
Abstract
Knots and entanglements are ubiquitous. Beyond their aesthetic appeal, these fascinating topological entities can be either useful or cumbersome. In recent decades, the importance and prevalence of molecular knots have been increasingly recognised by scientists from different disciplines. In this review, we provide an overview on the various molecular knots found in naturally occurring biological systems (DNA, RNA and proteins), and those created by synthetic chemists. We discuss the current knowledge in these fields, including recent developments in experimental and, in some cases, computational studies which are beginning to shed light into the complex interplay between the structure, formation and properties of these topologically intricate molecules.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Knots and entanglements are common topological features observed not only in the macroscopic world, but also at the molecular level (figure 1). In everyday life, they can be found in various useful applications, from applying surgical sutures to tying shoelaces. However, in some cases, knots can be a nuisance, for example, they can form spontaneously in electrical cables, headphones and garden pipes. They can also lead to undesirable outcomes such as the obstruction of blood circulation to the fetus when tight knots form in the umbilical cord during human pregnancy [1].

Figure 1. Examples of macroscopic (a)–(d) and molecular (e)–(g) knots. (a) Surgical suture knots used to close a wound [2]. (b) Tying a shoelace knot [3]. (c) Knots formed in entangled earphones. (d) A tight knot formed in an umbilical cord [4]. (e) Electron micrograph of a knotted DNA; figure taken with permission from reference [5]. (f) Ribbon diagram of a stevedore (61) knotted α-haloacid dehalogenase protein, PDB code: 4N2X. Inset: simplified view of the protein chain showing the knot. (g) Chemical structure of a synthesised organic trefoil knot. Inset: schematic representation of the knotted structure [6].
Download figure:
Standard image High-resolution imageRecently, the importance and prevalence of knots at a molecular level have become truly apparent and this has attracted increasing interest from scientists in different fields. In nature, molecular knots (including slipknots and pseudoknots) are found throughout biology and exist in three major classes of biopolymers: DNA, RNA and proteins [7–15]. Although it is still unclear as to whether these complex topologies are evolutionary advantageous, most natural knots are thought to play a significant role in the structural, dynamic and/or functional properties of the biological systems they are associated with. In addition, molecular knots are increasingly becoming targets of chemical synthesis [16, 17]. Understanding how knots form at a molecular level as well as how the properties of knotted molecular structures differ from unknotted ones is vital.
This review highlights some of the molecular knotted structures discovered in biology and chemistry. It focuses on the structural and mechanistic studies into which and how knots are formed, and summarises the recent developments made towards understanding their properties and potential functions. The review begins with a brief introduction to the classification and detection of knots, followed by an overview of knotted DNA, RNA pseudoknots, protein knots and slipknots, as well as synthetic molecular knots.
2. Classification and detection of knots
Concepts from the mathematical field of knot theory have been applied in almost all branches of science, providing tools essential for the detection and classification of different knotted structures. Mathematically, a knot (sometimes termed as a 'true knot') is defined as a topological state of a closed loop that is impossible to untie without being spliced [18]. Technically, this means that knots cannot be defined in open chains. However, many knots such as those found in biological systems are open chains. In the case of a simple linear string, one considers it knotted if it does not disentangle itself after being pulled at both ends. This idea is usually applied to open chains and is analogous to their ends being unambiguously connected with a loop to produce a corresponding closed curve.
Detecting knots in topologically complex systems is often not straightforward and requires mathematical methods to both detect and classify the knot type. To identify knotted structures, various algorithms can be employed. One of the simplest knot detection algorithms, known as the Alexander polynomial, can detect and classify a knot according to the minimum number of crossings in a projection of the chain onto a plane [18]. Each knot type is labelled in accordance with the Alexander–Briggs notation, where the first number is the crossing number (usually a measure of knot complexity) and the subscripted index number denotes the knot's order amongst all knots with that crossing number. A simple ring with zero crossings is referred to as the unknot (01) or the trivial knot whilst the simplest, non-trivial knot type is the trefoil knot (31) with three crossings. Other common knot types include the figure-of-eight knot (41) that has four crossings, two knots with five crossings (51, 52) and three knots with six crossings (61, 62, 63) (figure 2). In addition to the Alexander polynomial, the Jones and HOMFLY polynomials are more advanced algorithms that can discriminate between increasingly complex knot types. Further details of these polynomials are provided elsewhere [18–21].

Figure 2. Common knot types with up to six crossings denoted by the Alexander–Briggs notation. Knots were generated using KnotPlot (http://knotplot.com/).
Download figure:
Standard image High-resolution imageIt is important to note that amongst these knot polynomials, the HOMFLY polynomial is a powerful method for detecting the chirality of knots. However, even HOMFLY can not characterise chirality in all cases [22, 23]. Most knots are not equivalent to their mirror images and they are usually known as chiral knots. The simplest chiral knot is the trefoil knot (31), which comes in a left and a right-handed form, as shown in figure 3. In contrast, achiral knots are knots that can be converted to (or are indistinguishable from) their mirror images. Examples include the trivial (01) and figure-of-eight (41) knots. In knot theory, knots can also be classified as either torus or twist knots. Torus knots are a family of knots that can be drawn as closed curves on the surface of a torus (equivalent to a holed-doughnut) and include the 31, 51, 71 knots, etc. Twist knots, on the other hand, are knots that can be formed by linking together the ends of a repeatedly twisted, closed loop and comprise the 41, 52, 61 knots, etc.

Figure 3. The two distinct chiral trefoil knots; left and right-handed trefoil knots are illustrated on the left and right, respectively. Knots were generated using KnotPlot (http://knotplot.com/).
Download figure:
Standard image High-resolution imageAlthough the polynomials are useful for analysing simpler knots, they cannot differentiate knots with projections of many crossings or detect knots in extensively knotted systems, as these tend to be computationally challenging. In order to solve this problem, an alternative smoothing algorithm, sometimes referred to as the KMT reduction, was developed such that complex knotted structures are simplified by omitting regions of the chain unnecessary for maintaining the knot [24, 25]. This method produces highly reduced configurations of the original chain and, thus allows efficient computation of the polynomials. In the case of protein structures, this reduction algorithm is very useful for depicting the knotted chain in a simplified manner so that knots can be detected directly and easily visualised [26, 27]. Additionally, as proposed by Taylor, the method can also simultaneously pinpoint the location and depth of the knotted core by calculating the smallest number of residues that can be removed from each side before the structure becomes unknotted [26]. 'Shallow' knots tend to disappear when a few amino acids are deleted from each terminus whilst 'deep' knots remain until a significant amount of the chain (more than 20 amino acid residues on either side of the knotted core) have been removed. However, depending on how the chain is reduced, this method can result in the classification of different knot types. Millett and co-workers have introduced a relatively simple, unbiased method known as the uniform closure method, in which the free ends of a linear open chain are connected to random, uniformly chosen points on a large sphere surrounding the chain [28]. The procedure is repeated many times and a spectrum of knots is obtained, in which the knot type that is dominant is labelled as the knot type of the chain.
3. DNA
DNA (deoxyribonucleic acid) is a molecule that encodes the genetic information required for the development and functioning of all living organisms and many viruses. It is not only used as a template for replication but it is also involved in RNA synthesis, which, in some cases, leads on to protein synthesis. Based on the Watson–Crick model, DNA consists of two complementary polynucleotide chains that are intertwined around each other, forming a right-handed double helix [29] (figure 4(a)). DNA can exist as a linear or a closed circular form and is typically tightly packaged. As a result of the structure and metabolism of the double helix, DNA molecules can form three topological states: knotted, catenated or supercoiled (figure 4(b)). In this section, we briefly discuss knots in naturally occurring DNA, mainly focussing on the knotting mechanism and its biological consequences.

Figure 4. (a) Double-helical structure of a DNA molecule, PDB code: 3BSE. Cartoon representation generated using Pymol (www.pymol.org/). (b) Different topological forms of a DNA molecule, formation of which is catalysed by type II topoisomerases: (i) supercoiled, (ii) catenated and (iii) knotted. A single line represents a double strand of DNA.
Download figure:
Standard image High-resolution image3.1. Knots in DNA: structure and formation
A DNA knot is defined as the self-entanglement of a single DNA molecule, therefore this excludes catenane structures that are formed by more than one chain (figure 5(a)). In 1976, Liu and co-workers first discovered that single-stranded DNA chains in bacteriophages could knot when treated with Escherichia coli omega protein, a type I topoisomerase [30]. This was subsequently followed by the discovery of knots in double-stranded DNA chains in 1980 when a supercoiled plasmid was incubated with excess amounts of type II topoisomerase from bacteriophage T4 [31]. Since then, various knotted structures formed in nicked, circular duplex DNA molecules by E. coli topoisomerase I have been identified in vitro, ranging from simple trefoil knots to more complex higher order and composite knots (figure 5(b)) [9]. With the use of electron microscopy imaging and agarose gel electrophoresis, Dean and co-workers characterised these topologically different knotted DNA structures in detail [9].

Figure 5. (a) Schematic diagram of a trefoil knot, 31, in double-stranded DNA generated using KnotPlot (http://knotplot.com/). (b) Left panel: agarose gel electrophoresis of knotted DNA plasmids, where the mobility increases with the number of knot crossings, reflecting more compact species. Lane 1: unknotted DNA; Lanes 2–7: knotted DNA species. I and II indicate the mobilities of nicked circular and linear DNA, respectively. Right panel: number of crossings in knotted DNA based on electron micrographs of DNA gel bands. Adapted with permission from [9]. (c) Illustration of a site-specific recombination reaction, where arrows indicate the recombination sites. Reprinted from [40], with permission from Elsevier. (d) Schematic representation of the topological consequences of two actively transcribing genes with the origins of replication in convergent orientation. Reprinted from [43], with permission from Elsevier. (e) Schematic diagram of the topological conformation caused by the head-on collision of transcription and replication. Reprinted from [43], with permission from Elsevier. (f) Conformations of packed P4 phage genomes as determined by coarse-grained molecular dynamics simulations. Reprinted from [49], with permission from Elsevier. (g) Left panel: knotted DNA from bacteriophage P4 capsids separated by agarose gel electrophorosis. Middle panel: magnified portion, highlighting knot populations of low crossing number. Right panel: Knot populations and subpopulations contain three to nine crossings (labelled 3–9) and six or more crossings (labelled 6'–9'), respectively [7]. Copyright (2005) The National Academy of Sciences, USA.
Download figure:
Standard image High-resolution imageIn the last three decades, an increasing number of studies of DNA knots have been undertaken [32–35]. As discussed above, knots in DNA can form in vitro when DNA strands are cut and re-joined with the help of topoisomerases. DNA topoisomerases control the topology of DNA by introducing transient breaks in DNA strands then re-ligating them to different ends [36, 37]. They are classified into two types: type I or type II. Type I topoisomerases mediate the passage of a single strand of duplex DNA through a nick in the complementary strand. In contrast, type II topoisomerases introduce a transient double-stranded break in one segment of the DNA, allowing a second segment of duplex DNA to pass through before the strands are chemically ligated. A variety of knotted DNA products can also form when recombinases act on supercoiled circular DNA substrates (an example is shown in figure 5(c)) [38–40]. Recombinases are involved in changing the topology of DNA by a complex process called site-specific recombination [41]. In this case, they mediate genome rearrangement such that a DNA segment is inserted, excised or inverted in accordance with the appropriate recombination sites [41].
DNA knots can also arise in vivo during replication and transcription, as these processes require the action of topoisomerases to release accumulated torsional stress in the DNA [42]. In partially replicated bacterial plasmids with two origins of replication in head-to-head orientation, it has been observed that topoisomerases induce knot formation within replication bubbles that are helically wound (figure 5(d)) [35]. Olavarrieta and co-workers have also shown that complex knotting of the duplex DNA in small pBR322-derived plasmids can be initiated by a head-on collision of replication and transcription, resulting in plasmid instability in E. coli (figure 5(e)) [43]. Recently, the Schvartzman group has suggested that if the progression of the replication forks in DNA synthesis is impaired, sister duplexes can become loosely intertwined and this can lead to the introduction of knots by the action of topoisomerase IV (Topo IV) [44]. It should be noted, however, that these observations are made on small bacterial plasmids and whether they are applicable to large bacterial or eukaryotic chromosomes is still uncertain.
Several studies have also previously reported that linear viral genomic DNA can cyclise and form knots upon extraction from P4 bacteriophages (figure 5(f)) [31, 45]. Furthermore, it was found that the probability of DNA knotting was enhanced in intact P4 deletion mutants [46] and tailless P4 phages [47]. In a series of experiments, Arsuaga and co-workers showed that most viral DNA molecules (>95%) are highly knotted due to the tight confinement and writhe bias of their packing geometry within the phage capsid (figure 5(g)) [7, 33]. Writhe is the amount a piece of DNA is deformed to form coils as a result of torsional stress, which leads to the phenomenon of DNA supercoiling. Although the specific mechanism of knot formation is still unclear, characterisation of the complex knot spectrum of bacteriophage P4 genome by high-resolution gel electrophoresis revealed that chiral and torus knots were favoured by confinement over achiral and twist ones [7]. Results from recent simulations also showed that there was a preference for chiral knots, albeit no significant bias of torus over twist knots was found [48]. As yet, it remains to be seen what factors actually determine viral genome organisation in terms of its knot types and distribution.
3.2. Biological consequences of DNA knotting
How does DNA knotting affect its biological activity within cells? As discussed above, several processes such as DNA compaction, topoisomerisation, site-specific recombination, replication and transcription can result in the formation of DNA knots in cells. However, the presence of knots in DNA has potentially detrimental effects in several cellular processes such as transcription and replication [50–52] and, if unresolved, can lead to mutational defects in the genome or even cell death. To overcome these problems, cells express and produce essential, ubiquitous enzymes called topoisomerases, which can remove knots promptly and efficiently [53, 54]. Contrary to this, it has to be noted that these enzymes also play a role in creating DNA knots. As a result of their presence and dual-functionality, cells have evolved and taken advantage of the topologically constrained nature of their DNA. Lopez and co-workers demonstrated that Topo IV in bacteria can not only form knots in DNA during replication but it is also responsible in unknotting them later on so that DNA can get correctly segregated to every daughter cell [44].
In the case of bacteriophages, recent simulations have revealed that the organization and topology of packaged DNA in capsids are important in how fast the DNA gets ejected into an infected bacterial cell [55]. Marenduzzo and co-workers observed that ordered DNA spools in the capsid, favoured by DNA cholesteric interactions, were ejected at a faster rate than disordered, entangled DNA [55]. It was also shown that torus knots exited the capsid more easily than twist knots, which can halt the ejection process.
3.3. Summary
DNA is an extremely long biological polymer, and it is no surprise therefore that linear and circular, single- and double-stranded DNA molecules are all known to form a wide range of knotted structures from simple trefoil (31) knots to more complex knots such as those with nine crossings. Whereas there are examples of DNA forming both chiral and achiral knots as well as torus and twist knots, there is some evidence, at least in the context of highly packaged viral genomic DNA, that there is a preference for chiral and torus knots. In many cases, it is well established that DNA becomes knotted as a direct result of biological processes such as recombination, replication and transcription. In these cases, knotting is problematic and, consequently, numerous enzymes exist (topoisomerases) which catalyse the unknotting of a DNA chain through a 'cut and paste' mechanism in which the DNA is first cut, then moved/rotated and subsequently religated. Effectively, this breaks the chain into small segments and rearranges them to eliminate the knot. The biological consequences of not removing the knot can be severe, e.g. cell death. In contrast, there may also be benefits of knotting, such as the case of highly packaged viral genomes. Here, knotting may aid in the tight packing and it can also affect the rate at which the genomic DNA is ejected from its viral carrier/storage compartment, the capsid.
DNA can also form a range of other topologically complex states including catenane structures such as Hopf and higher-order links. For a comprehensive overview on the various topological forms of DNA, interested readers are directed towards the following references [8, 37, 56–58].
In addition to all of the studies discussed above on knotting in naturally occurring DNA, there is also considerable literature on knotting in synthetic single-stranded DNA. In particular, Seeman and co-workers have been able to rationally design and build synthetic forms of DNA with a range of knot types and links. A detailed discussion of this work is out of the scope of this review, however, a summary of the different types of structures that have been synthesised is given in table 3, and interested readers are directed to the references provided in the Table.
4. RNA
RNA (ribonucleic acid) is a single-stranded, linear polymer made up of four different types of nucleotides that are linked together by phosphodiester bonds. With the help of complementary base pairing and other types of hydrogen bonds between nucleotides in the same chain, RNA molecules can fold into various complex 3D structures and thus achieve diverse biological functions within cells; from mediating the transfer of genetic information from DNA into protein, to catalysis [59, 60]. In addition to these, many viruses have RNA as their genetic material.
Among the most common RNA structures is the pseudoknot motif, which was first discovered in turnip yellow mosaic virus (TYMV) in 1982 [61]. Although pseudoknots are not true topological knots, they fold into complex 3D conformations where there are a number of topological crossings of the chain. Here, we describe the main structural features of RNA pseudoknots and discuss how they have been intimately linked to the biological properties of naturally occurring RNAs.
4.1. Pseudoknot structures
A pseudoknot is generally defined as an RNA structure that consists of at least two helical segments linked together by single-stranded regions or loops [62]. Although pseudoknots can possess several distinct folding topologies, the best characterised to date is the so-called H (hairpin)-type or classical pseudoknot. As illustrated in figure 6, this is the simplest type of pseudoknot structure that results from the base pairing of a single-stranded segment of RNA in the loop of a hairpin to a complementary sequence outside the loop region. It comprises of two base-paired stem segments (S1 and S2) and, depending on the number of loop bases involved in the pseudoknotting interaction, two or three single-stranded connecting loops (L1, L2 and L3) [63]. However, in most classical pseudoknots (>85% [64]), L2 is missing and thus S1 and S2 can coaxially stack on top of each other to form a quasi-continuous helix. Figure 6(d) depicts this arrangement in the H-type pseudoknot structure of the 3'-terminus of the TYMV RNA, where L1 spans S2 and crosses the deep groove of the helix whilst L3 spans stem S1 and crosses the minor groove. In addition to coaxial stacking, pseudoknots can also be further stabilised by hydrogen bonds formed between single-stranded loop regions and the adjacent stem segments. As the connecting loops and stems can vary in length, and the interactions between them can differ, RNA pseudoknots represent a structurally diverse group. Hence, it comes as no surprise that these structures are associated with various vital roles in biology. These include forming functional domains within ribozymes [65] and telomerase [66] as well as inducing ribosomal frameshifting in many viruses [10, 67] and regulating translation [68].

Figure 6. Formation of an H-type RNA pseudoknot. (a) Linear organisation of the base-pairing elements (indicated with dashed lines) within an H-type RNA pseudoknot. (b) Formation of an initial hairpin prior to pseudoknotting. Bases from the loop are paired to bases outside the hairpin, as indicated with dashed lines. (c) A classical, H-type pseudoknot motif. (d) A ribbon representation of the acceptor arm pseudoknot structure of the 3' end of the turnip yellow mosaic virus genomic RNA is shown based on the NMR structure, PDB code: 1A60. Loops L1 (pink) and L3 (cyan) cross the deep and shallow groove of the helix, respectively. S1 is purple and S2 is blue. L2 is not present in the example shown.
Download figure:
Standard image High-resolution image4.2. Functional roles of the pseudoknot motif
The RNA pseudoknot is a ubiquitous folding topology that has been identified in almost all organisms [14]. Below, we describe well-characterised examples of pseudoknots involved in catalysis, ribosomal frameshifting and translational regulation, highlighting how the structures are related to their function. In most cases, it has also been shown that the function of pseudoknots is associated with their position along the RNA sequence [63, 69, 70]. For example, pseudoknots located at the core of the tertiary fold of RNAs tend to be crucial in catalysis whilst those found at the 5' end of mRNAs are typically involved in translational control. In addition, in non-coding regions (NCRs) of viral RNAs, pseudoknots play a role in the regulation of initiation of protein synthesis and in template recognition by viral replicases.
4.2.1. Catalytically active pseudoknots.
Catalytic RNAs, or ribozymes, are RNA molecules that can catalyse specific biochemical reactions. It has been shown that most ribozymes fold into similar 3D structures that are essential for their function [71]. As a model to understand the mechanism of catalytic RNAs, extensive studies have been done on the hepatitis delta virus (HDV) ribozyme, the fastest known naturally occurring self-cleaving ribozyme [72–74]. HDV is a satellite RNA virus of hepatitis B virus, which together can cause severe infection in humans [75]. The host RNA polymerase II replicates the circular genome of HDV through a double rolling-circle mechanism, producing long RNA transcripts that must be cleaved for viral replication. The processing of the HDV RNA is performed by the self-cleaving HDV ribozyme encoded in the RNA [76]. As illustrated in figure 7(a), the HDV ribozyme has a characteristic 'nested' double pseudoknot that not only forms the active site necessary for the specificity of substrate binding and catalysis but also stabilises the overall RNA structure [77]. This pseudoknot motif has also been discovered in other small self-cleaving ribozymes, particularly in the core of glmS ribozymes in many Gram-positive bacteria [78, 79] and mammalian cytoplasmic polyadenylation element-binding protein 3 ribozymes [80]. As a result, these RNAs are able to achieve an overall complex and stable conformation.

Figure 7. Sequences and structures of RNA pseudoknots. Loops and stems are colour-coded in reference to figure 6, where L1 is pink, L3 is cyan, S1 is purple and S2 is blue. (a) Hepatitis delta virus ribozyme, PDB code: 1DRZ. For simplicity, only the largest of the two pseudoknots is shown colour-coded. In this example, L2 exists and is shown in red. The grey loop is the U1A RNA binding domain, which is used to aid crystallisation of the ribozyme. (b) Human telomerase, PDB code: 1YMO. (c) Mouse mammary tumour virus, PDB code: 1RNK. (d) Simian retrovirus 1, PDB code: 1E95. (e) The base of domain (dom) III of the Hepatitis C virus internal ribosome entry site, PDB code: 3T4B, where a double pseudoknot (PK1 and PK2) structure surrounding a four-way helical junction is shown. In PK1, L2 (red) and a third base-paired stem, SII/J (orange) exists, in addition to L1, L3, S1 and S2. PK2 is formed between the IIe tetraloop (green) and the main helix of dom III (yellow).
Download figure:
Standard image High-resolution imageEukaryotic chromosomes possess telomere ends that protect themselves from loss of genetic material due to successive DNA replication events [81]. Maintenance of the telomeres is performed by the ribonucleoprotein telomerase, an RNA-dependent DNA polymerase made up of a specialised reverse transcriptase and a telomerase RNA (TR) [82, 83]. Although telomerase activity is essential for highly proliferative cells such as stem cells, it is also known to be elevated in ~90% of cancer cells [84, 85] and may play a role in aging [86]. TRs not only provide the template for DNA synthesis but also contain a highly conserved classic H-type pseudoknot within the core domain, which is needed for telomerase assembly and activity [87–90]. Figure 7(b) shows a structure of the human TR pseudoknot, where triple nucleotide interactions U—A-U between L1 and S2 in the deep groove form a triple helix important for telomerase repeat addition processivity [66]. Studies have also shown that the conformational switch that exists between the pseudoknot and a less stable hairpin might be crucial for telomerase activity [91, 92]. Mutations in the TR pseudoknot have also been associated with inherited human disorders such as aplastic anemia and autosomal dyskeratosis congenital [86, 93, 94].
4.2.2. Ribosomal frameshift-inducing pseudoknots.
Besides catalysis, RNA pseudoknots are also commonly involved in inducing ribosomes to move into alternative reading frames, a process known as frameshifting. RNA viruses, in particular, exploit the programmed -1 ribosomal frameshifting (-1 PRF) mechanism to regulate gene expression, which enables a single mRNA to get translated into two proteins at a defined ratio [95]. Importantly, this translational mechanism is known to be essential for the replication and proliferation of all retroviruses. Frameshift signals encoded in mRNAs consist of two essential elements: a heptanucleotide 'slippery' sequence X XXY YYZ and a downstream RNA structural element, typically a pseudoknot [96, 97]. It was discovered that even though the slip-site alone can increase frameshifting efficiency by 1%, it is the pseudoknot that is responsible in significantly stimulating the frameshift event, in some cases, by up to 30–50% [10, 98]. As such, pseudoknot structures in the coding regions associated with frameshifting are potential targets for the development of antiviral therapeutics.
The actual molecular mechanism as to how pseudoknots promote efficient -1 frameshifting still remains unclear. It has been suggested that the downstream pseudoknot structure causes the ribosome to pause on the 'slippery' sequence and forces it to shift back one nucleotide and continue mRNA translation in the -1 reading frame [99]. Studies have shown that this could be due to the unusual topology of the pseudoknot, which makes it resistant to unwinding by the ribosome's helicase activity [100–102].
The first -1 PRF stimulatory RNA element extensively studied in terms of its structure and function was the mouse mammary tumor virus (MMTV) frameshift-inducing pseudoknot [103]. Figure 7(c) shows the NMR structure of the MMTV pseudoknot, which has a characteristic unpaired adenine intercalated between two helical stems rich in guanine/cytosine. Consequently, this induces a pronounced bend of approximately 60° between the two helices, thus preventing them from being coaxially stacked. Through mutational analysis, structural and functional studies have revealed that the wedged nucleotide and subsequent bending between the helical stems strongly correlate with efficient frameshifting [104]. However, this does not seem to be the case for the simian retrovirus 1 (SRV-1) pseudoknot, where the S1 and S2 helices are coaxially stacked as a result of the base pairing between the adenine nucleotide found in between S1 and S2 with the last uridine nucleotide in L3 (figure 7(d)) [105]. Instead, subsequent structural studies revealed that favourable interactions between L3 and S1 in the helical junction might be responsible for the frameshifting efficiency in SRV-1.
4.2.3. Pseudoknots involved in translational regulation.
Pseudoknot structures have also been shown to regulate translation in viruses and bacteria. In the case of the hepatitis C virus (HCV), its genomic RNA consists of an internal ribosome entry site (IRES) in the 5' untranslated region, where the ribosome is recruited and translation initiated [106, 107]. The HCV IRES is made up of three main structural domains that adopt a tertiary conformation [106, 108]. The core domain of the HCV IRES consists of a four-way helical junction at the base of domain III, where a double pseudoknot is formed (figure 7(e)). The structural integrity of this domain has been found to be essential in positioning the mRNA start codon correctly on the 40 S ribosomal subunit during translation initiation [109]. As the pseudoknot domain is highly conserved and is crucial for viral translation, it represents a potential target for HCV therapeutics. Pseudoknots have also been found in the 3' NCR of many viral positive-strand genomic RNA, where they are associated with translational control, replication and genome packaging. Further details of the structure-function relationship of these 3'-NCR pseudoknots can be found elsewhere [69, 110].
A domain in the bacterial transfer-messenger RNA (tmRNA) has also been shown to consist of four pseudoknots [111]. tmRNAs remarkably possess dual tRNA- and mRNA-like structural and functional properties. They recognise and recycle stalled ribosomes, add a short proteolysis-inducing tag to incomplete growing polypeptide chains and assist degradation of the aberrant mRNAs lacking a stop codon [112]. Although the actual roles of each pseudoknot is still unclear, collectively, they have been suggested to aid in the folding of tmRNA, slow down tmRNA degradation and serve as binding sites for proteins that assist the functioning of tmRNA [68].
4.3. Computational prediction of RNA pseudoknots
The function of an RNA molecule can often be inferred from its 3D structure. Since RNA structures are hierarchical and the structural determination of their 3D conformation using experimental methods is difficult, RNA secondary structure prediction is important in elucidating the potential structures and therefore, functions of RNAs. A number of different approaches to RNA pseudoknot structure prediction have been developed over the last decade. These are described below.
Most pseudoknot-free structure prediction programs are based on determining a minimum free-energy (MFE) conformation from the primary nucleotide sequence. However, the prediction of RNA pseudoknots is computationally complex as the search for a MFE structure, in these cases, has been shown to be a Non-deterministic Polynomial-time (NP)-complete problem with respect to sequence length [113]. Dynamic programming (DP)-based methods, which use free energy minimization, can only predict limited classes of pseudoknots. For example, in the case of PKNOTS, the algorithm accurately predicts structures for RNA sequences of length up to 100 bases [114]. Other programs that also use the DP-method include NUPACK [115] and pknotsRG [116]. These approaches, however, are effective only for short sequences, as computation time can increase as the third to sixth power of sequence length, depending on the algorithm used [114, 115, 117].
To overcome this issue, heuristic prediction methods such as FlexStem [118] and HotKnots V2.0 [119] have been developed. Although the predicted structure is not necessarily the MFE, such approaches can handle a wider class of pseudoknots and longer sequences. In another case, the IPknot method, developed by Sato and co-workers, can predict pseudoknotted structures from sequences up to 1000 bases with increased speed and accuracy [120]. Based on integer programming (IP), this method breaks down the pseudoknotted structure into pseudoknot-free substructures and approximates a base-pairing probability distribution that considers pseudoknots. In addition, it can also use multiple aligned sequences to predict a consensus pseudoknotted structure [120].
Another algorithm that can predict the MFE RNA pseudoknot structure is TT2NE, which is based on classifying RNA structures according to their genus [121]. Although it can only predict structures for sequences up to 200 bases, it has been shown that the quality of predictions is significantly improved when compared to other state-of-the-art algorithms [121]. Based on the same concept, the same group recently developed McGenus, a Monte Carlo algorithm [122]. Here, the method stochastically searches the MFE structures from sequences of up to 1000 bases. More recently, Jabbari and co-workers have developed an iterative-based method called Iterative HFold, which uses a pseudoknot-free structure to predict pseudoknotted structures rather than a sequence as input [123].
Pseudoknotted structure prediction programs are a valuable resource; examples of some of these recent programs and webservers are listed in table 1. Further details of currently available pseudoknot structure prediction programs can be found elsewhere [124–126]. In general, most of the approaches have been developed with the aim of predicting pseudoknotted structures with increased speed and accuracy. However, it remains clear that these algorithms are still restricted by the lack of understanding of pseudoknot thermodynamics and the capacity to cope with pseudoknots containing stem regions with bulged residues or non-Watson-Crick pairs. In addition, steric constraints and the contribution of entropy to the free energy are often ignored, as there is limited information on the full 3D geometry of pseudoknots. Environmental factors such as ions, solvent, protein and other RNAs are also important in the structure and function of RNA; and ideally these also need to be accurately incorporated into the predictions.
Table 1. Examples of RNA pseudoknot prediction programmes.
| Programme | Year | Task | URL |
|---|---|---|---|
| Pseudobase [127] | 1999 | Pseudoknot database | http://pseudobaseplusplus.utep.edu |
| HotKnots [128] | 2005 | Pseudoknot prediction from short sequences | www.cs.ubc.ca/labs/beta/Software/HotKnots/ |
| PseudoViewer [129] | 2006 | Webserver for pseudoknot visualisation | http://pseudoviewer.inha.ac.kr |
| PknotsRG [116] | 2007 | MFE pseudoknot prediction from short sequences | http://bibiserv.techfak.uni-bielefeld.de/pknotsrg/welcome.html |
| McQFold [130] | 2008 | Pseudoknot prediction by Markov-chain Monte-Carlo (MCMC) sampling | www.cs.uni-frankfurt.de/~metzler/McQFold |
| ProbKnot [131] | 2010 | Fast prediction of pseudoknots of any topology | http://rna.urmc.rochester.edu/RNAstructure.html |
| HotKnots V2.0 [119] | 2010 | Pseudoknot prediction from short sequences | www.cs.ubc.ca/labs/beta/Software/HotKnots/ |
| IPknot [120] | 2011 | Pseudoknot prediction from single or aligned sequence(s) with <1000 bases | www.ncrna.org/software/ipknot/ http://rna.naist.jp/ipknot/ |
| TT2NE [121] | 2011 | Pseudoknot prediction from short sequences (⩽200 bases) | http://ipht.cea.fr/rna/tt2ne.php |
| McGenus [122] | 2012 | Pseudoknot prediction from sequences ⩽ 1000 bases | http://ipht.cea.fr/rna/mcgenus.php |
| Iterative HFold [123] | 2014 | Pseudoknot prediction based on an inputted pseudoknot-free structure | www.csubc.ca/~hjabbari/software.php |
4.4. Summary
In contrast to DNA, naturally occurring RNA, strictly speaking, does not form knotted structures. However, it frequently adopts structurally complex conformations in which there are a number of topological crossings of its chain. These structures are known as pseudoknots and are widespread in terms of the different classes of RNA in which they are found. They vary in the length and presence/absence of loop regions and therefore represent a structurally diverse group. It is perhaps, therefore unsurprising that the pseudoknot structure is associated with a range of different biological processes, including catalysis, ribosomal frameshifting and regulation of translation. Although it is not completely understood how their structure results in their specific activities, it is clear that the pseudoknot structure is stable (although it can be in equilibrium with other conformations such as hairpins), may be particularly stable with respect to unwinding by helicases, or degradation. Prediction of the structure of pseudoknots in RNA has rapidly developed over recent years, and, although it is still challenging for very long sequences, a number of different approaches can be used which are increasing in speed and accuracy. Interested readers are directed towards the following references for a more detailed discussion of all of these topics [10, 11, 14, 63, 68, 69]. It is interesting to note that RNA sequences have been designed to form a synthetic trefoil knot [132], see Discussion for further details.
5. Proteins
Proteins are linear biopolymers composed of different amino-acid residues covalently linked together by peptide bonds. They play a crucial role in almost all biological processes including cell signalling, catalysing metabolic reactions and structural support. In order to perform their function, most proteins have to fold to a compact 3D structure (native state), which is ultimately dictated by its unique amino-acid sequence.
Many thousands of proteins with a diverse array of structures and functions are known. Due to their structural variation and complexity, proteins have been shown to possess a wide range of intricate topological features (figure 8). Inter-molecular non-covalent interactions can lead to interlocked, oligomeric rings of protein subunits, where the two rings form a Hopf link and therefore become inseparable (figure 8(a)) [133]. In other cases, covalent bonding such as disulphide bonds or metal-side chain interactions can also result in covalent links or knots formed either during or after folding. Figure 8(b) illustrates a Hopf link structure formed as a result of intra-molecular disulphide bonds within each subunit of a dimeric protein [134]. In addition, the recently discovered pierced lasso bundle (PLB) topology is an example of a knot-like motif where the disulphide bond creates a covalent loop through which part of the polypeptide chain is threaded (figure 8(c)) [135]. 'Cysteine knots' can form when a disulphide bond between two segments of a polypeptide chain pass through a ring formed by two other disulphide bonds and their connecting backbone segments (figure 8(d)). Examples include the cyclotide family of naturally occurring plant-based miniproteins and the superfamily of growth factors and toxins [136–138]. In all of these cases, the link or knot is created by a covalent bond or oligomeric structure.

Figure 8. Different types of topologically complex protein structures. In each panel, the protein structure produced using Pymol (www.pymol.org/) is shown on the left, with a simplified representation of the topology of the system on the right. (a) The crystal structure of bovine mitochondrial peroxiredoxin III forms a Hopf link, PDB code: 1ZYE. In the simplified representation, the blue and red filled circles represent a single chain subunit which associate together to form a higher-order oligomeric ring structure. (b) P. aerophilum dimeric citrate synthase is topologically linked by two intramolecular disulphide bonds (black bars), PDB code: 2IBP. Each protein chain is coloured separately, in this case, blue or teal. (c) A pierced lasso bundle topology of the native structure of leptin, where a disulphide bridge (black bars) creates a covalent loop through which part of the polypeptide chain is threaded, PDB code: 1AX8. (d) The crystal structure of nerve growth factor contains a cysteine knot motif defined by three disulphide bonds (black bars), PDB code: 1BET. (e) The polypeptide backbone chain of E. coli methyltransferase YbeA contains a trefoil knot (31), PDB code: 1NS5. (f) The crystal structure of human phosphatase has a slipknotted topology, PDB code: 1EW2. For (c)–(f), both structures and reduced representations are coloured from blue (N-terminus) to red (C-terminus). Cysteine residues in (b)–(d) are represented as sticks and lines in the structure and simplified representation, respectively.
Download figure:
Standard image High-resolution imageComplex topologies such as linking or knotting can also be manifested within the protein backbone chain itself. Figure 8(e) illustrates an example of a class of proteins that possess a knotted topological feature in their structures formed by the path of the polypeptide backbone alone [13, 15, 139]. In another case, protein slipknot structures also arise when a protein chain forms a knot but then folds back upon itself to completely untie the knot, thus rendering the structure unknotted when considered in its entirety (figure 8(f)) [140–142]. This section of the review focuses on the structure, function and, in particular, the folding of these types of knotted and slipknotted proteins. Proteins that have knots formed by covalent bonds such as disulphides are not discussed here and readers who are interested in these structures are directed to other publications on these systems [136, 137, 143–146].
5.1. Knotted and slipknotted proteins
For a long time, it was thought that it was highly unlikely, if not impossible, for a polypeptide chain to 'knot' itself to form a functional folded protein. This was, in part, due to the fact that, at that time, no examples of deeply knotted proteins were identified within the protein data bank (PDB) [147]. In this study, a very shallow knot was discovered in carbonic anhydrase by Mansfield [147]. One of the challenges in the search for protein knots was the difficulty in determining whether a knot is present within a complex structure. Thus, for many years, knots in protein structures went undetected. As various computational and mathematical tools were developed to detect and identify knots, it became clear that topologically knotted protein structures do exist, even some with extremely deep knots [24, 26, 148, 149]. Now there are a few web-servers that have simplified the task of knot identification in proteins and can determine quickly whether a structure contains a knot and, if so, what type [150, 151]. In addition, the recent KnotProt database (http://knotprot.cent.uw.edu.pl/) created by Sulkowska and co-workers classifies knotted proteins and represents their knotting complexity (knot type and depth of knot) as a 'knotting fingerprint' in the form of a matrix diagram [142, 152, 153]. Matrix diagrams, which are an excellent method for visualising knots and slipknots in proteins, were originally used in the analysis of slipknots in proteins by the Yeates group [140].
To date, over 750 knotted proteins have been discovered within the PDB, equivalent to approximately 1% of all entries [152]. A current list of examples of these structures is provided in table 2. It is worth noting that the KnotProt database is updated regularly [152]. Over the years, a growing number of knotted proteins have been observed in all three domains of life [15, 142, 154]. These include structures that contain a trefoil (31), figure-of-eight (41), Gordian (52) and stevedore (61) knot with three, four, five and six projected crossings of the polypeptide backbone, respectively (figure 9).
Table 2. Examples of knotted and slipknotted proteins. For each fold, the PDB code for the structure of the protein or a typical protein in the family is given. + and − indicates right and left-handed knots and slipknots, respectively.
| Protein family or Protein | PDB code | Knot type |
|---|---|---|
| RNA methyltransferase (α/β knot) | 1NS5 | 31 + knot |
| Carbonic anhydrase | 1LUG | 31 + knot |
| SAM synthetase | 1FUG | 31 + knot |
| Transcarbamylase fold | 1JS1 | 31 + knot |
| Sodium/calcium exchanger membrane protein | 3V5S | 31 + knot |
| Zinc-finger fold | 2K0A | 31 − knot |
| Ribbon-helix–helix superfamily | 2EFV | 31 − knot |
| Artificially knotted protein | 3MLG | 31 − knot |
| Class II ketol acid reductoisomerase | 1YVE | 41 knot |
| Chromophore binding domain of phytochrome | 2O9C | 41 knot |
| Ubiquitin C-terminal hydrolases (UCHs) | 2ETL | 52 − knot |
| α-haloacid dehalogenase I | 3BJX | 61 + knot |
| Alkaline phosphatase | 1ALK | 31 + slipknot |
| Thymidine kinase | 1P6X | 31 + slipknot |
| Glutamate symport protein | 2NWL | 31 + slipknot |
| Sulfatase | 4TN0 | 31 + slipknot |
| STIV B116 | 2J85 | 31 + slipknot |
| Apoptosis inducing factor | 1GV4 | 31 − slipknot |
| Sodium:neurotransmitter symporter family | 2A65 | 31 + & 41 slipknot |
| Betaine/Carnitine/Choline Transporter (BCCT) family | 4AIN | 31 + & 41 slipknot |
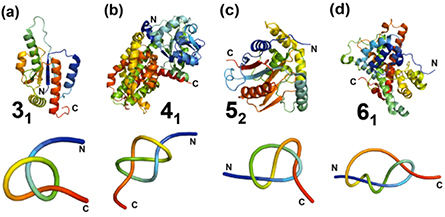
Figure 9. Structures of knotted proteins that contain the four different types of knots (31, 41, 52, 61) in the polypeptide backbone. (a) YbeA, a trefoil-knotted (31) methyltransferase from E. coli, PDB code: 1NS5. (b) E. coli class II ketol-acid reductoisomerase, containing the figure-of-eight (41) knot, PDB code: 1YRL. (c) Human ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), containing a knot with five projected crossings (52), PDB code: 2ETL. (d) α-haloacid dehalogenase containing a stevedore (61) knot, PDB code: 4N2X. Top panel: ribbon diagrams of the polypeptide chains produced using Pymol (www.pymol.org/). Lower panel: simplified view of the protein chain showing the knot, generated using KnotPlot (http://knotplot.com/). Both structures and reduced representations are coloured from blue (N-terminus) to red (C-terminus).
Download figure:
Standard image High-resolution imageTrefoil knots are the most prevalent and simplest type of knot discovered in proteins. The first protein trefoil knot to be identified was that found in carbonic anhydrase—a family of proteins involved in catalysing the reaction of carbon dioxide to hydrogen carbonate and H+ [147]. This trefoil, however, is rather shallow as the C-terminus extends through a wide loop by only a few residues. A few years after Mansfield's 1994 study, a much deeper trefoil knot was detected in E. coli S-adenosylmethionine synthetase, an enzyme that catalyses the reaction between methionine and ATP [155, 156]. By far, the largest and most well-studied family of deeply knotted proteins is the trefoil α/β knot fold—a class of methyltransferases (MTases) which are members of the SpoU family [157, 158]. These knotted proteins share common structural features and it is highly likely that all are MTases that catalyse the transfer of the methyl group of S-adenosyl methionine (AdoMet) to carbon, nitrogen or oxygen atoms in DNA, RNA, proteins and other small molecules [159]. In solution, all form dimers with the knotted region comprising part of the AdoMet binding site and forming a large part of the dimer interface [157, 160–163]. Trefoil knots have also been found in two homologues of N-succinylornithine transcarbamylase; the AOTCase from X. campestris catalyses the reaction from N-acetylornithine and carbamyl phosphate to acetylcitrulline [164], and SOTCase from B. fragilis promotes the carbamylation of N-succinylornithine [165]. Besides being found in enzymes, trefoil knots have also been identified in Rds3p, a eukaryotic metal-binding protein essential for pre-mRNA splicing [166] and more recently, in the family of sodium/calcium exchanger membrane proteins [152].
More complex knots have also been identified in proteins that catalyse various enzymatic reactions. A deeply embedded, figure-of-eight protein knot has been found in plant ketol-acid reductoisomerases, which are involved in the biosynthesis of branched-chain amino acids [167, 168]. In addition, a Gordian knot has been identified in the family of mammalian ubiquitin carboxyl-terminal hydrolases (UCHs); the proteins are deubiquitinating enzymes that catalyse the cleavage of the isopeptide bond formed between ubiquitin and lysine side chains of protein and other adducts, and thus are involved in the ubiquitin-proteasome system [169–171]. The most complex protein knot known to date is the 61 stevedore knot discovered in DehI, a α-haloacid dehalogenase that catalyses the removal of halides from organic haloacids [154]. Apart from these enzymes, it has been shown that the figure-of-eight knot also exists in the chromophore-binding domain of a red/far-red photoreceptor phytochrome from bacterium D. radiodurans [172, 173].
Slipknotted structures have also been found in a number of proteins (figure 8(f)) [140]. They cannot be identified using the standard methods for knot detection in proteins as, in these cases, the knot becomes undone when the chain is pulled at both termini. As such, it comes as no surprise that these structures had been overlooked until relatively recently. In 2007, Yeates and co-workers first discovered a number of protein slipknots by using an approach based on the fact that slipknots become real knots at some point when the polypeptide chains are shortened [140]. At present, over 450 protein slipknots have been identified [152] and a list of examples of these structures is listed in table 2. It is worth noting that the KnotProt database is the first, and currently only, database that provides details on slipknotted structures [152].
Alkaline phosphatase is the largest family of proteins that contain deep slipknots [15, 140, 152]. In the case of E. coli alkaline phosphatase, 30 residues have to be deleted from the C-terminus before a knotted conformation results. Similar to that of knotted proteins, many of the protein slipknots discovered to date are also found in other enzymes such as thymidine kinases and sulfatases [15, 140, 152]. Interestingly, slipknots have also been found in transmembrane proteins that span the entire cell membrane to which they are permanently embedded [15, 140, 152]. Examples include the families of sodium:neurotransmitter transporters, betaine/carnitine/choline transporters (BCCT) and proton:glutamate transporters [142].
Further details of knotted and slipknotted protein structures can be found in other recent reviews [12, 13, 15, 174] and the KnotProt server [152]. It should be noted that the KnotProt database also provides extensive key information about the biological functions of proteins with knots and slipknots [152].
5.2. Potential roles and implications of the knot and slipknot
Topologically knotted proteins have been found to be conserved across different families [142], suggesting that the knot itself may be advantageous and important to the function of the protein. It has been speculated that a knotted topology could play a key role in increasing catalytic activity or ligand binding affinity (potentially by decreasing dynamics) or enhancing stability (thermodynamic, kinetic and mechanical) of a protein. As yet, relatively little is known about the functional advantages, if any, of these complex knotted structures over their unknotted counterparts. However, various experimental and computational studies have been undertaken to address this question.
Many reports have shown that the knotted regions of knotted proteins play crucial roles in enzymatic activities and ligand binding. As discussed in section 5.1., it has been observed that the knotted regions of the proteins in the α/β-knotted SpoU MTase family comprise part of the active site to which the ligand binds (two examples of α/β knot MTases are illustrated in figure 10(a)) [159–162]. In the case of the N-succinylornithine transcarbamylase, Virnau and co-workers have demonstrated through a computational study that the presence of the knot in the knotted homologue AOTCase may structurally modify its active site and subsequently, may alter its enzymatic activity (in terms of substrate specificity) compared to its unknotted homologue OTCase (figure 10(b)) [149]. In addition, structural studies of the D. radiodurans phytochrome revealed that the deeply embedded knot in the chromophore-binding domain is in contact with the chromophore [172, 173]. A recent study on the conservation of knotting fingerprints in UCHs also showed that there was a correlation between the locations of active site residues and points characterising its knotted topology (i.e. the knotted core) [142]. Despite these examples, there is still little direct experimental evidence that a knotted structure can influence the activity of a protein.
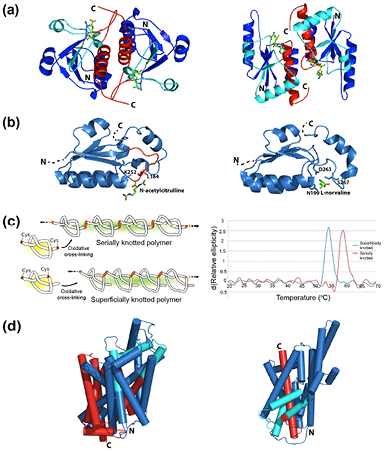
Figure 10. Examples highlighting the potential roles of knots and slipknots. (a) Dimeric structures of the α/β-knot MTases YibK, PDB code: 1MXI (left) and YbeA, PDB code: 1NS5 (right), coloured to show the knotting loop in cyan and the knotted chain in red. S-adenosyl homocysteine, an MTase co-factor, is shown as a stick model. (b) Structures of the knotted section (residues 171–278) of AOTCase with the reaction product N-acetylcitrulline and interacting side chains represented as sticks, PDB code: 3KZK (left), and corresponding (unknotted) section (residues 189–286) in OTCase with the inhibitor L-norvaline (analogous to its L-ornithine ligand) and interacting side chains shown as sticks, PDB code: 1C9Y (right). The knot containsf a rigid proline-rich loop (residues 178–185, coloured red) through which the chain is threaded. (c) Left panel: engineered knotted and unknotted ('superficially knotted') polymers using two different protein constructs. Right panel: first derivative melting curves obtained for the knotted and unknotted polymers. Adapted from [179], by permission of Oxford University Press. (d) Structures of transmembrane proteins LeuT(Aa), PDB code: 2A65 and Glt(Ph), PDB code: 2NWL, where the slipknot loop is coloured cyan and the slipknotted chain in red. Helices are represented as cylinders to ease visualisation. All structures are produced using Pymol (www.pymol.org/).
Download figure:
Standard image High-resolution imageThe question of whether knots have any effect on the conformational dynamics of proteins has also been raised. In the phytochrome protein, it has been noted that the figure-of-eight knot sits where increased rigidity could be important in driving conformational changes that occur when light energy is absorbed by the chromophore [172, 175]. Recent computational approaches using simple lattice models have shown a narrow and less extended native basin for a 52-knotted structure relative to a similar but unknotted one, suggesting enhanced rigidity [176]. However, experimental studies by Andersson et al, which measured 15N spin relaxation parameters using NMR experiments for the 52-knotted UCH-L1, reported no significant differences between the relaxation properties of the knotted protein relative to unknotted proteins of a similar size [177]. Thus, it remains to be clearly established, particularly experimentally, whether knotted structures can influence the conformational dynamics of a protein.
Much research effort has been undertaken to address the question of whether a knot can provide additional thermodynamic, kinetic or mechanical stability to a protein structure. Sulkowska et al performed coarse-grained simulations of the thermal and mechanical unfolding of the knotted (AOTCase) and unknotted (OTCase) variants of the transcarbamylase-like proteins as well as a synthetic construct of the knotted parent protein rewired so as to remove the knot [178]. In this case, the knotted structure was found to have longer unfolding times than the other two unknotted proteins, which were attributed to topological and geometrical frustration [178]. In an attempt to investigate the potential thermal stabilities of knotted proteins in an experimental study, Yeates and co-workers engineered a knotted and an unknotted ('superficially knotted') polymer [179]. They showed that the knotted chain had a higher thermal stability than the unknotted one (figure 10(c)), although it is important to note that the unfolding in both cases was not fully reversible and therefore only apparent melting temperatures were reported. However, computational studies using Monte Carlo simulations of a simple lattice model using Gō-like potentials showed that a trefoil knot did not have any effect on the thermodynamic stability of a simple protein structure [180]. Instead, it was found that the knot enhances kinetic stability as the knotted protein unfolds at a distinctively slower rate than its unknotted counterpart [180]. Further studies by the same group demonstrated that a more topologically complex protein knot, the 52 knot, clearly enhanced the protein's kinetic stability in comparison to that of a protein containing a 31 knot [176].
The resistance of knotted proteins to mechanical unfolding has been examined by atomic force microscopy (AFM). The first system to be studied was the shallow trefoil-knotted carbonic anhydrase B. In this particular case, an extremely high resistance to unfolding was observed when the protein was pulled from its termini in contrast to a considerably lower resistance when the molecule was pulled from other positions resulting in the untying of the knot [181, 182]. Although these initial studies suggested a dramatic effect of a knot on mechanical stability, the results have not been observed in AFM studies of other knotted systems [175]. In the case of carbonic anhydrase B, recent simulations have shed light on the possible reasons for its remarkable mechanostability [183]. These studies revealed that after an initial, rather limited unfolding event, the knot is wrapped around an inner β-sheet structure in the core of the protein. Thus, the knot is tightened but effectively locally captured by a structural obstacle in the chain. This is aided by the stabilising effects of a zinc ion, which coordinates to the region that becomes entangled by the knot. The simulations explain why in the AFM experiments, the contour length observed is so much smaller than that expected for a fully stretched polypeptide chain containing a tightened knot. In an interesting extension of their initial work, Ikai and co-workers made a tandem repeat of carbonic anhydrase B. Combining AFM with biochemical measurements of activity and binding, they were able to establish that the C-terminal knotted region was essential for activity [184].
The mechanical stability of the 41-knotted phytochrome protein has also been investigated by Bornschögl and co-workers using AFM [175]. In this case, however, they did not observe any enhanced resistance when the knot was tightened as the extension force for unfolding (73 pN) was within the range found for other unknotted proteins. It appears that whether a knot contributes to mechanical stability or not, may depend upon a number of factors including other aspects of the protein's structure and potentially pulling speed/force etc. Several computational studies have suggested that knotting might increase a knotted protein's mechanical stability, thus making it more resistant to cellular translocation and degradation pathways [149, 178, 185, 186]. Again, whether knotting confers any advantageous stabilising effect to a knotted protein over its unknotted counterpart is still inconclusive and thus remains to be tested with more experimental and computational studies.
The significant number of protein slipknots that have now been identified has also posed the question of whether such topologies have any functional or structural role in the protein. In the case of the homodimeric E. coli alkaline phosphatase, Yeates and co-workers engineered cysteine residues at various positions in the protruding loop of the slipknot such that inter-molecular disulphide bonding between the two subunits resulted in a knotted system [140]. Using thermal denaturation, the results showed that the knotted mutants were more thermally stable than either the wild-type or other control mutants. This suggested that the slipknot in the structure may play a role in the enzyme's thermostability [140]. It is also worth noting that the slipknotted B116-like protein is found in a virus that infects thermophilic Sulfolobus archaebacteria [140]. In another study, knotting fingerprint analyses of transmembrane transporting channels from five different families of proteins showed that the slipknotted topology is conserved. This has led to speculations that the slipknot loop, which straps together several transmembrane α-helices, may stabilise their location inside the membrane during their transporter and symporter action [142] (see figure 10(d) for examples of the structures of two slipknotted transmembrane proteins).
5.3. Experimental and computational insights into how knotted and slipknotted proteins fold
The study of how proteins achieve their unique 3D conformation (native state) has been the focus of many researchers in the field of protein folding. For many decades, extensive folding studies focussed on small, monomeric proteins and thus mechanisms of how they fold are now relatively well established [187–191]. These include the framework, nucleation-condensation and hydrophobic collapse mechanisms, which can be viewed as points on a spectrum of a unified mechanism [187, 188]. Current folding theories have shown that small, monomeric proteins, which fold efficiently and rapidly, can achieve their low-energy native configuration from an ensemble of denatured polypeptide chains in a highly cooperative manner and traverse relatively smooth, funneled energy landscapes [192, 193]. However, it is still unclear how these concepts and mechanisms are applicable to larger proteins with more complex topologies including the classes of knotted and slipknotted proteins. Not only do such proteins have to avoid kinetic traps but they also have to overcome significant topological barriers during folding. This section summarises recent developments made towards understanding the mechanisms involved in the formation of these types of complex structures.
5.3.1. Experimental studies on knotted proteins.
Although the elucidation of how knotted proteins fold using experimental approaches remains challenging, in recent years, some significant progress has been made. Most of the experimental folding studies on knotted proteins have focussed on the trefoil-knotted α/β MTases, YibK from H. influenzae and YbeA from E. coli [194–201]. Both proteins are homodimers, which bind to the co-factors AdoMet and S-adenosyl homocysteine (AdoHcy) and contain a trefoil knot at the C-terminus in which at least 40 residues pass through a similarly sized loop (figure 10(a)) [160, 202]. Extensive biophysical techniques have been employed to probe the knotting and folding mechanisms of purified, recombinantly expressed YibK and YbeA. Both unfold reversibly in vitro upon addition of chemical denaturant with a concomitant loss of secondary and tertiary structure [195, 198]. Kinetic studies demonstrated that YibK and YbeA fold similarly via sequential mechanisms that involved one or more monomeric intermediate states and a slow rate-limiting dimerization step [196, 198].
To probe chain knotting events during the folding of YibK and YbeA, Mallam and co-workers constructed a set of knotted fusion proteins in which A. fulgidus This, a stable 91-residue protein, was fused to the N-, C- or both termini of both MTases [201]. This was used as a 'molecular plug' in an attempt to disrupt threading events or to prevent the chain from knotting altogether. Remarkably, these experiments established that both proteins can withstand the fusion of additional domains to both their N- and C-termini and are able to fold to native or native-like states capable of binding cofactor. The fusion proteins created in this study represent some of the most deeply knotted proteins known, the C-terminal fusions requiring some 140 or more residues to pass through a loop to form the knotted native state. Surprisingly, all the fusion proteins showed unfolding and refolding kinetics very similar to the parent MTase giving the first hint that the polypeptide chain might remain knotted even in a highly unstructured chemically denatured state. This was subsequently shown to be the case through in vitro folding experiments on circularized variants of YibK and YbeA, Mallam and co-workers discovered that the denatured ensembles, even in high concentrations of chemical denaturant under which conditions there was little or no secondary or tertiary structure, contained kinetically trapped knotted polypeptide chains [194]. It was then concluded that all the previous in vitro folding experiments on these recombinantly expressed and chemically denatured proteins actually probed refolding from an unfolded but knotted denatured state to a knotted and folded native structure. This unexpected result suggests that there are interactions in the denatured state that kinetically stabilize the knot. Although far-UV CD measurements indicate that there is no significant secondary structure present in the denatured state, recent backbone NMR assignments and chemical shifts of urea-denatured YbeA, show that, in fact, some residual secondary structure still remains under these conditions [203]. The fact that the knot can persist in the denatured state over a long period of time was also confirmed by another group who shared that equilibrium unfolding and refolding transitions of a structurally homologous MTase displayed apparent hysteresis [204]. This behaviour was speculated to be consistent with the uncoupling of the unfolding and untying events of the knotted protein [204]. Recently, single-molecule fluorescence resonance energy transfer (FRET) experiments were performed to characterise the denatured state of TrmD, another trefoil-knotted MTase [205]. Results suggested that the knot was not only retained under denaturing conditions (similar to that of YibK and YbeA) but also slid towards the C-terminus of the polypeptide chain during the unfolding process [205].
Up until recently, there have been no experimental studies into how the knot is first formed from an unknotted linear polypeptide chain. However, with the use of a coupled in vitro transcription-translation system and kinetic pulse-proteolysis experiments, Mallam and Jackson were able to specifically probe folding of nascent chains of YibK and YbeA after they were first synthesised by the ribosome (figure 11(a)) [199]. The results showed that the nascent chains could fold correctly to their trefoil-knotted structure, albeit very slowly. Moreover, a significant lag period between chain synthesis and emergence of a proteolytically stable native state was observed. The results were consistent with the protein knotting and folding from an initially unknotted nascent chain, thus demonstrating that a process associated with the knotting step is rate limiting. Additionally, the GroEL-GroES chaperonin was found to have a dramatic effect on the folding rate of the newly translated polypeptide chains, thus establishing that chaperonins are likely to be important in the post-translational folding of these bacterial knotted proteins in vivo.

Figure 11. Experimental characterisation of the folding of the trefoil-knotted methyltransferases, YibK and YbeA. (a) A schematic representation of the folding and knotting pathways that have been experimentally observed. (b) A schematic diagram illustrating a possible active mechanism for the bacterial GroEL-GroES chaperonin action on the folding of bacterial trefoil-knotted methyltransferase. D, denatured; I, intermediate; N, native.
Download figure:
Standard image High-resolution imageVery recently, we have investigated the knotting and folding behaviour of the nascent chains of the different N- and C-terminal This fusions of YibK and YbeA with the use of the coupled in vitro transcription-translation system and kinetic pulse-proteolysis experiments [206]. The results demonstrated that these multi-domain proteins with extremely deep knots can be synthesized in vitro and spontaneously knot without the help of any molecular chaperones, albeit very slowly. In addition, it was concluded that the C-terminus of these proteins is critical to the threading of the polypeptide chain to form the knot, thus providing the first experimental insight as to the mechanism of knotting for this class of bacterial knotted MTase. Further experiments with the GroEL-GroES chaperonin demonstrated that it actively assists the folding of knotted proteins by a mechanism that may involve the unfolding of kinetically trapped unknotted and misfolded intermediates (figure 11(b)). These key observations provide not only vital information into the complex folding pathway of trefoil-knotted proteins but also further insights into how topologically knotted proteins have withstood evolutionary pressures and achieve efficient folding in vivo.
In 2010, the Yeates group engineered an artificially trefoil-knotted protein by covalently linking together two monomers intertwined in the dimeric structure of HP0242 from H. pylori [207]. An in vitro experimental characterisation of this designed knotted protein and an unknotted monomeric variant of the HP0242 dimer was undertaken. Results showed that, although the knotted variant was more stable than the unknotted one, it folded at a considerably slower rate (approximately 20-fold), indicating that knotting, or some event associated with it, is likely rate-limiting.
AFM has also been used to study the mechanical unfolding of the shallow trefoil-knotted carbonic anhydrase B. In this case, the polypeptide chain was found to extend to a distance much shorter than its theoretical stretching length, indicating that the knotted structure is tightened but retained [182, 208]. Similarly, AFM mechanical unfolding experiments on the figure-of-eight knot in the chromophore-binding domain of the phytochrome also resulted in a tightened knot of approximately 17 residues [175]. Although these experiments do not necessarily provide extensive information on the folding pathways of these proteins, they were critical in demonstrating that the knots were present in the structure and in determining the minimum length of polypeptide chain required for knotting.
In addition to the trefoil-knotted proteins described in detail above, the other family of knotted proteins for which there has been any substantial experimental characterisation of their folding pathways are the 52-knotted UCHs [177, 209]. The unfolding of two human UCHs- UCH-L1, a neuronal form of the enzyme, and UCH-L3, ubiquitously expressed in many cell types, have been determined and, in both cases, the in vitro unfolding/refolding with chemical denaturants was shown to be fully reversible [177, 209]. In the case of UCH-L3, equilibrium unfolding data were fitted to a simple two-state model [209] whilst that for UCH-L1 were consistent with a three-state model in which an intermediate state is populated [177]. Using NMR hydrogen-deuterium exchange (HDX) experiments, the intermediate state was characterised indirectly and it was found that the central β-sheet core of the protein remains structured whilst many of the surrounding α-helices have unfolded [177]. Although a more complete analysis of the folding pathway of UCH-L1 has yet to be published, the folding is similar to UCH-L3, such that, both have multiple unfolding and refolding phases that indicate parallel pathways and the population of at least two, metastable intermediate states (Luo et al unpublished results).
5.3.2. Computational studies on knotted proteins.
Many computational studies have shed considerable light on the folding of knotted proteins. Coarse-grained simulations have been excellent at revealing the possible mechanism(s) and generic features of how knotted proteins fold [210, 211]. Wallin et al performed the first such simulation using a Cα model representation of YibK and, similar to experimental studies, observed two parallel folding pathways [210]. They also concluded that specific, non-native interactions involving residues in the C-terminal region of the chain were needed for the protein to knot and fold successfully. In contrast, Sulkowska and co-workers showed that native interactions alone are sufficient for simulating the folding of YibK and YbeA using a coarse-grained structure-based model, although the number of successful trajectories was only 1–2% [211]. These simulations also illustrated that partial unfolding (backtracking) events were needed because the order in which native contacts are formed is critical for the correct folding of the knotted structure and that folding frequently occurred through a slipknotted intermediate (figure 12(a)). Importantly, in the same study, simulations of a rewired, unknotted variant established that there are significant topological barriers in the folding of the knotted structure [211]. Using a similar model, initial results from recent kinetic unfolding simulations of a structurally homologous MTase revealed that unfolding of the protein to a fully unfolded, unknotted state occurs in a stepwise process [204]. In addition, the simulations showed that unknotting of the chain is slow compared to the initial unfolding [199].

Figure 12. Computational simulations of the folding pathways of knotted proteins. (a) Structure-based model used to simulate the folding of trefoil-knotted MTase where the folding route that leads to the native knotted conformation occurs through an intermediate 'slipknot' configuration. Incorrect configurations have to use a 'backtracking' mechanism in order to escape kinetic traps which act as topological barriers. Adapted from [211]. (b) Snapshots taken from the folding simulation of the 61-knotted protein, DehI. Copyright 2010 Bölinger et al [154]. (c) An all-atom structure-based molecular dynamics simulation of the folding pathway of MJ0366. The protein forms a loop with the correct chirality (I), from which it follows two routes to the native state (N): a 'plugging' or 'slipknotting' route. T is an example of how the protein may be kinetically trapped and thus unable to proceed to N. Adapted from [141]. (d) Schematic representations of pulling a trefoil-knotted protein in different points (indicated by the circles) and their resulting final conformations.
Download figure:
Standard image High-resolution imageSimilar computational approaches were also employed in the folding simulations of the 61-knot in DehI [154]. Although the probability of successful folds was low, the study revealed that the complex knotted structure can be formed by a simple tying process. In this case, two unknotted loops, a small loop and a larger loop (which includes a proline-rich unstructured region) are aligned and a knot can be formed by two alternative routes (figure 12(b)) [154]. In the first route, the C-terminus is threaded through the smaller loop (S-loop) via a slipknot conformation before the larger loop (B-loop) flips over the smaller loop. In the other route, the order of the two steps is reversed.
In contrast to very small proteins with simple architectures (which generally have fast unfolding and folding rates), all-atom molecular dynamics (MD) simulations have not been extensively applied to knotted systems, as they are frequently too large for such atomistic approaches to be used. However, it has been possible to use this method in a few cases on small, shallow knotted proteins, such as for MJ0366 from M. jannaschii, one of the smallest trefoil-knotted protein discovered to date [141]. Data from a thermodynamic analysis of the unfolding/folding revealed that the system is three-state, and an intermediate is first formed by twisting of a loop, followed by a rate-limiting step associated with the threading of the C-terminus through the loop. At temperatures near the folding temperature, two folding mechanisms were observed for the formation of the knotted native structure, whereby threading can occur via (i) a plugging route (the C-terminus goes through the knotting loop first) or (ii) the formation of a slipknot (figure 12(c)) [141]. Interestingly, lowering the temperature of the simulation resulted in mechanistic changes. These include a knotting via threading of the N-terminus and the 'backtracking' of misfolded proteins in topological traps. More recently, simulations on VirC2, a protein that has the same fold as MJ0366 but which possesses a deeper knot, also showed that it has a similar free energy profile, suggesting that topology plays a major role in the folding mechanism [212]. A Gō-like potential in which there is minimal energy frustration was also used to simulate the folding of a truncated mutant of another trefoil-knotted MTase [213]. Results from this study suggested a pathway in which the N-terminal region of the protein folds first and that threading of the C-terminus through the structure to form the knot is a late and rate-limiting step [213].
Molecular dynamics simulations were also used to simulate the high temperature unfolding of YibK [214]. The simulations revealed up to four intermediate states on the free energy landscape consistent with the parallel pathways and multiple intermediates observed in experimental studies. In addition, it was found that the denatured state of YibK only untied at very high simulation temperatures, when the C-terminus threads out of the knotting loop via a slipknot conformation. Other unfolding simulations have also been used to investigate the mechanical stability of knotted proteins and the effect of pulling position, pulling speed and temperature on the unfolding/untying of two other MTases [215]. It was shown that pulling the chain at both termini leads to the tightening of the knot whilst pulling at other positions can result in the unknotting of the chain (figure 12(d)).
Various computational studies have also employed Monte Carlo simulations on lattice models using Gō-like potentials to understand the folding mechanism of knotted proteins. In these cases, a potential based on a generic polymer model is used and additional attractive interactions are included for residues that are in contact with each other in the native state. Faisca and co-workers demonstrated that the folding of a model deeply knotted trefoil protein was much slower than a structurally similar but unknotted variant, and that knotting was a late event and concomitant with folding [216]. Using the same model, Soler and Faisca examined the effect of surface tethering on the folding of the system [217]. In this case, it was shown that the mobility of the terminus closest to the knot is critical for successful folding and hindrance results in a decrease in the folding rate and a change in the knotting pathway such that it involves threading of the other terminus. Recently, the same group extended these studies and used the same model to investigate in further detail the effect of knots, knot depth and motif on folding properties of 31-knotted proteins [180]. The results revealed that deeply knotted proteins have a higher probability of retaining their knots in the denatured ensemble, consistent with experimental studies. Furthermore, it was shown that specific native contacts within the trefoil-knotted core are crucial in maintaining the knot in the denatured state, and that threading occurs in the late stages of folding [180]. Most recently, Soler and co-workers extended their studies to investigate the folding mechanism of the more complex 52-knot [176]. Similar to the trefoil knots, it was shown that the chain terminus that is closest to the knotted core is important for the threading movement to form the knot and in no cases was a mechanism that involved the initial formation of a 31-knot observed. However, it was discovered that the probability of concomitant knotting and folding of 52-knotted proteins is significantly smaller than that for trefoil knots as threading to form the 52 knot is a particularly late conformational event [176].
Monte Carlo simulations of a Cα model of trefoil-knotted AOTCase showed that non-native contacts between the C-terminus and other regions in the protein are critical to form the knotting loop through which the chain is threaded [218], consistent with the study by Wallin and co-workers [210]. The importance of non-native interactions in promoting the folding of the native knotted topology of AOTCase and MJ036 was also recently highlighted in simulations employing protein models with different structural resolution (coarse-grained or atomistic) and various force fields (from pure native-centric to realistic atomistic ones) [219]. Again, it appears that these contacts were found to be between the C-terminus and a loop, through which the chain is threaded.
5.3.3. Experimental and computational studies on slipknots.
Numerous simulation studies have shown that a slipknot may be an important intermediate configuration in the folding of knotted proteins [141, 142, 211, 212] and thus, understanding the mechanisms involved in their formation could offer insights into how deeply knotted proteins fold. Using structure-based coarse-grained simulations, Sulkowska and co-workers investigated the folding of thymidine kinase and found that its slipknotted structure can be achieved by a simple 'flipping' mechanism in which a slipknot loop rotates over the unknotted native core of the protein [211]. The rotation of the loop is most likely assisted by the presence of glycine and proline residues in the hinge regions [211]. However, the low success rate of folding events observed suggests that other factors may be needed to overcome the topological barrier or that the barrier is large. The same group extended these studies and used the same model to analyse the mechanical unfolding of the slipknot in the same protein [220]. Weak stretching forces resulted in the smooth untying of the slipknot whilst a metastable intermediate with a tightened knot was observed at sufficiently large pulling forces. It is worth noting that this behavior of slipknotted structures is different to that observed for uniformly elastic polymers [220]. Recently, He and co-workers used AFM to study experimentally the mechanical unfolding of AFV3-109, a protein which has a relatively simple slipknotted structure [221, 222]. Results showed that the slipknot untied and the polypeptide chain was fully extended when mechanical forces were applied at both termini as expected [221]. In contrast, applying forces at the N-terminus and the threaded loop resulted in the tightening of the slipknot into a trefoil knot involving ~13 amino acid residues [222]. In both cases, the unfolding process was found to proceed via multiple parallel pathways in either a two- or three-state fashion, and is consistent with a kinetic partitioning mechanism for mechanical unfolding [221, 222].
5.4. Evolution and conservation
Despite the fact that there are now a considerable number of topologically knotted proteins in the PDB, it is worth noting that most proteins are unknotted. This suggests that evolution has, in general, avoided such structures. However, a recent study by Sulkowska and co-workers has established that, when they do occur, that both knotted and slipknotted topologies are conserved across different families despite very low sequence similarity [142]. Unsurprisingly, the parts of proteins which are strongly conserved are found within the knotted core and potential hinge regions which it has been speculated are important in the threading of the chain to form a knot or slipknot [142].
For some families of proteins, where there are a sizeable number of knotted and unknotted variants, it has been possible to undertake a phylogenetic analysis of the sequences, and thereby identify how knotted structures may have evolved from unknotted ancestors. Potestio and co-workers generated a phylogenetic tree of transcarbamylase-like folds [223]. In this case, it was known that some knotted and unknotted variants had different degrees of sequence identity suggesting pathways where structures and therefore sequences had diverged at different times. For example, the two knotted enzymes AOTCase and SOTCase share only 35% sequence identity [224] whilst the knotted AOTCase has 41% sequence identity with unknotted OTCase [225]. Reconstruction of the phylogenetic tree demonstrated that all the knotted homologues populate a sub-branch of the tree and that they differ from unknotted homologues by the presence of additional loop segments [223]. Thus, it has been suggested that some knotted structures have evolved from unknotted ones by the insertion of a 'knot-promoting' loop, which effectively encompasses another part of the chain thus forming the knot.
Loops have also been implicated in the formation of knotted structures from other studies. Virnau and co-workers used computational approaches to show that the knotted transcarbamylase AOTCase possesses a rather rigid proline-rich loop, which is lacking in the unknotted OTCase (figure 10(b)) [149]. Interestingly, the stevedore knot in α-haloacid dehalogenase DehI is also partly formed by a large proline-rich loop that links two unknotted regions within the structure [154].
Using a completely different approach, the group of Yeates have also demonstrated another route to knotted structures through the rational design of a novel knotted structure. In this case, a monomeric knotted protein was created by fusion of C- and N-terminal chains of a homodimer that forms a highly entangled but unknotted structure. This study demonstrated that the genetic fusion and tandem repeat of a gene of an unknotted dimeric protein could lead to trefoil-knotted structures [207].
It is clear that, once formed through some evolutionary pathway, knotted and slipknotted protein structures are highly conserved. However, through both experimental and computational studies, we also know that these types of structures have more complex folding pathways than their unknotted counterparts. This suggests that the knotted and slipknotted motifs within protein families may, in some way, be advantageous and important to either the function, or regulation, of the protein.
5.5. Summary
In summary, both experimental and computational studies have made significant progress in establishing some of the key general features of the folding pathways of topologically complex proteins. In contrast to small monomeric proteins with simple folds, it is clear that proteins with topologically knotted or slipknotted structures have much more complex energy landscapes with many intermediate states and parallel pathways. Computational studies have provided insights into the folding process, which may involve formation of a twisted loop followed by threading via an intermediate slipknot configuration, a plugging route or a 'flipping' mechanism, in which the knotting step may be rate-limiting [141, 211, 226]. In addition, it seems that non-native interactions may play a more important role for these types of structures with complex architectures than for the folding of smaller proteins with relatively simple folds [227–229]. Moreover, the formation of transient misfolded species that results in kinetic traps in the free energy landscape of topologically knotted proteins highly likely requires backtracking events and potentially the action of molecular chaperones so that the native structure can be both rapidly and efficiently achieved [199, 206, 211]. Such a 'frustrated' folding energy landscape is in contrast to the relatively smooth folding funnels proposed for smaller, simpler proteins [192, 230].
A number of recent studies have shown that knotted and slipknotted proteins are conserved suggesting that the knot, or slipknot, potentially play a role in the structure, stability, function or regulation of the protein. Despite this finding, it still has to be unambiguously established whether there are any advantageous properties of a knotted structure over an unknotted one. Indeed, whether there are any chemical or physical properties of such structures that are fundamentally different from unknotted ones. Understanding and identifying such properties will potentially provide key insights for future protein engineering applications and therapeutic developments.
6. Synthetic molecular knots
Over the past few decades, the importance of knots and entanglements in naturally occurring biological systems has played a significant role in motivating chemists to develop synthetic strategies for creating topologically complex molecules [16, 17]. Although molecular knots and links have proven to be challenging targets for synthesis, the formation of such fascinating structures has acquired much interest, not only for their beauty, but also for the potential of discovering novel properties. Moreover, the knowledge gained from the synthetically engineered knots may shed light on the folding mechanisms and properties of natural topologically complex structures.
The first synthesis of a non-trivial topology goes back to the [2] catenanes (interlocked rings) reported by Wasserman in 1960 [231]. Since then, many higher order molecular links such as Solomon links [232, 233], Borromean rings [234, 235], a Star of David catenane [236] and a range of [n]catenanes [17, 237–240] (n denoting the number of interlocked rings) have been successfully synthesised (figure 13). Molecular knots are also increasingly becoming targets for chemical synthesis [16, 17, 241, 242]. Despite the fact that Frisch and Wasserman had first suggested the possibility of using Möbius strips to direct trefoil knot formation in 1961 [243], it was not until 1989 that the first molecular trefoil knot was synthesised [244]. Excluding DNA-based knotted molecules [245, 246] and composite knots [247], to date, only three different types of knots have been synthesised. These are the trefoil knot (31), figure-of-eight knot (41) and pentafoil knot (51). This section of the review focuses on the different synthetic approaches used to produce these molecular knotted structures, their mechanism of formation and physical properties. Molecular links are not discussed here and readers who are interested in these structures are directed to the following [17, 238, 241, 248].
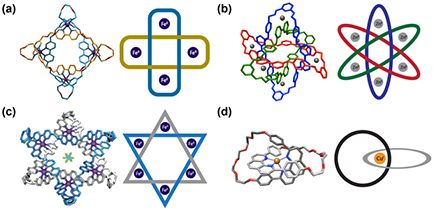
Figure 13. Examples of synthesised higher order molecular links: (a) a Solomon link [232], (b) a Borromean ring [234], (c) a Star of David catenane [236] and (d) a copper-templated [2]catenane [249]. In each case, an x-ray crystal structure (left) and a schematic representation of the link (right) are shown. X-ray crystal structures in (a) and (c) were reprinted with permission from [232] and [236], respectively.
Download figure:
Standard image High-resolution image6.1. Molecular knots: synthetic approaches and mechanism of formation
The synthesis of molecular knots is challenging, as it requires defined pathways and (usually) entropically demanding transition states to achieve a specific knotted structure. Many early experimental efforts (albeit unsuccessful) and proposed synthetic routes towards molecular knots have provided significant insights into the problems of assembling such systems [250, 251]. Over the past two decades, the field of chemical topology has seen various synthetic strategies and approaches being employed for the preparation of different knotted molecules, many of which rely on template effects related to non-covalent interactions identified from supramolecular and coordination chemistry [16, 17, 248]. Here, we discuss these approaches and, in particular, compare the different mechanisms of knot formation using stepwise synthetic approaches to those of 'all-in one' strategies.
6.1.1. Metal template-based synthetic approaches.
Using an extension of Sauvage's original strategy for assembling [2] catenanes [239], Dietrich–Buchecker and Sauvage reported the first successful synthesis of a molecular trefoil knot in 1989 [244]. In this case, the end-groups of a dimetallic, double-stranded helicate, composed of two bisphenanthroline ligands and two copper(I) ions, were connected using Williamson ether synthesis. This generated the three crossing points needed for a trefoil knot; however, it was isolated in 3% yield only. A separate study by Dietrich–Buchecker and co-workers later showed that different spacers linking the phenanthroline groups were critical in determining the yield. In particular, the use of a rigid 1,3-phenylene spacer was found to assist in the stabilisation of the helicate assembly thus resulting in a yield of 29% [252]. However, it was not until the introduction of efficient catalysts for ring-closing olefin metathesis (RCM) that the best yield for a molecular trefoil knot (74%) was achieved (figure 14(a)) [253]. This successful approach was then extended to the preparation of composite knots, details of which can be found in [247]. In another case, the same group used octahedral iron(II) ions reacted with terpyridine-based ligands to template the synthesis of a trefoil knot [254]. The yield achieved, however, was significantly lower (20%), probably because the macrocyclisation was not as effective as that of the previous ligand-metal ion system. Through collaborative work, the groups of von Zelewsky and Sauvage were able to synthesise the first diastereospecific molecular trefoil knot in 74% yield by fusing chiral groups to a 2,2'-bipyridine ligand, thus controlling the stereochemistry of the two copper(I) ions to which the ligands were coordinated [255].

Figure 14. Schematic representations of metal-template based approaches for the synthesis of molecular knots. (a) Synthesis of a phenanthroline molecular trefoil knot in which copper(I) ions are used as templates for the linear helicates to generate the crossings necessary [253], (b) synthesis of a molecular trefoil knot in which an octahedral zinc(II) ion acts as a template for folding and subsequent threading of the ligand [258], (c) active-template synthesis of a molecular trefoil knot [259], (d) lanthanide-template synthesis of a molecular trefoil knot [260]. Metal ions are represented as circles.
Download figure:
Standard image High-resolution imageIn 2001, Hunter and co-workers reported the synthesis of a stable, 'open-knotted' structure, wherein a single linear tris-bipyridine ligand was coordinated around an octahedral zinc(II) ion [256]. This strategy directly relates to that published by Sokolov in 1973, when he first proposed that a trefoil knot motif could be achieved by arranging three bidentate ligands around an octahedral metal centre to generate the necessary crossings [257]. However, it was not until a decade later that the same group was able to produce the closed trefoil-knotted structure in 68% yield by trapping the acyclic complex through RCM and subsequent removal of the metal template (figure 14(b)) [258].
Active metal template strategies have also played a significant role in the preparation of interlocked compounds [238]. In this case, a metal ion acts simultaneously as a template as well as a catalyst for the synthesis of an entangled structure. In 2011, Leigh and co-workers used this strategy to synthesise the smallest molecular trefoil knot to date (a 76-atom long closed structure) in a yield of 24% [259]. A tetrahedral copper(I) ion acts as a template to coordinate a single polypyridyl ligand and form the crossing points, while another copper(I) ion binds to the functional end groups of the ligand, threads the loop through its coordination geometry and subsequently catalyses the covalent bond formation to create the trefoil knot motif (figure 14(c)).
Up until recently, the synthesis of molecular knots via a metal-based template strategy has been mainly performed with transition metals. However, recently, with the use of a lanthanide (Ln3+) ion, Leigh and co-workers demonstrated that it can template three 2,6-pyridinedicarboxamide ligands to which subsequent cyclisation by RCM resulted in an 81-atom loop trefoil knot molecule isolated in 58% yield (figure 14(d)) [260].
In these metal-based template approaches, the molecular knots are clearly formed in a stepwise manner, whereby the ligand(s) are first coordinated to metal ion(s). In some cases, this step results in a molecule in which the single ligand assembles around a central metal ion in such a way that there are a number of crossings of regions of the ligand. Alternatively, a number of ligands preassemble around the central ion(s) resulting in crossings of the individual building blocks. In other cases, there is a threading event through a loop created by the initial metal-ligand complex. In all cases, covalent linkage of either the termini of a single ligand or the monomeric units results in a closed knotted structure.
6.1.2. Hydrogen-bond template approaches.
Although not as frequently used as the metal-based template strategies, amide–amide hydrogen bonding interactions have also been shown to be important in the synthesis of molecular knots. In 1994, Hunter and co-workers used this approach to produce what they thought was a [2] catenane from the reaction of a diamine and a diacyl chloride [261]. However, several years later, Vögtle and co-workers repeated this one-step synthesis and with the use of x-ray crystallography, discovered that the resultant molecule was, in fact, a trefoil knot [262]. It was then suggested that it was highly likely that the linear diamine, composed of three units of the diamine and two units of diacyl, forms first, then folds into a helical loop which subsequently self-threads its remaining part through the loop. A reaction between the remaining carboxylic acid chloride unit and the terminal amino groups of the open loop then results in the closing of the loop to form the trefoil knot in 20% yield (figure 15(a)) [242, 262]. This synthetic approach highlights the importance of intra-molecular hydrogen bonding in the loop for subsequent knot formation.

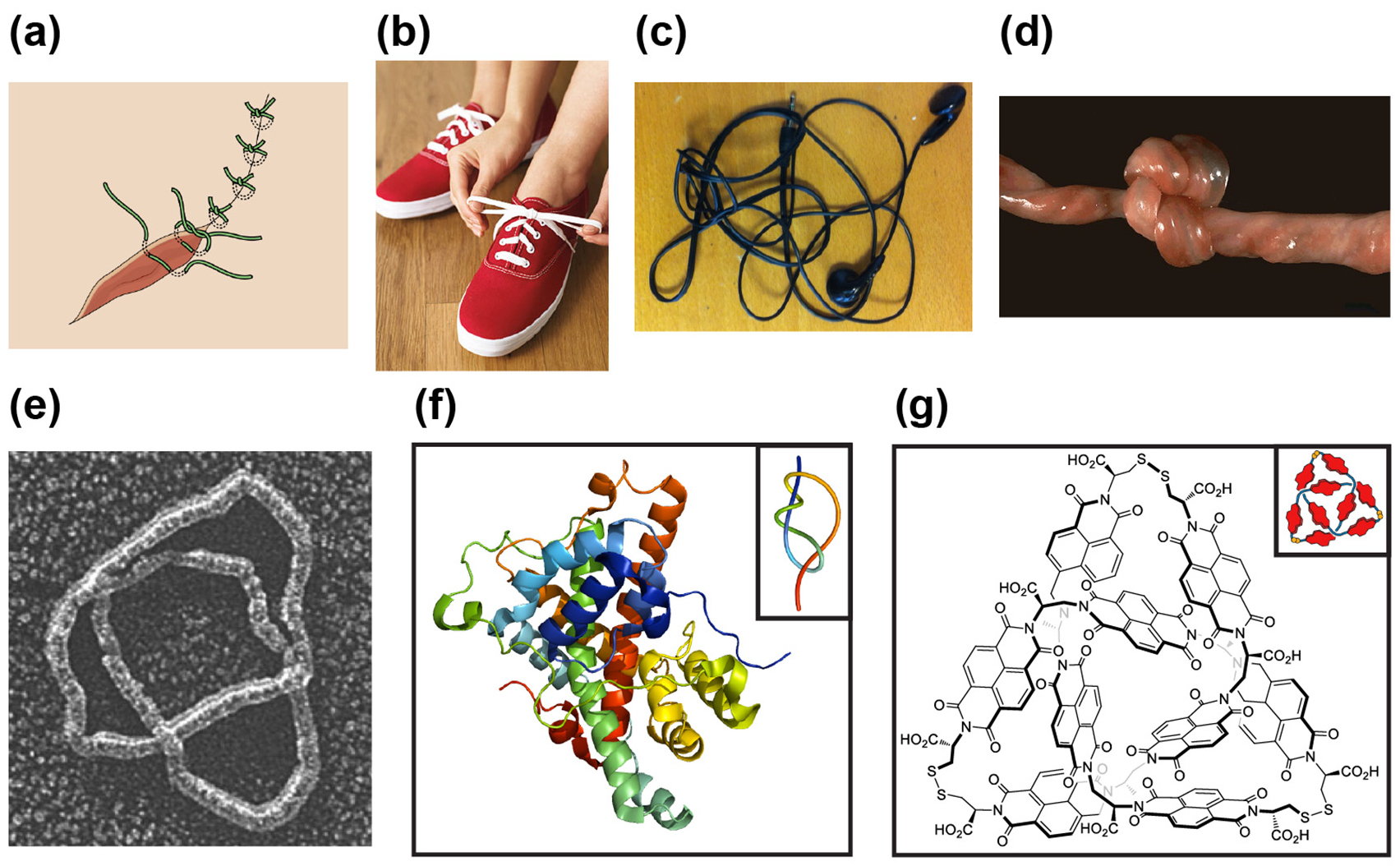{kind=link}
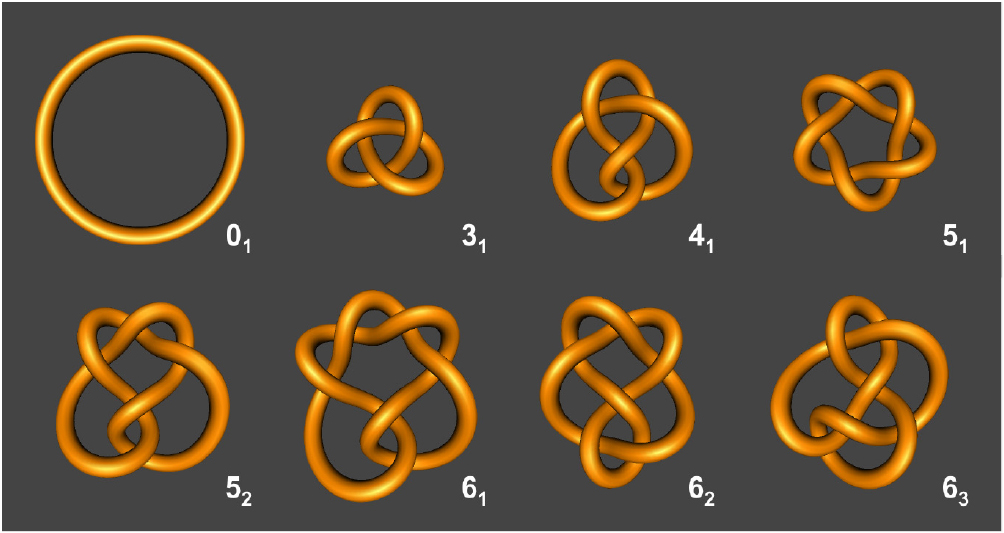{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
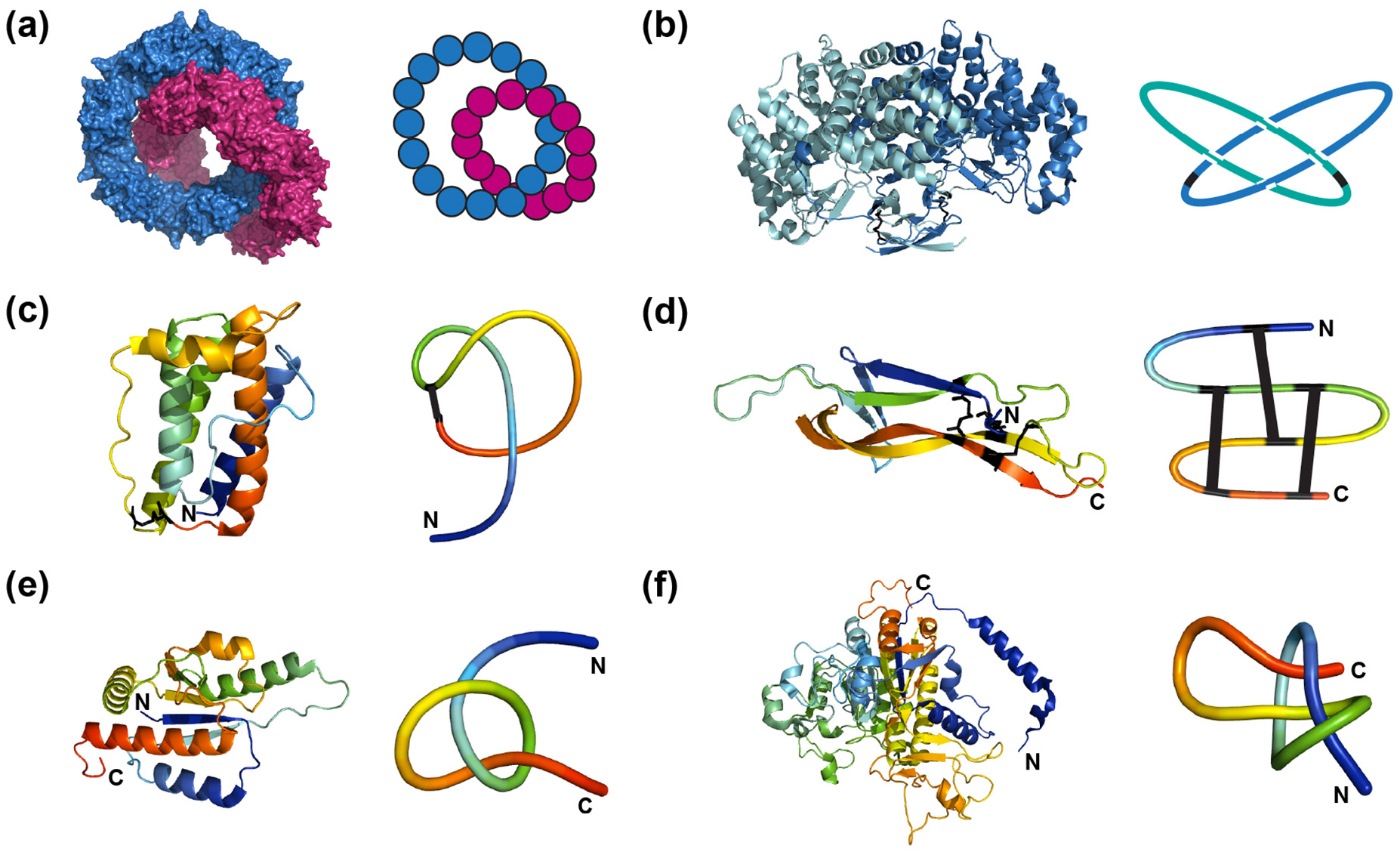{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 15. Schematic representations of self-assembly approaches used in the synthesis of molecular knots. (a) Synthesis of a molecular trefoil knot via amide-amide hydrogen bonding [262]. (b)–(c) NDI-based aqueous disulphide DCL approaches resulting in the synthesis of: (b) a trefoil knot [6], (c) a Solomon link and a figure-of-eight knot [264]. Figures were adapted with permission from [264]. Copyright (2014) American Chemical Society. (d) Synthesis of a molecular pentafoil knot [269].
Download figure:
Standard image High-resolution image{kind=link}
In 2006, Feigel and co-workers reported the synthesis of a molecular trefoil knot in 21% yield, which also made use of amide-amide hydrogen bonding interactions [263]. In this case, the trefoil knot was formed unexpectedly during the amide coupling reaction of 3-α-aminodeoxycholanic acid with L-valine. Similar to the previous synthesis, this is a one-pot procedure in which no external templating agent was needed to form the knotted architecture.
6.1.3. Dynamic combinatorial library (DCL) approaches.
Recently, Sanders and co-workers reported the self-assembly of a trefoil knot from a naphthalenediimide (NDI)-based aqueous disulphide dynamic combinatorial library (DCL) (figure 15(b)) [6]. In brief, the DCL approach allows the molecules themselves to discover different conformations in solution until those, which are thermodynamically the most stable, persist in the mixture once equilibrium is reached. In this study, knot formation was found to occur after an open linear trimer is formed. This then folds into a structure in which the hydrophobic NDI surfaces are buried and the terminal thiols are close in space to allow disulphide bond formation and ring closure. It was concluded that hydrophobic interactions play an important role in driving the folding of the linear molecule into a thermodynamically favoured knotted structure. It is worth noting that the chirality of the building blocks in the DCL resulted in stereoselectivity of the knotted conformation.
The Sanders group have also used different homochiral NDI-based building blocks, resulting in the formation of two thermodynamically favourable species in water, a topologically chiral Solomon link (60% of the library) and a topologically achiral figure-of-eight (41) knot (18% of the library) (figure 15(c)) [264]. Solomon link formation is not discussed here and can be found in the cited [264]. As with the previous study, it has been suggested that hydrophobic interactions are the driving force needed for a linear open tetramer to form a thermodynamically stable 41-knotted molecule. Interestingly, however, it was found that by using a racemic mixture of the same building blocks, another more stable structure, a topologically achiral meso 41 knot, can be formed (90% of the library). Although the formation of this meso 41 knot is significantly slower than that of the homochiral 41 knot, it is more thermodynamically stable. From this study, it was concluded that chirality and the number of rigid components in the building block can affect the major structure formed.
6.1.4. Other synthetic approaches to molecular knots.
Several other synthetic approaches have also been investigated. In 1997, Stoddart and co-workers isolated a trefoil knot in low yield, wherein a double helical precursor chain is formed with the help of π-donor/ π-acceptor interactions [265]. In another case, Siegel's group was able to synthesise a trefoil-knotted precursor in which the crossing points were generated by the combination of a covalently bonded organic scaffold and the coordination of copper(I) ions [266, 267]. Glaser couplings were then used to close the open chain, leading to 85% yield. Although the metal template can be removed with KCN treatment, the final removal of the organic template to create a formal trefoil knot has yet to be achieved. More recently, Trabolsi and co-workers reported the self-assembly of a trefoil knot, amongst other products (a [2] catenane and a Solomon link), with the use of a hybrid metal template-based/ dynamic covalent chemistry (DCC) strategy [268]. A pair of chelating imine ligands were combined with zinc(II) ions in a one-pot reaction to form the three structures, and the trefoil knot was precipitated and isolated by filtration.
In 2012, Leigh and co-workers reported the synthesis of the most complex molecular knot created to date, a pentafoil (51) knot, in 44% yield [269]. In this one-pot synthesis, iron(II) cyclic double helicates, which create the crossover points, are templated about a chloride anion in a cyclic array and the helicate monomers joined by reversible imine bond formation to form a 160-atom loop pentafoil-knotted structure (figure 15(d)).
6.2. Properties of molecular knots
Chirality is ubiquitous in chemistry, and knots are often chiral species. If the pure topological enantiomers of such can be obtained from the resolution of racemates, they will have specific optical properties. In many cases, it has been possible to isolate enantiomerically pure species. For example, enantiomers can be separated with the use of chiral HPLC [270, 271]. In another case, Sauvage and von Zelewsky were able to specifically form a single enantiomer by controlling the stereochemistry of the chiral helicate precursor [255]. Sanders and co-workers were also able to stereoselectively synthesise a trefoil knot by constraining the chirality of the building block in the DCL approach [6]. Recently, Leigh and co-workers who reported the synthesis of a lanthanide-templated molecular trefoil knot speculated that its chirality may influence the photophysical properties of the encapsulated lanthanide ion [260].
The study of the conformational properties of intertwined molecules is also of great interest due to their potential applications in the assembly of molecular switches. As molecular knots are increasingly becoming targets of chemical synthesis, it is important to understand what kind of motion is expected from the knotted topology. A study by Sauvage's group compared the dynamics of two different types of molecular trefoil knots formed by the metal-template based approach, in which the phenanthroline units were linked either by oligomethylene or m-phenylene spacers. In both cases, the molecular knots which still contained copper(I) ions were found to be generally rigid in solution [272]. However, removal of the metal ions led to rearrangement of the knotted backbone and, in both cases resulted in different dynamic behaviour. Those molecules containing the oligomethylene linkers had significantly greater conformational mobility in solution in comparison to those with m-phenylene spacers [244]. This study also showed that the conformational rigidity of partially or fully demetalated molecular knots can be restored again after re-complexation [272].
How do the conformational dynamics of the amide molecular knots formed via hydrogen bond interactions compare to those of the phenanthroline molecular knots? Based on 1H- and 31P-NMR spectroscopic measurements, Vögtle and co-workers reported that the amide molecular trefoil knots retain relatively rigid, non-symmetrical structures in DMSO, even though no metal ion is present [242]. However, addition of other solvents to the solution of these knots rapidly resulted in conformational change, and, in some cases, led to increased flexibility or increased rigidity [242]. Such changes in dynamics brought about by change in solution conditions makes these systems interesting for the development of molecular switches. In another case, the organic trefoil knot synthesised using the DCL approach exhibited sharp NMR signals in water demonstrating that the molecule was relatively rigid under these conditions. The signals remained unchanged upon increasing the temperature (from 298 to 358 K) or adding acetonitrile (from 0 to 50%), indicating that the structure is sufficiently stable such that it does not undergo gross conformational change upon a change in conditions [6], in contrast to the trefoil knots synthesised and studied by the Sauvage and Vögtle groups. Such conformational rigidity was also observed in the highly symmetric, achiral figure-of-eight knot synthesised using the DCL approach [264].
6.3. Summary
Although challenging, recently, chemists have successfully developed a number of different experimental strategies for the creation of molecular knots. These approaches have been used to synthesise a number of linked species, including Solomon links, Borromean rings, and a Star of David catenane. However, they have also been employed to make true knotted molecules including a 31, 41 and 51 knot. The different synthetic strategies can generally be considered as either a template-based method (for example the metal-based templates), or those which use hydrogen bonding or π − π interactions to first preassemble the building block(s) in such a way that covalent linkage of either the termini or of the monomer units results in a knotted structure. Alternatively, DCL-based approaches utilise the fact that a number of building blocks can come together to form chains of different lengths which can then fold to a thermodynamically stable state. In the first case, there need not be any threading event, but preassembly is crucial, whilst in the DCL approach, threading can occur. The properties of the molecular knots created using synthetic strategies are beginning to become established. Whereas, in some cases, the molecules are rigid in the presence of the templating metal ion, they can clearly undergo conformational change and their flexibility can alter when the metal ion is removed. The dynamics of such systems have also been found to vary depending upon environmental conditions. In other cases, such as those knotted molecules created by the DCL approach, which favours thermodynamically stable states, the evidence suggests these are relatively rigid molecules whose structures do not change significantly with environmental conditions.
As more and more new topologically complex structures are created, this raises the issue of whether knotting, linking, etc convey novel or important properties on the molecule. If they do, then it may be possible to exploit them in practical applications such as materials and pharmaceuticals. Readers who are interested in comprehensive discussion of these synthetic approaches are directed to the following reviews [16, 17, 241, 242, 248].
7. Discussion
In our macroscopic world, we are all very familiar with knots and other types of entangled objects. In many cases, knotted chains are incredibly useful at joining, securing and stabilising structures. However, in other cases, they are problematic and need undoing for something to function optimally. At the molecular level, this also appears to be the case. For some types of knotted or entangled molecules, the knot or entanglement conveys a particular and advantageous function to the chain, e.g. regulation of frameshifting in RNA. In contrast, knots can sometimes be hugely detrimental and need to be untied not only for optimal function but, in some cases, survival of an organism.
The mathematical field of knot theory has existed since the 19th century, and a great deal is known in terms of the identification and classification of knots, as well as polynomials to describe the different knotted topologies. In contrast, some molecular knots in Nature, the first of which was identified in DNA some three decades ago, were discovered in other biological polymers only recently, e.g. knots and slipknots in proteins and pseudoknots in RNA. These molecular knots remain relatively poorly understood in terms of their formation, properties and function. In addition, it is only recently that chemists have been able to synthesise knotted molecular species and they are only just beginning to study the properties of these types of structures.
In this review, we have attempted to summarise our current understanding of molecular knots in naturally occurring biological polymers, as well as those synthesised using a number of different chemical methods. Table 3 summarises a number of the key properties of the molecular knots that have been identified in the four different classes of molecules considered here: DNA, RNA, proteins and synthetic. This includes the types of knotted or otherwise topologically complex structures, such as slipknots, pseudoknots, linked chains (catenanes etc) that have either been found in Nature or synthesised. Compared to the knotted topologies that can be generated computationally, the molecular knots characterised experimentally are all relatively simple. Knotted DNA has been found with a crossing number up to 9 whilst for proteins, the highest crossing number identified to date remains a stevedore knot with 6 crossing points. Similarly, for synthesised knotted molecules, only a pentafoil knot with 5 crossings has been obtained for a single chain. Of interest is the observation that no truly knotted species have been found for naturally occurring RNA.
Table 3. Key properties of the molecular knots that have been identified in the four different classes of molecules.
| Type of molecule | Knot types | Other types of entangled species | Catalysis of knot-unknot transitions | Minimum number of monomers needed to create knot |
Persistence length | Chemical/physical properties | Mechanism of formation | Function of the knot or biological consequences of knot formation | |
|---|---|---|---|---|---|---|---|---|---|
| DNA |
|
|
Hopf links and other catenanes | Topoisomerases |
|
|
Compact |
|
|
| Synthetic: ssDNA |
|
Polyhedral catenanes, Borromean rings | Topoisomerases | 80 nt | 1.5–3 nm (ssDNA) |
Compact | Not characterised | N/A | |
| RNA | Naturally occurring | None | Pseudoknot | N/A | N/A | 72 nm (dsRNA) |
Not characterised | Not characterised | Linked to various functions: regulation of ribosome frameshifting |
| Synthetic |
31 | N/A | Topo III (E. coli) | 104 nt | 0.75–1.27 nm (ssRNA) |
Not characterised | Not characterised | N/A | |
| Proteins |
|
Hopf links (both formed by covalent bonding and also non-covalent interactions) and slipknots | GroEL-GroES (not essential) for a bacterial knotted MTase, otherwise not known |
|
0.7 nm |
|
Threading (through slipknot formation or via a plugging mechanism) |
|
|
| Synthetic molecule | Template-based approaches |
|
Solomon links, Borromean rings, Star of David catenane, [n]catenanes | None | 1 but typically 3 or more |
Not known | Not characterised | Pre-assembly and ligation of monomer units | N/A |
| DCL approach |
|
Solomon link, catenanes |
None | 1 but typically 3 or more | Not known | Not characterised | Creation of trimer and threading then closure (for the 31 knot) | N/A | |
aThis is the minimum length that has been observed to be knotted to date. In some cases, systematic studies suggest this will be the minimum length possible, whilst for other systems, shorter polymer lengths may yet be observed. bSize of the shortest genomic phage P4 DNA knot formed in the capsid [273, 274]. cThis value is estimated from optical tweezer pulling experiments on the basis of a tightened trefoil knot in dsDNA which has a length of 26 nm [275]. dReferences for the persistence length values: dsDNA [276], ssDNA [277], dsRNA [278] and ssRNA [279]. eAs shown by gel electrophoresis [280]. fFor reviews on the knotted and linked species of synthetic ssDNA, see [281–285]. gFor more details on the synthetic ssRNA knot, see [132]. hItzhaki, Jackson, Lim, Sivertsson (unpublished results). iA 76-atom loop trefoil knot is the smallest synthetic molecular knot prepared to date [259]. jFor more details on the catenanes that have been synthesised using the DCL approach, see [286–288].
Given that naturally occurring knotted forms of DNA and proteins are known, it is curious that there are no naturally occurring knotted forms of RNA. This is particularly so given the fact that synthetic RNA has been designed and shown to adopt a knotted circular form [132]. Recently, Micheletti et al conducted a systematic search for knots in the thousands of RNA structures in the PDB [289]. Although they found three examples of potentially knotted structures, all of these were low resolution. Comparing these with the structures from homologues that had been determined at much higher resolution, and also taking into account parameters that indicate how well the structure fits the experimental cryo-EM data, Micheletti et al concluded that it was unlikely that these knots were real. They speculate on why naturally occurring RNAs do not contain knots and suggest a number of possible causes: that RNA, which has a much smaller number of folds compared to proteins, has evolved to minimise geometrical complexity and therefore potential topological hindrance due to the need (at least for mRNAs) to translocate through the ribosome during translation. They also conjecture that because RNA structures are more modular in nature and that modular growth has led to longer RNAs, that this is incompatible with forming knotted structures. It is also recognised that the kinetics of folding may play a role: for RNA there is strong evidence that secondary structure forms early followed by tertiary structure, whereas, at least for relatively small proteins, we know that secondary and tertiary structure formation is generally concomitant. It is also noted that knotted forms of RNA may yet be found, for example, in the recently discovered eukaryotic circular RNAs [289].
Knots in homopolymers have been investigated in many experimental and computational studies over many years and much is known about how a number of key parameters such as chain length, solvent conditions, etc, affect the types of knots formed in these systems. On the other hand, much less is known about the factors influencing knot formation in heteropolymers, which includes all the molecular knots discussed here. It is clear, that in contrast to homopolymers, which can have high probabilities of knotting, heteropolymers, in general, have a much lower propensity to form knotted structures. In particular, proteins and RNA (discussed above). Although a number of knotted structures have been identified and characterised in proteins, with four different knot types in eleven different protein folds, it is clear that there are considerably fewer knotted proteins than one might expect for polymers of their size. A recent computational study by Virnau and co-workers has addressed why this might be by investigating the influence of sequence on the probability of knotting using a simple lattice model [290]. In this case, a basic hydrophobic-polar (HP) model was used in which there are favourable interactions between non-bonded H monomers. Chains of some 500 monomer units were studied, which had an overall composition of 50%:50% H:P but which varied in sequence. The introduction of just one more parameter into the model greatly decreased the number of knotted states found. Within the sequences assessed, some had a probability of knotting close to zero, whilst others had very high probabilities of knotting. Thus, this elegant study demonstrates that sequence has a very large impact on whether a chain is likely to form knotted species, or not. Consequently, these results suggest that Nature has favoured/selected protein sequences that are unlikely to form knotted structures. One way in which that might happen is to favour sequences where there is high local structural order, known to suppress knot formation. Given this, but also the fact that sequences are known which greatly increase the probability of knotting, this suggests that for the classes of knotted proteins that exist, that there may be some advantageous property of these systems which has allowed them to be selected and conserved.
Recent computational studies have also addressed the effects of how chain stiffness (or persistence length) influences the knottedness in single chain homopolymers [291]. This is more straightforward to study computationally than experimentally. These computational studies revealed non-trivial behaviour in which the extremes (i.e. a highly flexible chain or a rigid knot) both favoured the unknot. In between these extremes, there is an optimum chain stiffness which maximises the probability of knotting for any given chain length. At this point, the chain is semi-flexible in nature allowing loops to form through which other regions of the chain can thread to form the knot. It is interesting to see whether there is any evidence from experimental studies for this. table 3 reports the persistence length of the different biopolymers that can form topologically knotted or other entangled states. In addition, the number of minimum number of monomers within a chain known to form a knotted structure is given. As is expected, the persistence length of proteins, ssRNA and ssDNA (0.7–3 nm) [277, 279] is considerably smaller than for dsDNA or dsRNA (50–72 nm) [276, 278]. For those biopolymers that are relatively flexible (low persistence length), knotted structures have been characterised for minimum chain lengths of 82 amino acids [141], 104 [132] and 80 nt [285] for proteins, ssRNA and ssDNA, respectively. In contrast, biopolymers with much larger persistence lengths, such as dsDNA, form knots with chains that are considerably longer in length (5000 bp long) [273, 274]. It is interesting to note that RNA, which contains considerable secondary structure and has a persistence length of the order of 70 nm for dsRNA, are not known to form any knotted structures. As such, experimental results support the findings of the computational studies and establish that chain stiffness is an important property of a chain in determining whether it is likely to form a knotted structure.
At this stage, it is not possible to say much about whether such a correlation will be found for synthetic knotted systems created by chemists. For those molecules which are formed by a template-based method, the ligands first preassemble around a central metal ion or other template, and then the ligands are covalently linked. In this case, it is unlikely that there will be a correlation as the mechanism of knot formation does not involve loop formation and threading. In contrast, for knotted molecules synthesised using DCL approaches, there is evidence of an initial polymerisation of monomeric units to form a short chain and then threading of that chain to form the knot. Here, one might expect chain stiffness to play a role. However, with a single example of such, it is impossible, as yet, to say the degree to which persistence length influences knot formation.
For DNA, it is very well established that there are enzymes (topoisomerases), which catalyse both knotting and unknotting. The mechanism of action of this family of enzymes is known, and involves cutting of the DNA chain, movement of one part of the chain relative to another, and the ligation of the two ends of the cut chain to form a closed system. Thus, for DNA, no threading events are required for knot formation. However, there is some evidence that DNA can form knots with no cleavage of the chain. It is interesting to note that topoisomerases, known to work on DNA, can also catalyse knotting and unknotting events in synthetic forms of RNA, even though there is no known biological activity associated with this. For one family of knotted proteins, the bacterial methyltransferases, the chaperonin GroEL-GroES has been shown to significantly accelerate knotting and folding. However, the mechanism of action is not yet established and it is not known whether this chaperonin catalyses the folding of other classes of knotted proteins. So, for naturally occurring systems, catalysis of the knotting of the biopolymer is possible.
In a few cases, some of the physical properties of molecular knots have been characterised in some detail. For example, the electrophoretic mobility of unknotted, knotted and catenated forms of DNA, and to a much lesser degree of RNA, have been studied [280, 292]. Increasing the knot complexity, i.e. the average crossing number (ACN), is known to increase the electrophoretic mobility of DNA. This is due to the increased compactness of the molecule as the ACN increases. So clearly, in DNA and RNA, knots result in more compact states. It is unsurprising, therefore, that knotted DNA can be found in situations where it is densely packaged such as in viral DNA capsids. Is this also the case for knotted proteins and synthetic molecules? Comparing the size of families of proteins which have both knotted and unknotted variants, there is no evidence that the knotted structures are more compact or more densely packed. This may simply be due to the fact that, in many cases, the knotted region is associated with only part of the overall protein structure and a large amount of chain is often found in non-knotted regions. For the knotted small molecules that have been synthesized chemically, it is a little difficult to judge. Unknotted variants generally do not exist, however, the cyclised trefoil knotted species synthesised using DCL methods is extremely compact and certainly more compact than the linear trimer from which it is formed [6].
For proteins, there has been considerable speculation on how the properties of knotted species may differ from their unknotted counterparts. This has been of particular interest, given the evidence that knotted and slipknotted proteins are highly conserved. It has been suggested that changes in the dynamics and rigidity of the protein structure (especially close to active sites or binding sites), as a result of the knot, may play an important role in the activity/function of the protein. This was initially proposed at the time when all of the knotted proteins known were enzymes, however, there are now a number of knotted structures where the knotted region is not involved in any catalytic process. In general, there is relatively little evidence to support this hypothesis. Certainly, for the knotted proteins which have been investigated, there is no evidence that the dynamics of these structures is any different from unknotted ones [177].
It has also been suggested that thermodynamic and kinetic stability of knotted proteins may be greater than that for unknotted ones. Some computational studies have found small increases in thermodynamic stability for knotted proteins, however, other very similar studies have not. Although there is some experimental evidence for enhanced thermal stability of knotted structures, this has only been shown for one or two knotted and pseudo-knotted polymers of knotted proteins, and in this case, only apparent stabilities could be reported due to the irreversible nature of the thermal unfolding. Thus, it remains to be unambiguously established whether a knot increases the thermodynamic stability of proteins. Computationally, there is evidence that knots can decrease unfolding rates and, thus, the kinetic stability of the system. However, other elements of structure, such as the addition of stable beta motifs, also had a similar effect. Experimentally, there is no evidence to suggest that knotted proteins have significantly different unfolding rates compared to the range of unfolding rates measured for proteins lacking knots. This may not be a fair comparison, as unfolding rates can vary by orders of magnitude for proteins with the same unknotted topology but different sequences. This raises the real experimental and technical problem. In order to establish the effect of a knot on any physical property of a protein, it is essential to compare the knotted species with an unknotted species that is the same in all other respects other than the knot. So it is necessary that knotted and unknotted variants have the same overall secondary structure and overall packing of secondary structural elements to form a tertiary structure, and that the sequences corresponding to the secondary structural elements are the same, but only differs in the 'wiring', i.e. the order of the secondary structure in the overall sequence. Computationally, rewiring of a knotted structure to form an unknotted one is trivial. Experimentally, it can be achieved in a number of different ways: circular permutation where the original N and C- termini are joined with a linker and the new N- and C- termini of the circular permutant are in positions which remove the knot. Alternatively, it can be achieved by repositioning the regions of DNA corresponding to different elements of secondary structure (in general it is easiest to do this by making a synthetic gene). The Yeates group has taken a different approach by using disulphide binding to create chains of knotted and pseudo-knotted protein domains. Although the Jackson group has tried to create unknotted variants of known, and characterised knotted proteins, using the first two approaches, neither of them were successful and all attempts resulted in protein which aggregated and could not be studied (Jackson, Pina, Werrell, unpublished results). In order for the effect of the knot on thermodynamic and kinetic stability to be fully addressed experimentally, a system is needed where appropriate knotted and unknotted variants can be made, and where the unfolding is fully reversible.
An increased resistance to mechanical unfolding has also been proposed as a possible consequence of having a knotted structure. Despite early experiments on carbonic anhydrase, where results appeared to show a dramatic increase in mechanical stability in order to obtain full unfolding where high forces were used, later computational studies established that at such high forces the knot becomes wrapped tightly around an element of structure. At lower forces, one would expect the protein to unfold (in terms of its secondary and tertiary structure) to a state in which the chain is extended but still contains a tight knot. This has now been established for a number of other knotted structures and the forces required for mechanical unfolding are well within the range found for many other unknotted proteins. At this point, it is worth mentioning terminology, which can be confusing and can be used differently depending upon discipline. For a structural biologist, a protein can be considered unfolded or unstructured if it has lost all stable secondary and tertiary structure, but still contains a knot. For a physicist or mathematician, this may be not be considered a truly unfolded or unstructured state. For those outside this field, it should also be noted that some transient secondary and/or tertiary structure is known in a number of unfolded states of proteins populated under highly denaturing conditions (for unknotted proteins). This is referred to as residual structure in the denatured state.
Recently, the Jackson and Itzhaki groups have studied the resistance of knotted proteins, including the bacterial trefoil knotted methyltransferases and the 52-knotted UCH-L1 to degradation by the bacterial Clp degradation machine. In contrast to the trefoil knotted proteins which are rapidly degraded, UCH-L1 is extremely resistant to degradation (unpublished results). These results are similar to computational studies on translocation of a knotted protein through a pore, which show that, under certain conditions, the knot can tighten and prevent further translocation. Such translocation is necessary for a protein to be pulled inside the catalytic centre at the heart of the degradation machine. This is currently being investigated further.
How do knots form in heteropolymers? For DNA, there is a substantial amount of evidence for how it can knot and unknot. In vivo, topoisomerases are likely to play a dominant role and therefore the mechanism involves effectively cutting of the chain, movement of one part of the chain relative to another, followed by pasting of the two ends of the chain together to form a different topology. However, even for DNA, it has been shown that a standard threading mechanism involving formation of a loop through which another part of the chain passes can also occur. Such threading mechanisms also occur in proteins, and generally the terminus of the polypeptide chain closest to the knot undergoes the threading event. Considerable insight into loop formation and threading has come from computational studies, and a number of different pathways have been found. The formation of a slipknot, created by a β-hairpin like structure forming at the end of the chain, which is then pulled all the way through the loop, is frequently observed in simulations. However, a plugging pathway in which the end of the chain simply threads through the loop without forming any metastable structure has also been detected. With the synthetic knotted molecules, threading has also been shown to occur with a trefoil-knotted species that forms from a linear trimer using the DCL approach. In general, template-based synthetic methods do not require a threading event. However, relatively little is known about the mechanism of formation of these types of knotted structure.
For naturally occurring biopolymers such as DNA, RNA and proteins, one can ask the question as to whether the knot affects not only the physical properties of the system (discussed above), but also whether there is some biological function associated with the knot, or a biological consequence of knot formation. In the case of DNA, a great deal is known about how the mechanisms by which knots are introduced into the DNA chain. This can occur as the result of many cellular processes such as transcription, replication and recombination. In these cases, if not untied, the knots can go on to have severe detrimental consequences on the cell or organism, hence the ubiquitous nature of cellular topoisomerases which can remove knots promptly and efficiently. This suggests that knots in DNA are problematic. Given the principle of microscopic reversibility, topoisomerases not only catalyse the removal of knots from DNA but also introduce knots into the polymer. However, in another case, knots in DNA may be beneficial. For example, they are common in the densely packaged DNA found in viral capsids and can influence the rate of ejection of the DNA from the capsid. In this case, whether the knots just form as a natural consequence of the confined volume in a capsid, or whether they play an important biological role in influencing ejection rates remains to be established. In all of these cases, knots can be seen as the product of a cellular process or biological environment, and there is no evidence that the knot has any beneficial function on the system. In contrast, for RNA, although there are no naturally-occurring knotted RNA species, pseudoknots, which have a number of topological crossings whilst not being knotted, are abundant. In this case, it is very clear that there is a close link between the topological form of the RNA and its function, as pseudoknots are found to play a role in frameshifting, transcriptional regulation including the initiation of protein synthesis and template recognition by viral replicases. There is also some evidence that the pseudoknot structure is required for optimum catalytic activity of a number of ribozymes or riboproteins.
For proteins, the role of the knot in the function of the biomolecule remains very unclear. Certainly, knots in polypeptide chains, may influence a number of the physical properties of the protein, such as stability (thermodynamic, kinetic, mechanical) or dynamics (rigidity) that could affect the half-life or the function of the protein in vivo. However, although some experimental and computational studies have demonstrated that there may be some effect of the knot on stability and/or dynamics, we are still far from understanding the role of knots in proteins, and definitive experiments, in particular, have yet to be performed. This is, of particular interest and importance, given the conservation of knotted and slipknotted protein structures.
8. Summary and future perspective
In summary, it is now clear that, just as in the macroscopic world, molecular knots are abundant in Nature. In addition, we are now able to design and synthesise different knotted species, be they based on nucleic acid building blocks (DNA and RNA), amino acid building blocks (proteins), or a very wide range of building blocks available to synthetic chemists.
For DNA, we already know a considerable amount on how knots can be introduced into DNA chains, how knots can be removed from chains, what biological processes result in knot formation, and some of the biological consequences of knotted structures. For RNA, we can design and make knotted RNA structures even though no naturally occurring species have yet to be found, which raises the fascinating possibility that RNA knots may still yet be found in Nature. In contrast to DNA, we know much less about how knots are formed in polypeptide chains, and the role of those knots in the structure and function of proteins. This is an area where both computational and experimental research programmes are beginning to reveal some of the facets of these systems, both in terms of how the knotted structures are formed, and to a lesser degree, how the knot might influence function. There remains considerable work to be done to fully address these questions and understand these systems. Recently, after decades of little progress, synthetic chemists have developed strategies for the design and synthesis of a number of molecular knots. This really now opens up the field and hopefully will allow many more knotted and other types of topologically complex species to be made and characterised in the near future. Characterisation of these synthetic molecular knots is crucial in order to understand how their properties differ from unknotted ones, and whether these types of molecule will be of use in nanotechnology, biotechnology, medicine, etc.
In the past few decades, interest in knotted systems has moved away from the realm of just the mathematicians and physicists, and chemists and biologists are now equally fascinated by these types of structure. Over the past ten years, it has been a great pleasure to see how these very disparate communities and disciplines have come together, to share knowledge and solve problems within the field. Going forward, we need to continue to do this and to combine a whole range of computational and experimental approaches on different knotted systems, in order to address some of the big questions in the field and to understand the formation and behaviour of knots both on a theoretical basis, but also, on a molecular basis, and in challenging heteropolymeric systems where different regions of the chain can interact with each other in complex ways, and where non-trivial behaviour can be expected.
Acknowledgments
We would like to thank Dr Sarah Harris, Prof Henri Orland, Prof David Leigh, Augustinas Markevicius and Dr Guzman Gil-Ramirez for their helpful comments on the manuscript. We would also like to thank Dr Gokhan Barin and Prof J Fraser Stoddart for sending us the x-ray crystal structures in figures 13(b) and (d), and Dr G Dan Pantoş for sending us the structures shown in figure 1(g). We would also like to thank many people in the field for interesting discussions and useful advice. N C H Lim is supported by a UBD Chancellor scholarship from the Brunei Government. S E Jackson would like to dedicate this review to the memory of her mother, Ursula Margaret Jackson, who taught her a great deal even though she did not know any chemistry.