is a commensurate reduction in the appearance time (53 fs) of the equivalent
in conjunction with the CH3 co-fragment in its vibrational ground state continues to be the dominant product channel. The appearance time was recently measured to be shorter than that observed at 266 nm standing at approximately 83 fs and there is a commensurate reduction in the appearance time (53 fs) of the equivalent  channel [19]. This is perhaps unsurprising given the extra kinetic energy available upon dissociation at this higher energy but is contrary to the extended excited state lifetime observed in our photoelectron spectroscopy experiments. The product state measurements also show that vibrationally excited fragments are produced with higher yield at 243 nm and the appearance times increase with the level of vibrational excitation observed. This is more in line with the supporting simulations, however, the magnitude of the observed increase is much larger than those calculated. The vibrationally excited fragments typically appear within 10 fs of the vibrationless product in the calculation while experimental measurements suggest the delay can be as much as 50–60 fs for some fragments [19]. We suggest that the higher prevalence of the vibrationally excited products and their increased lifetimes would result in an effective increase in the excited state lifetime observed in our photoelectron spectroscopy measurements explaining why the measured lifetime was ∼20 fs longer than lifetime measured for 269/279 nm. The increased lifetime therefore reflects the different vibrational dynamics associated with the various dissociation channels. This discussion highlights clear issues around comparing time frames obtained from various measurement techniques. The selectivity provided by REMPI ionization and product fragment recoil detection, allows measurement of product fragment appearance times with full vibrational and spin–orbit state-resolution but is limited in only measuring the dynamics of the system at the end of the dynamic process. In contrast the photoelectron spectroscopy probes provide a more general probe of the system over all geometries but this is necessarily averaged over all quantum states. A combination of all techniques will clearly be required to disentangle the dynamics of systems where multiple product states are populated.
channel [19]. This is perhaps unsurprising given the extra kinetic energy available upon dissociation at this higher energy but is contrary to the extended excited state lifetime observed in our photoelectron spectroscopy experiments. The product state measurements also show that vibrationally excited fragments are produced with higher yield at 243 nm and the appearance times increase with the level of vibrational excitation observed. This is more in line with the supporting simulations, however, the magnitude of the observed increase is much larger than those calculated. The vibrationally excited fragments typically appear within 10 fs of the vibrationless product in the calculation while experimental measurements suggest the delay can be as much as 50–60 fs for some fragments [19]. We suggest that the higher prevalence of the vibrationally excited products and their increased lifetimes would result in an effective increase in the excited state lifetime observed in our photoelectron spectroscopy measurements explaining why the measured lifetime was ∼20 fs longer than lifetime measured for 269/279 nm. The increased lifetime therefore reflects the different vibrational dynamics associated with the various dissociation channels. This discussion highlights clear issues around comparing time frames obtained from various measurement techniques. The selectivity provided by REMPI ionization and product fragment recoil detection, allows measurement of product fragment appearance times with full vibrational and spin–orbit state-resolution but is limited in only measuring the dynamics of the system at the end of the dynamic process. In contrast the photoelectron spectroscopy probes provide a more general probe of the system over all geometries but this is necessarily averaged over all quantum states. A combination of all techniques will clearly be required to disentangle the dynamics of systems where multiple product states are populated.
4.3. 254 nm
As far as we are aware, there are no time-resolved product state measurements at wavelengths close to 254 nm which makes comparison difficult. In the work of Murillo-Sánchez et al [19], it was suggested that the extended lifetime observed in the MPI photoelectron spectroscopy measurements could be due to accidental resonances in the ionization step. Our one-photon measurements presented here rule this suggestion out. The accidental resonances appear to have enhanced the amplitude of the longer lived features when compared with the one-photon measurements but the extended lifetime measured, and the unusual profile of the excited state decay transient, persists when this effect is removed. The authors also suggested that the enhanced lifetime may be due to transfer of kinetic energy into product vibrational modes, as seen at 243 nm, as opposed to an enhanced contribution of the 1
Q1 state as originally suggested in reference [29]. Both of these options are possible. We observe the extension of the lifetime due to vibrational dynamics in the 243 nm measurements but the appearance of the time-delay transient at 243 nm is strikingly different to that measured at 254 nm. If a new region of the 3
Q0 potential energy surface became accessible at 254 nm where FC overlap with the methyl cation  state were reduced, this may explain the time-dependent modulation of the ionization cross section observed in this measurement. However, we would also expect to continue to see the effect of this on the shape of the delay transients at shorter wavelengths, yet there is no evidence of this in the 243 nm measurement. We also expected some vibrational excitation of the room temperature ensemble of methyl iodide, with about 7% in the ν3 vibration (C–I stretch) and 1% in the ν6 vibration (CH3 rock). This may have an effect on the observed photoelectron spectrum but why this would only be observed at 254 nm is not clear. Therefore, while we cannot rule out the possibility that the extended lifetime is due to vibrational dynamics, the stark difference in the appearance of the delay transient at 254 nm makes us lean towards an alternative theory, such as enhanced population of the 1
Q1 state at this wavelength, followed by geometric relaxation on the 1
Q1 surface, for the changes in the photoelectron spectrum. What is clear is that more measurements are required and we hope that our measurements encourage directly comparable measurements around 254 nm to allow a direct comparison of the quantum state distributions obtained with the excited state dynamics measured.
state were reduced, this may explain the time-dependent modulation of the ionization cross section observed in this measurement. However, we would also expect to continue to see the effect of this on the shape of the delay transients at shorter wavelengths, yet there is no evidence of this in the 243 nm measurement. We also expected some vibrational excitation of the room temperature ensemble of methyl iodide, with about 7% in the ν3 vibration (C–I stretch) and 1% in the ν6 vibration (CH3 rock). This may have an effect on the observed photoelectron spectrum but why this would only be observed at 254 nm is not clear. Therefore, while we cannot rule out the possibility that the extended lifetime is due to vibrational dynamics, the stark difference in the appearance of the delay transient at 254 nm makes us lean towards an alternative theory, such as enhanced population of the 1
Q1 state at this wavelength, followed by geometric relaxation on the 1
Q1 surface, for the changes in the photoelectron spectrum. What is clear is that more measurements are required and we hope that our measurements encourage directly comparable measurements around 254 nm to allow a direct comparison of the quantum state distributions obtained with the excited state dynamics measured.
5. Summary
We have presented new experimental measurements on the photodissociation dynamics of CH3I using a 22.6 eV probe in a time-resolved photoelectron spectroscopy measurement. The experiments have been performed at a series of pump wavelengths across the A-band absorption profile covering both the red and blue sides of the absorption maximum. The analysis of the resulting measurements has been compared with previously published MPI photoelectron spectroscopy measurements of the excited state population and vibronic state-resolved measurements of the products. At pump wavelengths of 243 nm, 269 nm and 279 nm, the results are consistent with rapid dissociation along the C–I bond while the 254 nm data shows a more complex dynamic profile and an extended excited state lifetime with a secondary feature appearing after 100 fs. These new one-photon ionization measurements are consistent with the results obtained with the MPI probe and with the results of product state measurements at wavelengths where direct comparisons are available.
The extended excited state lifetime with a secondary feature appearing after 100 fs is observed at pump wavelengths around 255 nm is present in both one-photon and MPI experiments. It had previously been suggested that the extended lifetime could be due to an accidental resonance occurring at some extended internuclear separation. The similarity in appearance and observed lifetime between the single and MPI measurements means that we can now confidently rule out the effect of accidental resonances on this extended lifetime. The use of a higher-energy single-photon probe also allows us to follow the excited state population away from the initial FC region. Again, the 254 nm data shows significant differences from the other pump wavelengths with an extended observation lifetime at higher binding energies, and an unusual excited state decay profile, indicative of more complex dynamics of dissociation. The origin of these dynamics is difficult to identify, particularly with no complementary time-resolved and vibronically state-resolved measurements of the products close to this wavelength. Quantum resolved final state product measurements similar to those performed by Bañares and co-workers [10, 19] but close to 254 nm could provide helpful insights into the dynamics at this wavelength. Improved potentials that can quantitatively reproduce the absorption spectrum could provide information on the role of the 1 Q1 state.
Acknowledgments
RSM thanks the Royal Society for a University Research Fellowship (UF100047, UF150655). RSM and JW thank the EPSRC for funding (EP/R010609/1). BDW thanks the Central Laser Facility and Chemistry at the University of Southampton for a studentship. EMW thanks Chemistry at the University of Southampton for a studentship. Access to Artemis at the Central Laser Facility was provided by the STFC. We thank John Dyke for very useful conversations and Phil Rice for technical assistance.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.