
Abstract
We show through first-principles calculations that the electronic properties of Pt4 clusters can be tuned by adsorption on substrates with different electronic valence characters. Pt clusters exhibit a metallic character on γ-Al2O3(111) and insulator properties on CaZrO3(001). The noted difference indicates the role of the electronic valence states of the substrate atoms that directly bond with Pt.
Export citation and abstract BibTeX RIS
The excellent catalytic behavior and exceptional chemical reaction properties of Pt clusters have stimulated significant research interest in the use of these nanostructures in important industrial applications such as catalytic converters for vehicles to reduce toxic pollutants such as CO, NOx, and hydrocarbons, and in hydrogen fuel cells [1–3]. An interesting characteristic of Pt clusters is that the supporting substrate can greatly affect their chemical reactivity and selectivity, which could originate from the modification of the electronic structure of Pt by the supporting substrate. The mechanism behind such a substrate-induced electronic modification of Pt clusters, however, is so far not well established. In this communication, we show that the electronic properties (whether metallic or insulator) of Pt4 clusters can be tuned by adsorption on substrates (γ-Al2O3 and CaZrO3) with different electronic valence characters. γ-Al2O3 and CaZrO3 substrates are industrially important catalysts for conventional automobile and fuel cell applications [4–6]. Both have insulating properties and wide band gaps, but the former is a p-block metal oxide while the latter has a strong valence d character.
The tetrahedral structure of the Pt4 cluster was chosen because of its relative stability over a planar rhombus isomer [7–9]. Also, this structure was shown to be more stably adsorbed on γ-Al2O3(111) than the planar structure [10]. The calculated gas phase isolated Pt4 cluster with a tetrahedral structure has an average Pt–Pt distance of 2.62 Å, in agreement with related studies [7–11]. For the γ-Al2O3 substrate, the Pt4 cluster was adsorbed on the (111) facet of a slab modeled as a 2 × 2 supercell with 32 Al and 48 O atoms and a vacuum space of ∼25 Å. This model is based on the proposed structure of γ-Al2O3(111) in similar density functional theory (DFT) studies [10, 12]. The adsorption of the Pt4 cluster on the (001) facet of the CaZrO3 substrate was modeled with an 84-atom (16 Ca, 16 Zr, 48 O and 4 Pt atoms) supercell with 0.25 ML adsorbate coverage and a vacuum layer of ∼16 Å. The CaZrO3 orthorhombic perovskite structure is based on the related literature [13, 14]. The CaZrO3(001) and γ-Al2O3(111) facets were chosen because of their stability [12]. The Pt4 cluster and the top 40 atoms of the slab were fully relaxed to obtain a stable adsorption structure of the cluster on the surface sites shown in figure 1. For the γ-Al2O3(111) surface, the a, b, and c sites correspond to O hollow sites, Al hollow sites, and Al atop sites, respectively. These sites are expected to be the most favorable adsorption sites since these configurations can promote a strong Pt–O bonding interaction. For CaZrO3(001), sites a, b and c correspond to the positions above Ca, O, and Zr, respectively. The adsorption energy was computed by taking the difference between the total energy of the Pt4 cluster–slab system in the lowest energy adsorption site and the summed energies of the relaxed clean surface and the gas phase Pt4 cluster.
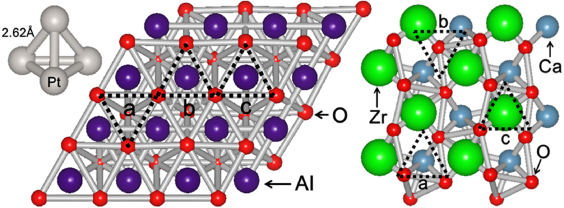
Figure 1. From left to right, respectively, the gas phase tetrahedral Pt4 cluster, γ-Al2O3(111), and CaZrO3(001) surfaces. The tetragonal gas phase isomer of the Pt4 cluster has an average Pt–Pt distance of 2.62 Å. The dashed triangles (labeled a, b, and c) are the adsorption sites considered. Each type of atom is labeled respectively.
Download figure:
Standard image High-resolution imageSpin-polarized density functional theory (DFT) calculations were implemented via the Vienna ab initio simulation package (VASP) [15–18]. The interactions between ions and electrons were described using the projector augmented wave (PAW) method [19, 20]. Plane-wave basis sets were employed with an energy cut-off of 400 eV. The exchange–correlation term was described using the generalized gradient approximation (GGA) based on the Perdew–Burke–Ernzerhof (PBE) [21, 22] functional. The surface Brillouin zone integrations were performed on a grid of 3 × 3 × 1 Monkhorst–Pack k-points [23]. The conjugate gradient algorithm was used to relax the ions into their ground state. An electric dipole correction layer in the vacuum area was used to cut the dipole interactions between the repeated image layer systems. Convergence of the numerical results with respect to the slab thickness, the kinetic energy cut-off and the k-point was established.
We first discuss the adsorption of Pt4 clusters on γ-Al2O3(111) and CaZrO3(001) surfaces. The Pt4 cluster adsorbs strongly on site b of γ-Al2O3(111) and site c of CaZrO3(001), with adsorption energies of −4.09 eV and −4.30 eV respectively, in excellent agreement with a previous study [10]. The bottom plane of the tetrahedral Pt4 cluster is parallel to the surface of γ-Al2O3(111). For CaZrO3(001), one of the surface Pt atoms goes deeper into the surface by ∼0.3 Å as compared to the other two atoms, making the bottom plane of the tetrahedral structure slightly inclined on the surface. This is because of corrugation of the CaZrO3(001) surface, as shown in figure 1. The average Pt–Pt distances of the Pt atoms directly bonded to the surface are 2.60 Å and 2.81 Å for γ-Al2O3(111) and CaZrO3(001) surfaces, respectively. The greater surface area of the bottom plane of the tetrahedral cluster on CaZrO3(001) than on γ-Al2O3(111) results in stronger binding of the cluster on the CaZrO3(001) surface.
We then report the difference in the electronic properties of the Pt4 cluster upon its adsorption on γ-Al2O3(111) and CaZrO3(001) surfaces. The density of states (DOS) projected on the Pt atom (denoted as 'Pt' in figure 3 for adsorbed systems) is shown in figure 2. The gas phase tetrahedral Pt4 cluster has metallic characteristics and exhibits a magnetic character, as shown by the spin polarization of the DOS (figure 2(a)) and the magnetization of 0.44 μB/atom. Upon adsorption on the surfaces, these states are broadened and shifted, due to the interaction of the adsorbate with the surface. For the Pt4 cluster adsorbed on the γ-Al2O3(111) surface, the presence of states in the Fermi level (figure 2(b)) shows that Pt has metallic characteristics. It is noted that the occupied regions from ∼ − 2.0 eV to the Fermi level are essentially unchanged. The same metallic characteristics are also noted for the other three Pt atoms (their DOS plots are not shown). Interestingly, Pt exhibits insulator properties on the CaZrO3(001) surface, with an energy band gap of 2.2 eV. Since the energy band gap in PtO2 has been found and calculated in both experimental [24, 25] and theoretical [26] studies, Pt exhibits oxide-like properties on CaZrO3. The same properties were noted for the other three Pt atoms (DOS plots are not shown). For both of these substrates, the spin polarization of the Pt d state is retained. Such a spin-polarized Pt d state was also observed for a Pt overlayer on a ferromagnetic substrate [27].

Figure 2. Density of states projected on the Pt atom for (a) a gas phase Pt4 cluster and a Pt4 cluster adsorbed on (b) γ-Al2O3(111) and (c) CaZrO3(001). In (a) and (b), Pt shows metallic characteristics. For (c), Pt has insulator characteristics with an energy band gap of 2.2 eV. Blue, red, and black curves correspond to s, p, and d states respectively.
Download figure:
Standard image High-resolution image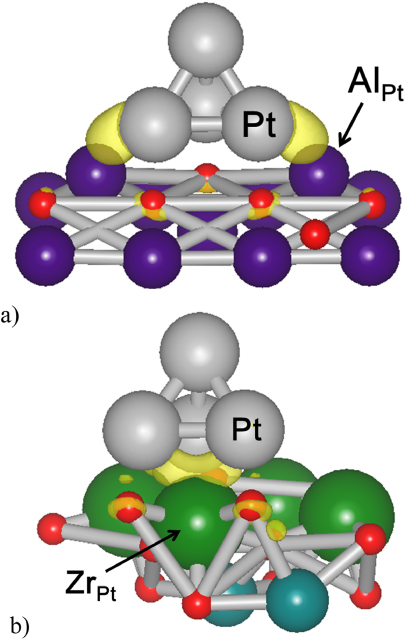
Figure 3. Charge density difference projected on a  isosurface value for (a) γ-Al2O3(111) and (b) CaZrO3(001) surfaces. A high accumulation of electron density is seen in the region between Pt and Al for γ-Al2O3(111) and above Zr for CaZrO3(001). These Al and Zr atoms are referred to in the text as AlPt and ZrPt respectively.
isosurface value for (a) γ-Al2O3(111) and (b) CaZrO3(001) surfaces. A high accumulation of electron density is seen in the region between Pt and Al for γ-Al2O3(111) and above Zr for CaZrO3(001). These Al and Zr atoms are referred to in the text as AlPt and ZrPt respectively.
Download figure:
Standard image High-resolution imageNext, we discuss the changes in the electronic properties of the substrate atoms which directly interact with the Pt4 cluster. Figure 3 shows the charge density difference plot projected on a  isosurface value. Strong interaction between atoms is shown by the accumulation of electron density (yellow). For the γ-Al2O3(111) surface, the Pt atoms near the surface interact mainly with the Al atoms which protrude out of the surface (which we now refer to as AlPt). This was also noted in a related study [10]. While for CaZrO3(001) surface, Pt atoms interact strongly with the nearest Zr (which we also now refer to as ZrPt). As shown in figure 4, the valence charge is largely associated with the O p state, suggesting the polar character of the Al–O bond. Such a property has been reported in the literature [12]. The adsorption of the Pt4 cluster on γ-Al2O3(111) dramatically decreases the energy band gap of AlPt from 4.1 to 0.6 eV. For the CaZrO3(001) surface, the adsorption of the Pt4 cluster results in downward shifting of the occupied and unoccupied states of ZrPt, but the energy band gap is essentially unchanged (figure 5). To understand these differences, we first note that for ZrPt, the d state participates mainly in the interaction with the Pt4 cluster. The DOS for s and p states is negligible, and too small to significantly alter the Pt d states. Therefore, the Pt d state has to adapt to the insulator characteristics of ZrPt to efficiently hybridize with the ZrPt d state. Thus, the strong hybridization between Pt d and Zr d states is promoted by the electronic switching of the Pt d state from metallic to insulator. For AlPt, however, the weak hybridization of the Pt d state with Al states allows the Pt d state to maintain its metallic electronic properties upon its adsorption on the surface. We note that the occupied and unoccupied AlPt states have shifted closer to the Fermi level, narrowing the energy band gap.
isosurface value. Strong interaction between atoms is shown by the accumulation of electron density (yellow). For the γ-Al2O3(111) surface, the Pt atoms near the surface interact mainly with the Al atoms which protrude out of the surface (which we now refer to as AlPt). This was also noted in a related study [10]. While for CaZrO3(001) surface, Pt atoms interact strongly with the nearest Zr (which we also now refer to as ZrPt). As shown in figure 4, the valence charge is largely associated with the O p state, suggesting the polar character of the Al–O bond. Such a property has been reported in the literature [12]. The adsorption of the Pt4 cluster on γ-Al2O3(111) dramatically decreases the energy band gap of AlPt from 4.1 to 0.6 eV. For the CaZrO3(001) surface, the adsorption of the Pt4 cluster results in downward shifting of the occupied and unoccupied states of ZrPt, but the energy band gap is essentially unchanged (figure 5). To understand these differences, we first note that for ZrPt, the d state participates mainly in the interaction with the Pt4 cluster. The DOS for s and p states is negligible, and too small to significantly alter the Pt d states. Therefore, the Pt d state has to adapt to the insulator characteristics of ZrPt to efficiently hybridize with the ZrPt d state. Thus, the strong hybridization between Pt d and Zr d states is promoted by the electronic switching of the Pt d state from metallic to insulator. For AlPt, however, the weak hybridization of the Pt d state with Al states allows the Pt d state to maintain its metallic electronic properties upon its adsorption on the surface. We note that the occupied and unoccupied AlPt states have shifted closer to the Fermi level, narrowing the energy band gap.
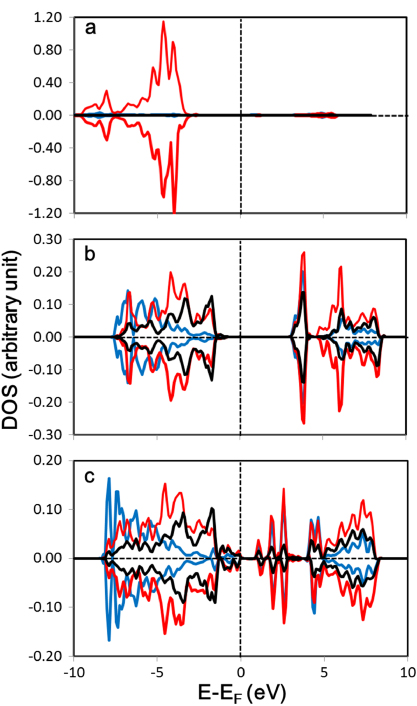
Figure 4. Density of states projected on the (a) surface O atom of γ-Al2O3(111) before the adsorption of Pt4, and the Al atom (which we refer to as AlPt) at the surface of γ-Al2O3(111) which directly interacts with Pt atoms (b) before and (c) after the adsorption of the Pt4 cluster. The band gap of Al has decreased from 4.07 to 0.63 eV. Blue, red, and black curves correspond to s, p, and d states respectively.
Download figure:
Standard image High-resolution image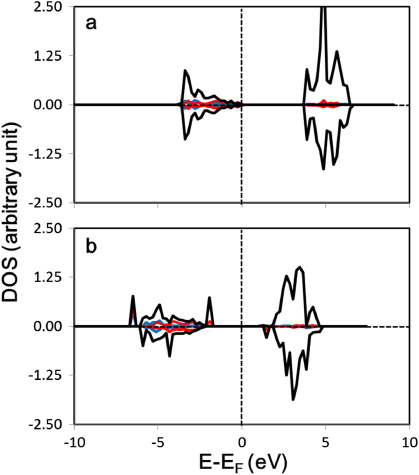
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Density of states projected on the Zr atom (which we refer to as ZrPt) at the surface of CaZrO3(001) which directly interacts with Pt atoms (a) before and (b) after the adsorption of the Pt4 cluster. The adsorption of the Pt4 cluster results in downward shifting of the occupied and unoccupied states, but the energy band gap is essentially unchanged. Note that the DOS of s and p states are relatively low compared to d states. Blue, red, and black curves correspond to s, p, and d states respectively.
Download figure:
Standard image High-resolution image{kind=link}
In conclusion, we have shown that the electronic properties of Pt4 clusters can be tuned by adsorption on substrates with different electronic structures. Pt4 clusters exhibit metallic characteristics on γ-Al2O3(111) and insulator properties on CaZrO3(001). The noted difference indicates the role of electronic valence states of the substrate atoms that directly bond with Pt. The strong hybridization between Pt d and Zr d states for Pt4/CaZrO3(001) is promoted by the electronic switching of the Pt d state from metallic to insulator. On the other hand, weak hybridization between Pt d and Al states allows the Pt cluster to retain metallic characteristics on γ-Al2O3(111).
Acknowledgments
This work is supported in part by MEXT (Ministry of Education, Culture, Sports, Science and Technology) through the G-COE (Special Coordination Funds for the Global Center of Excellence) program 'Atomically Controlled Fabrication Technology', a Grant-in-Aid for Scientific Research on Innovative Areas program (2203-22104008) and Scientific Research (c) (22510107) program, and JST (Japan Science and Technology Agency) through the ALCA (Advanced Low Carbon Technology Research and Development) program. Some of the calculations were done using the computer facilities of CyberMedia Center (Osaka University), ISSP Super Computer Center (University of Tokyo), and Large Scale Simulation Program KEK No. T10-12 of the High Energy Accelerator Research Organization. R L Arevalo, A A B Padama and J L V Moreno acknowledge MEXT for scholarship grants.