

Abstract
In order to study the practical means of a dissemination of the redefined kilogram, the mass laboratory of METAS has built a new and unique instrument, which combines high precision mass measurement and element-specific surface chemical analysis by x-ray photoelectron spectroscopy (XPS) under identical conditions. It is especially designed for the analysis of large samples such as real 1 kg standards and artefacts of up to 100 mm in diameter. In this paper, we first provide a detailed description of the apparatus. In a second step we demonstrate its performance by applying various processes, such as the transfer from ambient to vacuum, cleaning by low-pressure hydrogen plasma, transfer from vacuum to ambient and recontamination in air with time. Real 1 kg PtIr standards and surface artefacts have been monitored at individual steps gravimetrically, and in vacuum additionally by XPS. Values for the gain and loss of mass due to the application of different processes are provided. A model for the short-term recontamination after cleaning is presented, showing the initial growth of contaminants is self-limited.
Export citation and abstract BibTeX RIS
1. Introduction
The potential new definition of the kilogram based on fundamental constants is within reach. This challenge has triggered considerable activities [1–14]. Today, the kilogram is the last of the seven base units of the International System of Units (SI) which is based on a physical artefact, the platinum–iridium international prototype kilogram (IPK), conserved at the International Bureau of Weights and Measures (BIPM, Sèvres). The dissemination of the unit is realized by direct mass comparison of the IPK with platinum–iridium working standards and their copies owned by national metrological institutes.
The problems with this definition are the lack of a value which is invariant in time with respect to the fundamental constants of physics, and the limited stability of the mass of the standards caused by atmospheric contamination over time. While the problem of a definition based on fundamental constants could be solved by means of watt-balance [4, 5, 8–12] and Avogadro [1–3, 6] experiments, a few problems still persist. All experimental attempts related to a future new definition of the kilogram are realized under vacuum conditions. For the practical dissemination of the mass unit, the standards must be transferred from vacuum to ambient. During this transition the mass of the standards is significantly affected by sorption phenomena, caused by atmospheric gases and humidity. The underlying physics is not well understood, and a precise prediction of the gain in mass depends on the processes applied, wherefore a description is still missing. Furthermore, in between two primary realizations of the kilogram, the mass scale temporarily relies completely on the standards themselves, which are subject to limited stability with time.
The problem of the instability in mass of artefacts has been known since the General Conference on Weights and Measures (CGPM) decided in 1889 that the IPK should define the unit of mass. Consequently, a lot of investigations mainly on PtIr and stainless steel but also other materials were performed. A complete summary of accomplished studies about surface contamination and the stability of standard masses was reviewed by Davidson [15]. The studies did focus on various effects, such as the long-term stability, water-vapour sorption and the performance and reproducibility of cleaning techniques. Unfortunately, the data exhibit a huge variety of results with large discrepancies even for measurements performed under comparable conditions.
In addition to gravimetric measurements on kilogram standards, also surface analytical techniques, such as electron spectroscopies like x-ray photoelectron spectroscopy (XPS) [16–23] and Auger electron spectroscopy (AES) [22] or optical methods such as ellipsometry [23–25], have been used. But these state-of-the-art techniques are mainly restricted to small samples (XPS, AES), or the results are not element-specific (ellipsometry). Only very few investigations [23, 24] combined gravimetric measurements and surface analysis on the real mass standards. A correlation between chemical surface characterization of contaminants and gain in mass is almost completely missing [15].
2. Experimental layout
In order to find a direct link and a correlation between the different methods, we have built a new instrument which combines gravimetric measurements and surface analysis by XPS and AES. A mass spectrometer for residual gas analysis (RGA) and a quartz crystal microbalance (QCM) complete the instrument. Figure 1 shows an overview of the instrument. The multichamber UHV system can be operated at a base pressure below 5 × 10−7 Pa.

Figure 1. Overview of the instrument: XPS/AES instrument (left), storage (middle) and mass comparator (right). The transfer chamber is in front with a loadlock on the right side (red caps) and three forks perpendicular to the transfer chamber.
Download figure:
Standard imageThe main components are three individual UHV vacuum chambers, one for a high precision 1 kg mass comparator (right), one for the storage of 24 standards under UHV conditions (middle) and an XPS/AES chamber (left), and a transfer chamber which is connected to each of the three UHV chambers. The transfer chamber enables the transport of the mass standards between the different chambers under UHV conditions using a cable railway. The standards can be lifted gently from the cart and inserted into the individual chambers using forks that can be placed under the standards. Without breaking the vacuum in the main part, the mass standards can be inserted in a loadlock attached to the transfer (right). The main chambers can be separated from each other and are pumped individually by turbomolecular pumps (transfer and loadlock) and ion getter pumps (main chambers).
A plasma reactor for sample cleaning is integrated in the loadlock system which is attached to the transfer chamber (right).
For a better overview of the instrument, the main components are depicted in figure 2 with vacuum chambers removed for better illustration.

Figure 2. Schematics of the instrument showing the main components: high precision mass comparator (right), three platforms for storage (middle) and the XPS instrument (left). Three forks (not shown) allow the standards to be transported perpendicular to the railway and to be placed easily on the turntable in the comparator, on the finger-shaped locations of the three storage platforms, and on the tiltable XPS sample stage. X-ray gun and energy analyser are aligned at the magic angle.
Download figure:
Standard imageThe high precision mass comparator is built from a 1 kg M_One System by Mettler-Toledo AG. In collaboration with Mettler-Toledo, the weighing cell was slightly modified to make it fully compatible with our UHV vacuum system. The typical standard deviations achieved in our open, unshielded configuration are 0.1 µg in vacuum and 0.3 µg at ambient. Statistical analysis of our measurements yields uncertainties for the differences of mass for a comparison of less than 0.5 µg (k = 2). The six-place turntable was also modified in order to be compatible with our special sample transport and UHV vacuum system. The comparator chamber is additionally equipped with a QCM for in situ studies of sorption effects when transferring from ambient to vacuum and vice versa.
For surface chemical analysis by XPS we use an Alpha 110 hemispherical photoelectron analyser (Thermo Electron Corp.) normally operated in the constant energy mode (CAE) with pass energies of 50 eV for survey and 20 eV for higher resolution scans. The analyser was calibrated using the photoelectron peaks of Au 4f7/2 at 84.0 eV and Cu 2p3/2 at 932.6 eV. The samples were irradiated by Mg Kα (hν = 1253.6 eV) at 300 W/15 kV generated with a twin anode x-ray source (Thermo Electron Corp.). Additionally, an electron gun (EGL-2022, Kimbal Physics) is attached to the chamber for AES. Raw XPS data are analysed using the manufacturer's 'Avantage V 4.75' data processing software. For curve fitting we used published values of binding energies [25]. One quite unique feature of our XPS is the self-designed sample stage. It was especially designed for huge objects like real mass standards or Si spheres of 1 kg in mass and of up to 100 mm in diameter. The entire sample stage can be tilted for angle-resolved measurements (ARXPS). This special feature already proved to be very useful for surface chemical analysis of the single isotope silicon spheres from the Avogadro project, and consequently, contributed to the most accurate value of the Avogadro constant published up to now [1–3]. Furthermore, the XPS is equipped with a quadrupole mass spectrometer (Prisma, Pfeiffer Vacuum) for RGA.
For the cleaning of mass standards, a low-pressure plasma cleaning system is integrated in the loadlock. The electrodes, consisting of adhesive copper tapes, are placed on the air-side of two opposite viewports of the loadlock (red protective caps, furthest right). The high-frequency field is supplied by a 40 kHz/1 kW LFG 40 plasma generator from Diener Electronics (Germany). The chamber is equipped with a precision leak valve for the supply of high purity process gases.
3. Experimental procedures and results
For our first combined measurements both national PtIr prototypes of Switzerland were involved. No 38, one of the first 40 national copies of the IPK received in 1889 from the BIPM, was kept under ambient pressure and temperature in the clean room of the mass laboratory at METAS during the measurements, and served as a reference. The second prototype No 89 and a PtIr surface artefact, made of a stack of eight discs separated by spacers, were purchased from the BIPM in 2004 especially for research purposes. The three involved PtIr standards are shown in figure 3. Both artefacts were manufactured and cleaned at the BIPM. They have the same physical properties except for their surface area which is roughly 72 cm2 for the cylinder and 240 cm2 for the stack of discs. Consequently, surface effects such as sorption and desorption will affect the mass of the artefacts differently. Assuming the sorption coefficient is the same for both artefacts, the change in mass difference between them can directly be related to the difference in surface area.

Figure 3. Swiss national PtIr prototype kilograms. No 89 and the stack of discs were used as surface artefacts, No 38 served as a reference.
Download figure:
Standard imageThis set of surface artefacts was exposed to several processes and continuously monitored gravimetrically and additionally by XPS when under vacuum.
First the masses of No 89 and the stack of discs were compared against the reference No 38 at ambient constant pressure of 940 mbar (94 kPa) in the M_One comparator of the METAS mass calibration laboratory. Subsequently, the set of surface artefacts was compared against each other in the mass comparator of the new instrument, first at ambient constant pressure again for control of consistency of this comparator. After evacuating the comparator to UHV, a second mass comparison was performed, followed by the analysis of the surface chemical state.
Figure 4 shows the difference in mass between the cylinder and the stack of discs for each step of the applied process. The corresponding numerical values are summarized in table 1.
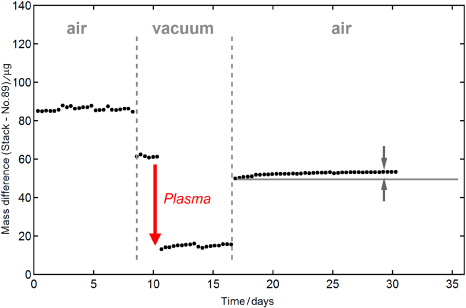
Figure 4. Evolution of the mass difference between the stack of discs and the surface artefact No 89 with time. The mass difference significantly decreases after air-to-vacuum transfer and decreases further by H-plasma treatment. The mass difference increases significantly after vacuum-to-air transfer. The gain in mass at ambient after cleaning is indicated by the horizontal line and arrows.
Download figure:
Standard imageTable 1. Gain and loss of mass upon different processes applied. The measured changes in mass difference between the stack of discs and No 89 are presented in the first column. Values for the cylinder and the stack were extrapolated using the effective surface areas. Uncertainties correspond to k = 1.
| Process | Change in mass | |||
|---|---|---|---|---|
| Difference (stack − 89)/μg | Cylinder No 89/μg | Stack of discs/μg | Per unit surface area/μg cm−2 | |
| Air vacuum transfer | −24.1 ± 0.3 | −10.2 ± 0.1 | −34.3 ± 0.4 | −0.14 ± 0.01 |
| H-plasma cleaning | −48.0 ± 0.3 | −20.6 ± 0.1 | −68.6 ± 0.4 | −0.29 ± 0.01 |
| Vacuum air transfer | +48.2 ± 0.3 | +20.7 ± 0.1 | +68.9 ± 0.4 | +0.29 ± 0.01 |
| Recontamination in air | +3.78 ± 0.12 | +1.63 ± 0.12 | 5.38 ± 0.15 | 0.0225 ± 0.0002 |
When transferred from ambient to vacuum, we found a change in the mass difference of 24 µg. The stack of discs lost more weight than the cylinder due to higher desorption of surface contaminants in vacuum. From the difference in surface area of 168 cm2 we calculated a natural desorption of 0.14 µg cm−2. Subsequent to the mass comparison, the surface of both artefacts was analysed by XPS for surface chemical state. In addition to the intense photoelectron lines of the bulk material we found remaining contaminants of hydrocarbons and oxygen compounds which are quite typical for polished surfaces stored in air. Both artefacts showed comparable amounts of contamination. In order to remove these contaminants we exposed both artefacts simultaneously to a low-pressure hydrogen plasma at a pressure of 70 Pa for 15 min. Figure 5 shows the plasma reactor in operation. H-plasma is proved to be very effective and non-abrasive for the cleaning of noble metal surfaces [26]. It was demonstrated that the surface of Au and PtIr can repeatedly and reproducibly be brought to the same chemical state by H-plasma treatment. An advantage of this process is its penetrating power. The gas even penetrates into small pores and cleans cavities and interspaces [27], and therefore is best suited for complex shaped objects such as the stack of discs. Figure 6(a) shows the survey scan spectra of artefacts before and after cleaning. The surface of both standards exhibits only little contamination. After the H-plasma treatment, all Pt photoelectron lines were increased in magnitude, while the carbon and oxygen peaks were reduced. This clearly demonstrates that carbon and oxygen compounds can be removed or reduced efficiently. For more detail, the narrow scan spectra of the platinum, carbon and oxygen are presented in figures 6(b), (c) and (d). Assuming a homogenous flat contamination layer, we calculated the thickness of the removed layer as (1.25 ± 0.5) nm, using the Pt doublet lines and published values for the attenuation length in polymer of 3.4 nm [27]. From the additional gravimetric measurements we found a further increase in the difference in mass of 48 µg between the artefacts, which corresponds to 0.29 µg cm−2 or 21 µg in total for the cylinder. From the calculated layer thickness and the related mass loss we found a density of 2320 kg m−3. This value is quite high and we assume the thickness determination could be affected by several influences. First, the structure of the contamination layer, which largely affects the attenuation length, is not known. Second, if the contaminants are present in the form of islands and clusters, the substrate intensities are too high and the calculated thickness too small.

Figure 5. Plasma reactor in operation. The standard, here an OIML knob weight, is cleaned by ionized H-gas at low pressure. The electrodes are placed under the red protective caps on the side of the loadlock.
Download figure:
Standard image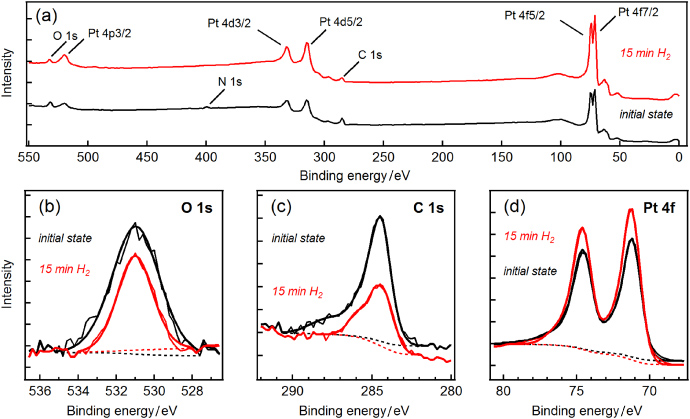
Figure 6. (a) Overview XPS spectra of the Swiss national PtIr prototype kilogram No 89. Black line: after weighing in vacuum (initial state); red line: after 15 min exposure to hydrogen plasma. (b) Oxygen signal, decrease in the peak area by −46% after cleaning. (c) Carbon signal and chemical compounds, decrease in the total peak area by −54% after cleaning. (d) Substrate signal, increase in the peak area by +25% for each photoelectron line after cleaning.
Download figure:
Standard imageThe cleaning of PtIr copies of the IPK has been the subject of several studies [29–33, 17]. The currently used procedure, nettoyage-lavage developed at the BIPM, consists of mechanical rubbing with solvent-soaked chamois followed by steam cleaning. From the mass loss due to this cleaning, the average contamination rate since the last cleaning can be evaluated, assuming a linear adsorption with time. Published values for this average contamination rate range from 1 µg/year for national prototypes cleaned at the BIPM [34], 1.4 µg/year for BIPM kilograms [35], 2.6 µg/year for UK standards [30] and up to 3.5 µg/year (1974–1984) for a German primary standard [29]. From our findings on H-plasma cleaning we calculated an average rate of 2.1 µg/year, which is in good accordance with the above results obtained by nettoyage-lavage. The broad range of data may at least partly arise from the nettoyage-lavage procedure itself. The mechanical rubbing depends on the operator and abrasion cannot be excluded. On the other hand, it is well known in surface physics that polishing procedures alone cannot remove all surface contaminants [25]. Furthermore, it was shown that mechanical treatment can increase surface reactions [36]. Other influences such as storage conditions or the frequency of use can also affect the stability in mass.
After the different vacuum procedures, the artefacts were transferred to ambient. The masses were determined again in the calibration laboratory using the national prototype No 38 as reference. For these instantly started measurements at ambient we did not use the new vacuum comparator because the humidity in the chamber needs some time to stabilize after a few days under vacuum. When transferred from vacuum to air, the mass difference between the stack and No 89 increases immediately by 48.2 µg, giving a resorption rate of 0.29 µg cm−2. Absolute mass values are extracted by comparison with the reference standard No 38. Due to the desorption, cleaning by H-plasma and resorption when exposed to ambient again, the surface artefacts show a loss in mass of 22.5 µg for the cylinder and 57 µg for the stack of discs. This corresponds to an overall desorption of 0.31 µg cm−2 and 0.24 µg cm−2, respectively. The results are in good agreement with previous findings [17, 30, 33, 34]. The difference between these values clearly demonstrates the limits to the use of surface artefacts. Both bodies were manufactured in the same way and initially cleaned by nettoyage-lavage. They exhibit comparable but not identical sorption coefficients.
The masses of the artefacts were repeatedly measured in order to monitor the gain of mass with time. Figure 7 shows the temporal evolution of mass with respect to both artefacts with time. Right after transferring the standards from vacuum to laboratory air, the increase in mass is very high with up to 1 µg/day for the cylinder and 3 µg/day for the stack during the first 3 days, but then the recontamination rate decreases very rapidly. We find our short-term recontamination data can be best described by an exponential function as shown in the graph. Following the concepts of Longmuir [36], this can be explained in a simple two-step model with a self-limited initial growth, followed by a small linear gain in mass in the long term. Contaminants have a certain probability S (e.g. S = 1) to stick on a clean surface. Once the surface is covered, the sticking probability decreases drastically (S ≪ 1). In this model, the loss of clean surface area with time δAc/δt, respectively the contamination rate δm/δt, is consequently proportional to the clean surface area Ac itself:


This can be described by an exponential function in time. Integration over time delivers the gain in mass Δm(t) with time, where Δmtot is the mass of the saturated contamination layer:


After initial accumulation the surface is completely covered and the sticking probability is changed. Fitting our data we found an initial contamination rate that starts at 1.1 µg/day for the stack and 0.25 µg/day for the cylinder, but diminishes exponentially with a half-life of (3 ± 0.5) days. For the total masses of the contamination layers, Δmtot, we find (5.38 ± 0.15) µg for the stack and (1.63 ± 0.12) µg for the cylinder. This corresponds to a gain in mass per unit area of (0.0225 ± 0.0002) µg cm−2. Further accumulation of contamination in the long term must be described by a second step of the model which can be superimposed. When the surface is completely covered by contaminants, the sticking factor does not change upon further contamination. The sticking factor for hydrocarbons on hydrocarbons remains constant. Therefore, we assume linear growth in the long term, which is in good agreement with published results [15, 34, 35]. Since the long-term contamination is reported to be as small as 3–9 ng/day [15], it cannot be detected in the short term.

Figure 7. Mass difference between the stack of discs and the reference No 38 (upper red points) and between the stack of discs and the surface artefact No 89 (lower black points) at ambient right after cleaning and vacuum. Errors for k = 1 are indicated. The solid lines in the upper graph are best fits using the exponential curve of the model described in the text. The lower graph shows best fit to the same data, using a root time dependence as proposed by Havard and Lewis [30] (dashed lines) and a power law (solid lines).
Download figure:
Standard imageOur assumptions are based on experimental results reported earlier. Ethylene, as an example of a hydrocarbon, adsorbs irreversibly and dissociatively on a clean platinum surface with a sticking coefficient of unity in the first monolayer [38–41]. Once the first monolayer is completed, the surface sticking coefficient has irreversibly changed. Additional adlayers are adsorbed reversibly [38], or can even be prevented. Other examples of self-limited growth are reported for oxygen on Pt [42–44, 48], Ir [45], Pd [46–48], Au [48–50] and also by numerous references in this literature. In some cases (Au, Pt) repulsive interactions between chemisorbed oxygen atoms prevent further sorption on the surface.
For sorption empirical models are very often used, which do not explain the underlying mechanism. For mass standards, it is reported that the contamination rate is highest right after cleaning [15]. A few models for the gain in mass after cleaning were presented earlier. From analysis of data, Quinn [51] concludes that the mass of a kilogram increases by 1 µg/month for 3–4 months, but then stabilizes at a rate of 1 µg/year. At least the short-term linear growth mode is in contrast with other findings [30] and our results. We found the maximum contamination rate for a clean surface is two orders of magnitude higher. Havard and Lewis [30] found the increase in mass for their standard is roughly proportional to the square root of time for the first 15 months after cleaning. Figure 7(b) shows such a root time dependence fitted to our data (dashed lines). The mismatch is obvious. In order to find a better accordance we applied a more general power law (solid lines), and found exponents of 0.33 (upper) and 0.37 (lower). The shape of these curves is in better agreement with our data, but still shows significant and larger residuals than our exponential approach. This Freundlich type mechanism [52] takes into account that for a partly covered surface the sorption rate decreases. This empirical model delivers very high sorption rates right at the beginning, where they increase to infinity. The non-physical nature of this sorption model is also demonstrated in the long term over several years, where it shows less agreement with gathered data [30]. This may also indicate that recontamination is a result of different mechanisms and cannot be described by one simple model. Therefore, the initial linear approach with an average contamination rate extracted from the mass of removed contaminants is quite crude and does also partly lead to the spread of previous results.
4. Outlook/conclusions
For the 'kg, mise en pratique' the underlying physics must be known. Effects such as sorption and cleaning are affecting the mass and need to be studied finally on real standards with complementary methods in addition to mass determination. In this paper, we have presented a new instrument combining gravimetric measurements with complementary methods such as XPS.
On a real PtIr standard and a surface artefact we have measured with complementary methods the natural desorption when transferred to vacuum, the mass loss due to H-plasma cleaning, the initial resorption when brought back to ambient and the mass-evolution due to resorption when stored in air. We have developed a simple model for the growth of contaminants, and demonstrated that this recontamination in the short term can be described by a self-limited growth mechanism.
Surface chemistry and physics are quite complex. They rely on the outermost atomic layer and can change drastically upon adsorption of an adlayer [38]. Impurities present on the surface can prevent or provoke chemical reactions [50, 53, 54]. Catalytic processes can take place, which were studied intensively in the past [38–41]. Gaining insight in the underlying physics can help us to improve the accuracy in mass metrology. In order to provide practical means of disseminating the redefined kilogram realized in vacuum to the mass scale at ambient, processes such as air–vacuum transfer, cleaning procedures and storage conditions for standards must be studied. For a pre-selection of best processes and materials studies can be carried out on surface samples. However, in order to prove their reliability, the means finally need to be tested by surface chemical analysis and also gravimetrically on real 1 kg standards. The presented instrument is designed to address the open questions in this field. First results clearly demonstrate that it is a powerful tool which may help to open the way to a practical and urgently needed means for the dissemination of the new defined kilogram.
However, for the stability of mass standards it is essential that an identical surface chemical state can be achieved repeatedly and periodically. The processes must be fully mastered. Consequently, modern processes like H-plasma cleaning can contribute to improve the stability of mass standards.
After submitting our paper, a highly relevant publication appeared in Metrologia [55]. The long-term stability of PtIr mass standards stored in vacuum and in air was studied by weighing and in parallel by XPS on small coupons stored under the same conditions. Over a four-year period the author found a linear mass gain of 3.7 µg/year when stored in vacuum and 4.4 µg/year when stored at ambient. The small and linear gain in mass in the long term is in perfect agreement with the prediction of our model.
Acknowledgments
The authors would like to thank Mettler-Toledo AG for continuous support. The authors are especially grateful to the METAS technical teams for expert technical design, construction and their outstanding mechanical work which created this new instrument. Scientific and technical support from S Wunderli and H-P Haerri is highly appreciated.