

Abstract
The problem of atoms and molecules caged inside fullerenes has attracted renewed interests since a new endohedral species has been experimentally realized (Bloodworth et al 2019 Angew. Chem., Int. Ed. 58 5038). In this sense, detailed theoretical studies on the spectroscopic properties of atoms and ions spatially confined in fullerene-like structures are convenient. Here we perform density functional theory (DFT) and time-dependent DFT (TDDFT) calculations to investigate the electronic, vibrational and optical properties of two-electron atomic systems, X, caged in C20 and C20H20 endohedral complexes; i.e. X@C20 and X@C20H20 (X = He, Li+, and Be++). Among these endohedral complexes, only the encapsulated Be++ ion gives rise to strongly bound complexes, whereas the encapsulated Li+ ion depends on the confining environment, and the encapsulated He atom seems to be highly repulsive in both types of cages. Our calculated excitation energies indicate that the lowest-lying singlet states strongly depend on both the nature of the endohedral atom/ion and the type of the carbon cage. Although He@C20H20 and He@C20 are obtained as repulsive complexes, they produce a small effect in the absorption spectra of the complexes. However, the presence of Li+ or Be++ in the endohedral complexes dramatically changes the electronic absorption profile of these cages. Overall, this study shows that the confinement of a Be++ ion in a very restricted space is energetically favorable, being its quantum states controllable by the confining environment.
Export citation and abstract BibTeX RIS
1. Introduction
More than thirty years have passed since the publication of the celebrated papers of Kroto et al [1, 2] and Kratschmer et al [3], with the discovery of C60 and also its synthesis in macroscopic quantities. Even after so long time, fullerene-like molecules and their derivatives still attract great interest in basic and applied research [4–16]. Most of this interest is mainly because of their fascinating properties and the number of application in different fields. There are applications, for example, in material physics [4–6], quantum information [7–9], biophysics and medicine [10–12], nanophotonics, and optoelectronics [13–15]. Recent studies have also suggested their use in photophysics, superconductivity, development of solar cells and other electronic devices [6, 16].
Because of the cage-like structure of fullerenes, the major challenge is to discover the feasibility of hosting atoms, ions and small molecules [17–21] inside. Resulting systems, known as endohedral fullerenes, define a new class of materials [22] and have increased the range of applications [23, 24], other related issue is that fullerene-like structures formed by different atoms of carbon have been proposed and investigated. Examples of these systems are the cage structures formed by boron [25, 26], bismuth [27–29], silicon [30–32], and bromine [33]. For these reasons, it is important to have a better understanding of the physical and chemical properties of X@Cn systems, being X the guest in the system.
As it is known, small Cn fullerenes with n ⩽ 40 are less stable than compounds with n ⩾ 60, since the pentagon rule is violated [34, 35]. However, the confinement of atoms or ions inside them can be a way of stabilizing such systems [36]. Other possibility of stabilizing these carbon clusters is to saturate the external structure with hydrogen or other appropriate atoms [37, 38].
In the carbon fullerene family, the smallest structure obtained is C20 [37, 39], with an approximately 4 Å diameter cavity. Due to its curvature and equilibrium geometry formed by 12 pentagons, in which each carbon atom makes three simple bonds with its neighbors, C20 is highly reactive and difficult to synthesize. Nonetheless, a possible stable related structure, which can be obtained as a step of the synthesis of C20, is the C20H20. Indeed, this structure has been extensively explored from both experimental and theoretical point of view [37, 40–43].
From the electronic point of view, many theoretical studies have been carried out to investigate the properties of endohedral C20H20 and also other related small cages [44–46]. In C20 case, the interaction with hydrogen and transition metal atoms seems to be a topic of most interest [23, 47]. In this context, recently, Salem [48] reported a study where the electronic properties of this molecule doped with H, F, and Cl atoms where examined. In general, C20 has been employed as the confined system in larger structures [49]. Such an application is interesting since endohedral C20 could be employed as a tunable doping of other carbon materials [50].
In order to understand the effects of spatial confinement of atoms and ions encapsulated in small carbon cages (C20 and C20H20) on its properties, of the encapsulated atoms and ions, and also on the properties of the whole systems, we have carried out a theoretical study based on density functional theory (DFT) [51] and time-dependent DFT (TDDFT) [52]. Here, we have investigated the cases of the X@C20 and X@C20H20 systems (X = He, Li+, and Be++). Within this prescription, we analyze their geometric features, electronic structure, vibrational spectra, and optical properties.
2. Computational details
We have employed computational procedures based on DFT [53] and TDDFT [52] to carry out the electronic structure calculations, as implemented in GAMESS-US Package [54]. The systems considered here are C20, C20H20, X@C20, and X@C20H20, with X = He, Li+, and Be++. Following the literature related to this issue [31, 45, 55], we adopt the hybrid functional B3LYP [56, 57], combined with the 6-31G(d, p) basis set to obtain the optimized structures of all considered systems in this study. We have avoided the application of symmetry constraints in order to find the minimum energy of each structure.
For the endohedral systems, we have initially considered the encapsulated X atom located in the center of the cages. After the relaxation process to obtain the equilibrium geometries, we have calculated their vibrational frequencies within the harmonic approximation. With this approach, we evaluate the structural and dynamic stability of each compound. We have also evaluated the sphericity parameter (SP) [58] of the resulting structures, which is defined as
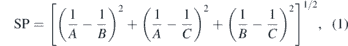
where A, B and C are the rotational constants (in GHz). For this parameter the structure is less spherically perfect if the SP exhibits a high value.
The inclusion energy of an atom/ion X inside the fullerene is calculated as the difference between the total energy of the endohedral system (Eendo) and the sum of the total energies of the pure cages (Ecage) and an isolated atom/ion (EX), i.e.
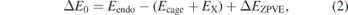
which is corrected for the zero-point vibrational energies (ZPVE) of the systems. Moreover, the basis set superposition error (BSSE) has been computed using the counterpoise method [59]. Another interesting energy parameter to be analyzed is the deformation energy, defined as:
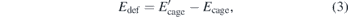
where E'cage is obtained with a single point calculation of energy of the pure cage, with the optimized geometry of the endohedral system, while Ecage is calculated with the pure cage optimized geometry.
Excited state properties of all these systems are also calculated using a time-dependent version of the B3LYP functional, implemented in GAMESS-US package [54, 60]. Thus, the excitation energies (ΔE) are calculated using the same basis set and functional, which is consistent from our methodological procedure.
For these systems we used the equilibrium geometries obtained, and calculated the excitations using the TDDFT/B3LYP/6-31G(d, p) method with the linear response approximation. To analyze transitions and absorptions, we have used the programs GaussSum [61] and QMForge [62]. An important quantity that we determined was the oscillator strength (f) which is directly related to the absorptions and intense transitions whose value is around 1.
The convergence criteria used in DFT and TDDFT calculations follow the standard implemented in GAMESS-US package. In this way, the precision of our results is in accordance with the precision of B3LYP exchange functional, which is widely used in the literature.
3. Results and discussion
3.1. Structural parameters and energetic characteristics
The fully optimized geometries of the considered systems at the B3LYP/6-31G(d, p) level of calculation are displayed in figure 1. As visually discernible, not all endohedral fullerenes contain the atom/ion in the center. This is clear in the cases of Be++@C20, Be++@C20H20 and Li+@C20H20, for example. The calculated equilibrium distances, rC–C, rC–X and rC–H, and also the average diameters,  , of all these structures are reported in table 1, where we indicate the smallest and largest (in parentheses) distances calculated. Additional informations about the rotational constants and the SP values, obtained from equation (1), are given in table 2.
, of all these structures are reported in table 1, where we indicate the smallest and largest (in parentheses) distances calculated. Additional informations about the rotational constants and the SP values, obtained from equation (1), are given in table 2.
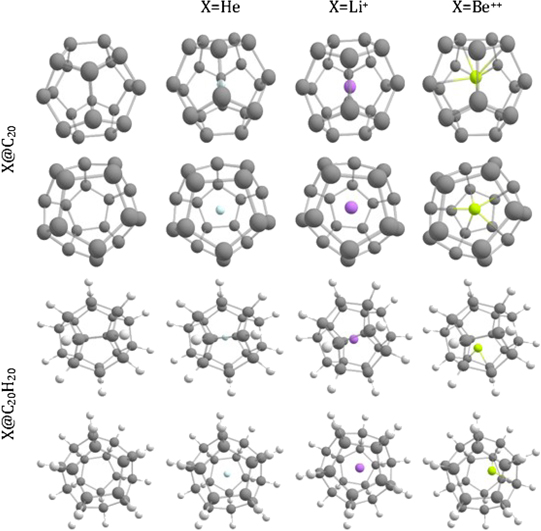
Figure 1. Optimized structures of the X@C20 and X@C20H20 systems.
Download figure:
Standard image High-resolution imageTable 1. Calculated distances between atoms and cage average diameters (in Å) for the optimized geometries of the systems, at the B3LYP/6-31G(d, p) level of theory. Our results are compared with those obtained by Zhang et al [63]a and Moran et al [45]b .
| Systems |
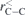 ( (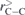 ) ) |
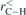 ( ( ) ) |
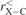 ( (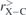 ) ) |
|
|---|---|---|---|---|
| C20 | 1.400 (1.536) | — | — | 4.06 |
| 1.41(1.52)a | — | |||
| He@C20 | 1.408 (1.535) | — | 1.984 (2.105) | 4.09 |
| 1.41 (1.53)a | — | — | ||
| Li+@C20 | 1.409 (1.533) | — | 1.964 (2.146) | 4.09 |
| Be++@C20 | 1.360 (1.398) | — | 1.799 (2.434) | 4.08 |
| C20H20 | 1.556 | 1.094 | — | 4.36 |
| 1.555b | 1.092b | — | ||
| He@C20H20 | 1.564 | 1.094 | 2.192 | 4.43 |
| 1.563b | 1.092b | 2.190b | ||
| Li+@C20H20 | 1.556 (1.580) | 1.094 (1.105) | 2.064 (2.347) | 4.42 |
| 1.570b | 1.087b | 2.200b | ||
| Be++@C20H20 | 1.563 (1.640) | 1.084 (1.090) | 1.722 (2.180) | 4.44 |
| 1.571b | 1.087b | 2.202b |
Table 2. Calculated rotational constants A, B and C (in GHz), moment of inertia IA , IB and IC (in a.u.), and sphericity parameter (SP) (in GHz−1) of the systems at the B3LYP/6-31G(d, p) level of theory.
| Systems | A | B | C | IA | IB | IC | SP |
|---|---|---|---|---|---|---|---|
| C20 | 0.782 | 0.775 | 0.730 | 2306.399 | 2326.643 | 2471.487 | 0.1215 |
| He@C20 | 0.767 | 0.763 | 0.724 | 2350.585 | 2364.507 | 2488.953 | 0.1050 |
| Li+@C20 | 0.764 | 0.757 | 0.720 | 2360.862 | 2381.368 | 2505.638 | 0.1056 |
| Be++@C20 | 0.763 | 0.754 | 0.713 | 2362.994 | 2392.575 | 2529.344 | 0.1205 |
| C20H20 | 0.558 | 0.558 | 0.558 | 3231.159 | 3231.493 | 3231.807 | 0.0000 |
| He@C20H20 | 0.553 | 0.553 | 0.553 | 3262.063 | 3262.710 | 3263.390 | 0.0000 |
| Li+@C20H20 | 0.549 | 0.548 | 0.547 | 3284.334 | 3291.609 | 3299.032 | 0.0082 |
| Be++@C20H20 | 0.541 | 0.540 | 0.539 | 3331.062 | 3341.402 | 3342.744 | 0.0084 |
We observe that our calculated values of rC–C for C20 and of rC–C and rC–H for C20H20 are in good agreement with those reported in the literature [45, 63]. In order to determine the average diameters of the cavities, we have determined the distance between two diametrically opposite carbon atoms. So, our calculated values are  Å and
Å and  Å, which are also comparable to the values determined by others [45, 63]. Notice that these values are higher than the van der Waals radii of the atoms and also their ionic radii, which indicate that some atoms/ions can be favorably encapsulated in these cavities. In C20 case, our calculated
Å, which are also comparable to the values determined by others [45, 63]. Notice that these values are higher than the van der Waals radii of the atoms and also their ionic radii, which indicate that some atoms/ions can be favorably encapsulated in these cavities. In C20 case, our calculated  exhibits an interesting decreasing order, i.e.
exhibits an interesting decreasing order, i.e.  , which is consistent with the number of electrons and ionization state in each guest atom. However, the same decreasing order does not hold for C20H20, and that is, in this case,
, which is consistent with the number of electrons and ionization state in each guest atom. However, the same decreasing order does not hold for C20H20, and that is, in this case,  .
.
Comparing the values of the smallest and largest distances between two neighbor atoms in C20 and C20H20, it is noticed that in C20 the difference reaches 13.6%, whereas in C20H20 the difference is only of 0.1%. As a consequence, C20 exhibits a less symmetrical structure than C20H20, which can be verified by the calculated SP values. We obtain 0.1215 and 0.000 GHz−1 for C20 and C20H20, respectively (see table 2).
The confinement of atoms/ions X = He, Li+, and Be++ in the C20 and C20H20 cavities is performed by geometry optimization of the centered complexes without symmetry constraints. In this case, when He atom is placed in the center of the carbon cages, we notice a small increase of 0.8% in the rC–C distances, in both cases, whereas the rH–C distances exhibit a small decrease of around 0.1%, in the case of He@C20H20. The increase of the rC–C distances leads to an increase of the average diameter of the cavities, while the He atom remains in the center. As it can be seen in table 2, the presence of the He atom does not alter the symmetry of C20H20, while in C20 there is a decrease in the value of SP, i.e. SP(He@C20) < SP(C20).
For confined Li+ and Be++ cases, the ions in the relaxed complexes exhibit off-center positions, and Be++ is more off-center in both types of cages. Moreover, although these ions are both two-electron systems, they deform more the cages than the He atom, what leads to a significantly less spherical structure in the case of X@C20H20, X = Li+ and Be++. Considering the endohedral systems, X@C20 and X@C20H20, the behavior in the SP values are similar, i.e. there is an increase in SP values. However, when comparing with pure systems, we noticed that SP(C20) > SP (X@C20) while SP (C20H20) < SP (X@C20H20).
At this point, we analyze the dynamic stability and the inclusion energy of the atoms/ions in the cages. Our results are reported in table 3. The calculated vibrational frequencies of these systems indicate that all these structures have found their stationary points as a true minimum. Thus, we report in table 3 only the lowest vibrational frequency (ω1) of each system, while vibrational spectra are discussed later. We realized that the inclusion of an He atom or a Li+ ion in the C20 cavity yields to a systematic blue shift in ω1 of 27 and 72 cm−1 respectively, whereas the inclusion of a Be++ ion yields to a red shift of 5 cm−1. On the other hand, in the case of X@C20H20, only the spatial confinement of an He atom yields a blue shift of 22 cm−1 in this vibrational mode. Now, the inclusion of Li+ and Be++ yields to red shifts of 5 and 295 cm−1. We compare our calculated values with those obtained by Moran et al [45].
Table 3. Calculated total energy (in a.u.), the lowest vibrational frequency (in cm−1), deformation energy (in a.u.), inclusion energy (kcal mol−1), taking into account the BSSE correction, for the systems at the B3LYP/6-31G(d, p) level of theory. Our results are compared with those obtained by Moran et al [45] for  and
and  symmetry.
symmetry.
| Systems | E | ω1 | Edef | ΔE0 | BSSE |
|---|---|---|---|---|---|
| C20 | −760.9984 | 113.6 | — | — | — |
| He@C20 | −763.7859 | 153.8 | 0.0034 | 74.65 | 3.785 |
| Li+@C20 | −768.2332 | 198.5 | 0.0058 | 29.51 | 2.710 |
| Be++@C20 | −774.9618 | 121.7 | 0.0092 | −200.68 | 2.284 |
| C20 H20 | −773.6979 | 479.3 | — | — | |
| −774.3596a | 479.6a | ||||
| He@C20 H20 | −776.5434 | 500.8 | 0.0009 | 37.90 | 2.714 |
| −777.2165a | 502.5a | 37.89a | |||
| Li+@C20 H20 | −781.0029 | 473.9 | 0.0045 | −15.16 | 2.352 |
| −781.6663a | 355.2a | −12.70a | |||
| Be++@C20 H20 | −787.7281 | 184.8 | 0.0178 | −254.50 | 1.527 |
| −788.3684a | 369.8ia | −236.29a | |||
| −788.3871b | 240.0b |
Naturally, systems that can be superimposed on a dodecahedron have Ih
symmetry type, and two good examples are fullerenes C20 and C20H20. However, in the case of endohedral systems, it must be observed whether the confined atom is located on a point or axis of symmetry. Among the systems presented by Moran et al [45], the one that contains encapsulated Be++, has no minimum with Ih
symmetry (imaginary frequency, see table 3). The minimum is obtained considering the symmetry C5v
, with the encapsulated atom located on the axis of rotation, close and centered to one of the C20H20 faces, with distances  . In our calculations, as we do not restrict the symmetry of the system, we have obtained a local minimum with Be++ offset from the center of the cavity, and slightly offset from the center of the nearest face. This difference in methodology leads us to different values for the fundamental frequency ω1, for both Be++@C20H20 and Li+@C20H20.
. In our calculations, as we do not restrict the symmetry of the system, we have obtained a local minimum with Be++ offset from the center of the cavity, and slightly offset from the center of the nearest face. This difference in methodology leads us to different values for the fundamental frequency ω1, for both Be++@C20H20 and Li+@C20H20.
In this context, we must remember that the imposition of symmetry represents a constraint in the variational process and, consequently, an increase in energy [64]. Thus, if we do not impose symmetry constraint on the geometry optimization process, we can obtain a lower energy and, therefore, a better variational value, being necessary, however, to verify if the obtained wave functions describe the spectroscopic states well. Note, however, that when placing the X atom or ion in the cage, the final configuration can change the system symmetry, and in this case, the optimization without the symmetry restriction can give this new configuration. In fact, for C20H20 and He@C20H20 systems cases, we could observe by comparing the values of the first vibration frequency ω1, a good agreement between our values and those obtained by Moran et al [45]; in particular, for the He@C20H20 case, the optimized structure without the symmetry restriction meets the He atom in the center of the cavity, which coincides with the optimized system using the Ih symmetry restriction. For Li+ and Be++ ions, our values differ from those presented by Moran et al [45] precisely because, in our geometry optimization process, these ions are located outside a point or axis of symmetry. It is also important to note that for the results obtained by Moran et al for Be++ using the Ih and C5v symmetry constraints (see table 3), the values of ω1 already differ considerably, and the Ih symmetry constraint does not lead to a global minimum structure.
To evaluate the inclusion energy of the atoms/ions inside the cavities, we have employed equation (2), taking into account both the BSSE effect and the ZPVE correction. From these results, we obtain that He@C20, He@C20H20 and Li+@C20 should not be energetically favorable. In these two series, only Be++@C20, Li+@C20H20, and Be++@C20H20 appear to be favorably formed, since their inclusion energies are, respectively, −200.7, −15.2, and −254.5 kcal mol−1. As we can see, Be++ exhibits the highest inclusion energies in both cavities, with values in the same order of typical ionic bonds.
Using equation (3), we have calculated the deformation energies (see table 3). We observed that in both structures there is an increase in Edef with the atomic number of the inserted atoms/ions. We can highlight that the He atom in C20H20, has the lowest Edef value, indicating a small deformation in the structure, as previously discussed, while Be++ in C20H20 presents the highest deformation energy among the studied systems, indicating a greater deformation in the structure.
3.2. Electronic structure and vibrational spectra
To examine the ground state electronic structure of the studied systems, we analyze first the frontier molecular orbitals energies − the highest occupied (HOMO) and the lowest unoccupied (LUMO) − and also the HOMO–LUMO energy differences ( HL), for each system, as reported in table 4. In addition, as an important parameter to investigate the electronic density of these quasi-spherical systems, we calculate the absolute hardness (η) [65], which can be estimated from these frontier energies and is also reported in table 4.
HL), for each system, as reported in table 4. In addition, as an important parameter to investigate the electronic density of these quasi-spherical systems, we calculate the absolute hardness (η) [65], which can be estimated from these frontier energies and is also reported in table 4.
Table 4. Frontier energies (HOMO, LUMO and HL, in eV) and hardness η (in eV) of the X@C20 and X@C20H20 systems with X = He, Li+ and Be++, at the B3LYP/6-31G(d, p) level of theory.
| Systems | HOMO | LUMO |
HL
| η |
|---|---|---|---|---|
| C20 | −4.97 | −3.02 | 1.95 | 0.98 |
| He@C20 | −4.99 | −3.16 | 1.83 | 0.92 |
| Li+@C20 | −10.00 | −8.22 | 1.78 | 0.89 |
| Be++@C20 | −14.89 | −13.13 | 1.76 | 0.88 |
| C20H20 | −6.99 | 0.98 | 7.97 | 3.99 |
| He@C20H20 | −7.01 | 2.07 | 9.08 | 4.54 |
| Li+@C20H20 | −11.27 | −2.92 | 8.35 | 4.18 |
| Be++@C20H20 | −15.25 | −9.54 | 5.71 | 2.86 |
It is interesting to notice that the inclusion of an He atom in C20 reduces HL by 0.1 eV, while in C20H20 it raises by 1.1 eV. Similarly, Li+ also reduces HL by ∼0.2 eV in C20 and increases by ∼0.4 in C20H20. On the other hand, the presence of Be++ reduces HL by ∼0.2 and ∼2.3 eV in both cages.
In figures 2 and 3, we display the calculated total state density (DOS) for the two series of systems, i.e. X@C20 and X@C20H20. We have presented a comparison of the DOS of the C20 and C20H20 systems (in pink) with the DOS of the compounds X@C20 and X@C20H20 where (a) X = He (in black), (b) X = Li+ (in blue) and (c) X = Be++ (in green). The dotted vertical lines indicate the positions of the HOMO and LUMO orbitals. For the He@C20 system it is noted that the pattern of the DOS graph changes little when compared to the graph of C20: the energy of the HOMO orbital is practically the same as that of C20 (0.4% lower) and the energy of the LUMO orbital shows a ∼4.6% decrease; as a result, we have a HL = 1.83 eV. In the case of the two systems Li+@C20 and Be++@C20 it is observed that there is a displacement of the HOMO and LUMO orbitals for smaller energy values; it is also observed that the value of HL decreases in both systems with HL = 1.78 eV for Li+@C20 and HL = 1.76 eV for Be++@C20. These HL values indicate that the compounds tend to be more reactive than C20.
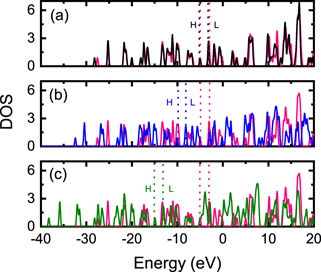
Figure 2. DOS for the X@C20 system, with X = He, Li+ and Be++. The dotted vertical lines represent the positions of the HOMO and LUMO orbitals. The pink curve refers to DOS from C20, (a) black for DOS of He@C20, (b) blue for DOS of Li+@C20 and (c) green for DOS of Be++@C20.
Download figure:
Standard image High-resolution image
Figure 3. DOS for the X@C20H20 system, with X = He, Li+ and Be++. The dotted vertical lines represent the positions of the HOMO and LUMO orbitals. The pink curve refers to DOS from C20H20, (a) black for DOS of He@C20H20, (b) blue for DOS of Li+@C20H20 and (c) green for DOS of Be++@C20H20.
Download figure:
Standard image High-resolution imageWhen we compare C20 and C20H20 we note that HL of C20H20 is four times greater than HL of C20, that is, C20H20 is less reactive than C20; it is also noted that the ionization potential of C20H20 is smaller than the ionization potential of C20 indicating a greater difficulty in donating electrons. For the He@C20H20 system, we have obtained that HL is 13.9% greater than in C20H20 case; this indicates that He@C20H20 is less reactive than C20H20. The energy of the HOMO orbital is practically the same as that of C20H20; the LUMO orbital, however, in the He@C20H20 system has energy 1.09 eV above the value found in the C20H20 system. In the case of compounds Li+@C20H20 and Be++@C20H20 the HOMO energies show a decrease of 4.28 eV and 9.26 eV, respectively, when compared to the value for C20H20; an increase in the ionization potential is thus noted with the increase of the atomic number Z of the ion. Regarding the value of HL, we notice that HL(Li+@C20H20) < HL(He@C20H20), HL(Li+@C20H20) > HL(C20H20) and Be++@C20H20 has the smallest HL of the studied systems.
The absolute hardness η is a very useful concept to understand the reactivity of a system. The higher its value is less reactive the system will be, thus being more stable. Our results indicate that, for compounds X@C20, the reactivity increases with the atomic number Z of the confined atom/ion, whereas for X@C20H20 systems, those with X = He and Li+ are less reactive than C20H20. In this series, the system with Be++ is the most reactive.
We have also calculated the vibrational frequencies and presented infrared spectra (IR) graphs as a function of frequency in figures 4 and 5. As a result, we can observe how the infrared spectrum profile is modified due to the insertion of the atom or ion in the C20 and C20H20 cavities; the rotational constants A, B, C, the moments of inertia IA , IB , IC and the SP are shown in table 2. As we can see in figure 4, the peaks located in the frequency range 1.200–1.500 cm−1 are attenuated for increasing Z of the atom/ion inserted in C20 cavity; this result may be associated with the increase in the moment of inertia of the studied systems (see table 2). A fact to note is that A ≠ B ≠ C which implies that the structure of X@C20 is deformed, i.e. the lengths rC–C are different.
Figure 4. Infrared (IR) spectra as a function of frequency for the X@C20 system where (a) C20, (b) He@C20, (c) Li+@C20 and (d) Be++@C20.
Download figure:
Standard image High-resolution image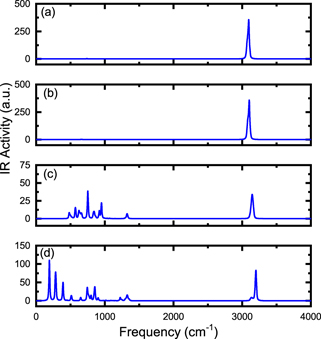
Figure 5. Infrared (IR) spectra as a function of frequency for the X@C20H20 system where (a) C20H20, (b) He@C20H20, (c) Li+@C20H20 and (d) Be++@C20H20.
Download figure:
Standard image High-resolution imageIn figure 5 we can analyze the behavior of the frequency when the atom/ion is inserted into the C20H20 cavity. The frequency range 3.000–3.500 cm−1 refers to stretch vibration of hydrogen atoms in X@C20H20 system; we can observe that there is a decrease in the infrared spectrum only when the Li+ and Be++ ions are inserted; the peaks that appear in the frequency range 0–1.500 cm−1 refer to the oscillations of the Li+ and Be++ ions inside the cavity. By analyzing the data in table 2, it can be seen that the values of the moment of inertia of X@C20H20 are higher than in C20H20, and considering the constants A, B, C and SP values we have that there is structural deformation for Li+@C20H20 and Be++@C20H20 systems.
3.3. Optical properties
In order to study the optical absorption of C20 and X@C20 systems, we have performed the calculation of the first 150 excitations, while for C20H20 and X@C20H20 systems, we have performed the calculation of the first 200 excitations.
Our results are presented in tables 5 and 6 for C20 series and in table 7 for C20H20. In these tables, we have presented the main transitions with the transition energy ΔE, the λ wavelength, oscillator strength f and the transitions with the respective weight (in percentages).
Table 5. Computed single excitation energies ΔE (in eV), corresponding wavelength λ (in nm), oscillator strength f, type of transition, weight (in %) and occurrence region for the C20 and He@C20 systems at the B3LYP/6-31G(d, p) level of theory.
| Systems | ΔE | λ | f | Transitions | Weight | Region |
|---|---|---|---|---|---|---|
| C20 | 4.159 | 298.0 | 0.1349 | H-2 → LUMO | 28.7 | UVB |
| HOMO → L + 6 | 17.0 | |||||
| H-3 → L + 4 | 14.9 | |||||
| 4.714 | 263.0 | 0.1151 | H-3 → L + 4 | 50.0 | UVC | |
| H-2 → L + 5 | 18.1 | |||||
| 5.362 | 231.2 | 0.5113 | H-4 → L + 2 | 20.0 | UVC | |
| H-5 → L + 1 | 19.8 | |||||
| H-3 → LUMO | 14.1 | |||||
| 7.823 | 158.5 | 0.0271 | H-7 → L + 6 | 28.5 | Vacuum UV | |
| H-1 → L + 10 | 12.7 | |||||
| 8.643 | 143.5 | 0.0504 | H-8 → L + 4 | 33.2 | Vacuum UV | |
| HOMO → L + 13 | 11.1 | |||||
| He@C20 | 4.088 | 303.3 | 0.1217 | H-2 → LUMO | 20.3 | UVB |
| HOMO → L + 7 | 17.9 | |||||
| 4.595 | 269.8 | 0.0933 | H-3 → L + 4 | 65.5 | UVC | |
| 5.264 | 235.5 | 0.4659 | H-5 → L + 1 | 18.2 | UVC | |
| H-3 → L + 2 | 12.3 | |||||
| H-4 → L + 2 | 11.5 | |||||
| 7.815 | 158.7 | 0.0193 | H-6 → L + 6 | 28.7 | Vacuum UV | |
| H-6 → L + 7 | 10.5 | |||||
| H-17 → L + 1 | 9.67 | |||||
| 8.434 | 147.0 | 0.0144 | H-3 → L + 11 | 24.2 | Vacuum UV | |
| H-10 → L + 7 | 23.9 | |||||
| H-9 → L + 7 | 11.6 |
Table 6. Computed single excitation energies ΔE (in eV), corresponding wavelength λ (in nm), oscillator strength f, type of transition, weight (in %) and occurrence region for the X = Li+@C20 and Be++@C20 systems at the B3LYP/6-31G(d, p) level of theory.
| Systems | ΔE | λ | f | Transitions | Weight | Region |
|---|---|---|---|---|---|---|
| Li+C20 | 4.288 | 289.2 | 0.1689 | HOMO → L + 8 | 31.7 | UVB |
| H-3 → LUMO | 12.6 | |||||
| 4.849 | 255.7 | 0.0648 | H-3 → L + 3 | 83.9 | UVC | |
| 5.431 | 228.3 | 0.1698 | H-4 → L + 5 | 43.7 | UVC | |
| H-4 → L + 3 | 23.4 | |||||
| H-5 → L + 4 | 11.7 | |||||
| 7.899 | 156.5 | 0.0235 | H-7 → L + 6 | 26.6 | Vacuum UV | |
| H-18 → L + 2 | 22.7 | |||||
| H-6 → L + 6 | 13.3 | |||||
| 8.642 | 143.5 | 0.0646 | H-8 → L + 8 | 25.8 | Vacuum UV | |
| H-9 → L + 7 | 15.3 | |||||
| H-20 → L + 2 | 10.1 | |||||
| Be++@C20 | 3.542 | 350.0 | 0.0221 | HOMO → L + 7 | 38.3 | UVA |
| H-2 → L + 1 | 12.8 | |||||
| H-1 → L + 2 | 10.4 | |||||
| 4.459 | 278.0 | 0.1938 | HOMO → L + 8 | 35.7 | UVC | |
| H-3 → LUMO | 17.9 | |||||
| H-2 → L + 2 | 9.06 | |||||
| 5.512 | 224.9 | 0.2402 | H-3 → L + 5 | 24.8 | UVC | |
| H-1 → L + 6 | 15.8 | |||||
| H-3 → L + 2 | 13.5 | |||||
| 5.601 | 221.3 | 0.0460 | H-4 → L + 3 | 29.2 | UVC | |
| H-5 → L + 3 | 17.2 | |||||
| H-5 → L + 4 | 16.1 | |||||
| 8.053 | 154.0 | 0.0191 | H-6 → L + 6 | 24.2 | Vacuum UV | |
| H-7 → L + 6 | 17.2 | |||||
| H-17 → L + 2 | 16.7 | |||||
| 8.759 | 141.5 | 0.0162 | H-19 → LUMO | 26.6 | Vacuum UV | |
| H-8 → L + 8 | 18.7 |
Table 7. Computed single excitation energies ΔE (in eV), corresponding wavelength λ (in nm), oscillator strength f, type of transition, weight (in %) and occurrence region for the C20H20 and X@C20H20 systems, with X = He, Li+ and Be++, at the B3LYP/6-31G(d, p) level of theory.
| Systems | ΔE | λ | f | Transitions | Weight | Region |
|---|---|---|---|---|---|---|
| C20H20 | 11.037 | 112.3 | 0.2125 | H-17 → L + 5 | 24.8 | Vacuum UV |
| H-16 → L + 6 | 11.7 | |||||
| H-14 → L + 4 | 9.87 | |||||
| 11.181 | 110.9 | 0.1164 | H-5 → L + 9 | 19.1 | Vacuum UV | |
| H-6 → L + 7 | 12.6 | |||||
| H-7 → L + 8 | 9.42 | |||||
| He@C20H20 | 10.992 | 112.8 | 0.2039 | H-16 → L + 4 | 24.6 | Vacuum UV |
| H-17 → L + 6 | 20.3 | |||||
| H-15 → L + 5 | 19.2 | |||||
| 11.153 | 111.2 | 0.1028 | H-5 → L + 8 | 17.0 | Vacuum UV | |
| H-7 → L + 7 | 11.7 | |||||
| H-6 → L + 11 | 9.48 | |||||
| Li+C20H20 | 9.884 | 125.4 | 0.0166 | H-14 → L + 3 | 34.7 | Vacuum UV |
| H-16 → L + 3 | 28.4 | |||||
| 10.316 | 120.4 | 0.1840 | H-14 → L + 6 | 23.6 | Vacuum UV | |
| H-17 → L + 5 | 18.7 | |||||
| H-17 → L + 4 | 14.4 | |||||
| Be++C20H20 | 7.411 | 167.3 | 0.0095 | H-18 → LUMO | 86.2 | Vacuum UV |
| 8.094 | 153.2 | 0.0115 | H-9 → L + 9 | 49.9 | Vacuum UV | |
| H-16 → L + 1 | 43.3 | |||||
| 9.344 | 132.7 | 0.0281 | H-24 → LUMO | 95.5 | Vacuum UV | |
| 9.348 | 132.6 | 0.0281 | H-25 → LUMO | 95.6 | Vacuum UV | |
| 10.364 | 119.6 | 0.0046 | H-9 → L + 5 | 44.8 | Vacuum UV | |
| H-10 → L + 4 | 39.5 | |||||
| 11.177 | 110.9 | 0.0259 | H-20 → LUMO | 40.2 | Vacuum UV | |
| H-3 → L + 12 | 18.1 |
For C20 and X@C20 systems, the absorption spectra have been shown in figure 6. In C20 spectrum, we have observed that, in the studied wavelength range, we can highlight 5 absorption peaks with allowed transitions; the highest intensity peak (f = 0.5113) occurs for λ = 231.2 nm and ΔE = 5.362 eV; we will adopt this peak as a reference. Introducing the He atom into the cavity, we can still identify 5 main peaks but shifted to longer wavelengths, that is, with smaller energies; in fact, the peak of greatest intensity in He@C20 appears at λ = 235.5 nm, with f = 0.4659 and ΔE = 5.264 eV. In the Li+ ion case, the peak displacement is for shorter wavelengths, i.e. for larger energies; thus, in the compound Li+@C20, the peak of absorption of greater intensity occurs for λ = 228.3 nm and ΔE = 5.431 eV. When the Be++ ion is introduced in the cavity, the absorption pattern changes more significantly and a new transition is allowed for λ = 350.0 nm, ΔE = 3.542 eV and f = 0.0221; the peak of greatest intensity in the Be++@C20 system has λ = 224.9 nm and ΔE = 5.512 eV. Comparing C20 with Be++@C20, in Be++@C20 the peak occurs for shorter wavelength values and therefore greater energy.

Figure 6. Absorption graphs, black curve with values shown on the left axis; the oscillator strength, vertical bars in gray with values shown on the right axis, as a function of the λ wavelength for the systems: (a) C20, (b) He@C20, (c) Li+@C20 and (d) Be++@C20.
Download figure:
Standard image High-resolution imageThe spectra for the C20 and X@C20 systems, show absorption in the ultraviolet (UV) range. The highest intensities occur in the UVC and UVB regions. With the encapsulation of atoms and ions, we can notice an increase in absorption in the UVA region, due to the atomic number Z. The less intense peaks occur in the vacuum UV region. Highlight for Be++@C20, which presents a significant peak, in the UVA region, close to the visible region (λ = 350 nm and f = 0.021).
Figure 7 shows that, in the studied wavelength range, the C20H20 system presents two absorption peaks with allowed transitions: one with λ = 112.3 nm, ΔE = 11.03 eV and f = 0.2125 and the other with λ = 110.9 nm, ΔE = 11.181 eV and f = 0.1164. When inserting the X = He atom in the cavity, the absorption spectrum remains with two peaks but the peaks are slightly shifted to values of greater wavelength; then we have, in He@C20H20, λ = 112.8 nm, ΔE = 10.99 eV, f = 0.2039 and λ = 111.2 nm, ΔE = 11.153 eV and f = 0.1028 respectively. We can associate this behavior of the systems with the similarity observed when comparing the electronic structure of C20H20 and He@C20H20 i.e. the state distributions (see figure 3).

Figure 7. Absorption graphs, black curve with values shown on the left axis; the oscillator strength, vertical bars in gray with values shown on the right axis, depending on the wavelength λ for the systems: (a) C20H20, (b) He@C20H20, (c) Li+@C20H20 and (d) Be++@C20H20.
Download figure:
Standard image High-resolution imageThe insertion of X = Li+ into the cavity shows a change in the absorption pattern; there is in Li+@C20H20 a peak of lower intensity for λ = 125.4 nm, ΔE = 9.88 eV, f = 0.184 and another with a higher intensity for λ = 120.4 nm, ΔE = 10.31 eV and f = 0.184. For the Be++@C20H20 system, there are 6 absorption peaks: two peaks with partially allowed transitions (λ = 167.3 nm, ΔE = 7.41 eV, f = 0.0095 and λ = 119.6 nm, ΔE = 10.36 eV, f = 0.0045) and the others highlighted in the figure 7 with permitted transitions. A fact to note is that in the Be++@C20H20 system we have absorption processes with lower intensities than in other systems, but the range in which this compound absorbs energy and presents transitions is greater. We can see in our calculations that, for the C20H20 and X@C20H20 systems, the absorption spectrum is found in vacuum UV region.
Next, in order to complete the analysis of the optical properties of the X@C20 and X@C20H20 systems we have used the partial state density (PDOS) graphs, i.e. we have highlighted the contribution of the atom or ion X in the total state density (DOS). In this way, we have identified in which system orbitals the trapped atom or ion contributes more significantly and consequently we have analyzed which transitions involve such orbitals.
The graphs of the PDOS for the X@C20 systems are shown in figure 8. For X = He it is noted that the helium atom contributes more significantly to the densities of the H-14, H-30 and H-40 orbitals; in the range of energy we use, there are no contributions from He to the unoccupied orbitals; comparing the data in figure 8 with those in table 5, the permitted transitions do not involve the orbitals of that atom. For ion X = Li+ we can see that the orbitals of that ion contribute to move the orbitals of the compound Li+@C20 to lower energies; analyzing figure 8, it can be seen that the contribution of this ion is greater in the density of the H-36 orbital and that the orbitals from H-1 to H-5 also contain contributions from the density of ion states; by analyzing it together with table 6, we can verify that the Li+ orbitals contribute directly to the electronic transitions related to ΔE equal to 4.288 eV, 4.849 eV and 5.431 eV. These excitations correspond to electronic transitions between the states of the lithium ion and carbon atoms in C20.
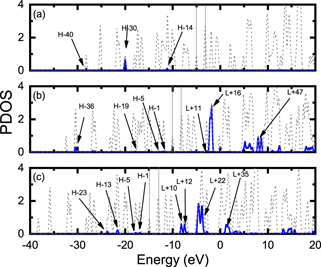
Figure 8. Contribution of the states of atoms confined in the partial state density (PDOS) to the X@C20 systems. Dotted line in gray represents the contribution of the total system X@C20, and solid blue line indicates the contribution of the atoms (a) X = He, (b) X = Li+ and (c) X = Be++.
Download figure:
Standard image High-resolution imageFor Be++, we can see that this ion's orbitals contribute to the system's orbitals: H-1, H-5, H13 and H-23. This way, we can verify that, among the six possible transitions presented for the Be++@C20 system, three of them present contributions of this ion: ΔE equal to 3.543 eV, 5.512 eV and 5.601 eV. So, we can infer that, there may be electronic transitions between the Be++ orbitals and the C20 orbitals.
The PDOS graphs for X@C20H20 systems are shown in figure 9; we can observe that when inserting the atom X = He in C20H20 cavity, the atom contributes more significantly in the densities of the occupied orbitals H-29, H-41, H-50 and in the unoccupied orbital L + 53; we also noticed that in the analyzed energy range there are no permitted transitions involving these orbitals. When the Li+ ion is inserted into the cavity, it can be seen that its orbitals contribute mainly to the densities of the unoccupied orbitals LUMO, L + 1, L + 2, L + 3, L + 4, L + 5 and L + 6; it is also noted that in the studied energy range these orbitals are involved in the allowed transitions; the same occurs with the insertion of Be++ in the C20H20 cavity: the ion orbitals contribute significantly to the densities of the LUMO, L + 1, L + 2, L + 3, L + 4, L + 5, L + 6, L + 7, L + 8, L + 9, L + 10 and L + 11 orbitals, and these orbitals are involved in the permitted transitions of Be++@C20H20. In both cases, transitions from the C20H20 orbitals to the encapsulated ions are possible.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Contribution of the states of atoms confined in the partial state density (PDOS) to the X@C20H20 systems. Dotted line in gray represents the contribution of the total system X@C20H20, and solid blue line indicates the contribution of the atoms (a) X = He, (b) X = Li+ and (c) X = Be++.
Download figure:
Standard image High-resolution image{kind=link}
One way to validate the results obtained for the optical properties discussed here, would be a comparison with spectroscopic data obtained experimentally. However, such data are still scarce in the literature. The comparison between theoretical and experimental results often requires an adaptation in theory, in order to obtain values that can be obtained via experiment. As an example, to obtain UV absorption spectra, we used here the usual TDDFT of the linear response approach (first order) of the system when it is subjected to a weak field, as implemented in GAMESS-US package [54]. Now, some authors [66, 67] have been working to develop methods, from the extension of TDDFT, for the description of non-linear optical phenomena and spectral properties of excited states; in this way, theoretical results can be compared with experiments of transient absorption spectroscopy. This type of analysis, together with an analysis of the electronic population of the states, can elucidate, for example, the behavior of the endohedral systems presented here and similar.
4. Conclusions
In this work we have performed an analysis of the C20, C20H20, X@C20 and X@C20H20 systems with X = He, Li+ and Be++. The calculations were performed using the DFT/B3LYP/6-31G(d, p) level of theory for the electronic structure, energetic characteristics, stability and analysis of the structural parameter of the systems and TDDFT/B3LYP/6-31G(d, p) theory level for analysis of optical properties.
Our results for X@C20H20 agree with those in the literature [45]; this fact provides quantitative arguments for the reliability of our results for X@C20 system. A point to note is that in our calculations we have optimized the position of all atoms; as a consequence, the Li+ and Be++ ions in the equilibrium geometry are located outside the center of the cavity. Another result is that, in the vibrational frequencies calculation, we have obtained only real values, which indicates that our equilibrium structures correspond to a minimal global energy.
Our results show that the energy HL decreases by inserting He, Li+ and Be++ in the cavity, i.e. HL(He@C20) > HL(Li+@C20) > HL(Be++@C20). It is also noted that there is an increase in the reactivity of the compounds compared to the C20 system. This can be verified also by analyzing the absolute hardness. Another result is that by including He and Li+ in the C20H20 cavity the value of HL increases compared to the value of HL(C20H20), i.e. HL(He@C20H20) > HL(Li+@C20H20). By including Be++ the HL(Be++@C20H20) value decreases in relation to the HL(C20H20); Be++@C20H20 is the one with lowest HL value.
The transitions, as expected, depend on the type of cavity and the atom/ion encapsulated in the cavity. We have also found that, when inserting Li+ and Be++ ions in the cavities of C20 and C20H20, the IR spectrum changes, indicating that some vibrations become less energetic. This can be due to the increase in the moment of inertia in the systems containing the confined ions.
For endohedral systems containing Li+ and Be++ ions, we verified that it is possible that there are transitions between the orbitals of the ion and the orbitals of carbon atoms that make up the fullerene type systems. We also note that the transitions in the C20H20 and X@C20H20 systems show absorption in the vacuum UV region. On the other hand, the C20 and X@C20 systems, the absorptions occur with greater intensity in the UVB and UVC regions. Highlight to the Be++@C20 system, which presents absorption in the UVA region, close to the visible.
In conclusion, among the studied systems, our results indicate that Li+ and Be++ ions can significantly modify the electronic and optical properties of fullerene-like systems. Our results are theoretical. However, as it is known, photoelectron spectroscopy is a widely used technique to analyze the electronic structure of complex systems [68, 69] and the advent of intense ultra-short laser sources has extended the range of applicability of this technique to a vast variety of non-linear phenomena [66, 67]. Furthermore, it turned attosecond time-resolved pump–probe photoelectron spectroscopy into a powerful technique for the characterization of excited-states dynamics in nano-structures. About this spectroscopy, as we have cited in the section 3.3, a methodology based on TDDFT has been developed in the literature [66, 67]. Thus, it is possible to measure properties of Li+@C20H20, Be++@C20 and Be++@C20H20 which have favorable forming energies and analyze, for instance, the modification in the distribution of the absorption signal with the inclusions of Li+ and Be++ in the cages. In fact, the knowledge and control of quantum states of systems due to confinement and how this confinement alters the electronic and optical properties of systems, is a topic of great interest for practical applications in areas such as material physics and development of optical and electronic devices.
Acknowledgments
This work has been supported by the Brazilian agencies Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB). We also thank Dr. R Rivelino for discussions and suggestions.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).