

Abstract
Far-field optical microscopy using focused light is an important tool in a number of scientific disciplines including chemical, (bio)physical and biomedical research, particularly with respect to the study of living cells and organisms. Unfortunately, the applicability of the optical microscope is limited, since the diffraction of light imposes limitations on the spatial resolution of the image. Consequently the details of, for example, cellular protein distributions, can be visualized only to a certain extent. Fortunately, recent years have witnessed the development of 'super-resolution' far-field optical microscopy (nanoscopy) techniques such as stimulated emission depletion (STED), ground state depletion (GSD), reversible saturated optical (fluorescence) transitions (RESOLFT), photoactivation localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), structured illumination microscopy (SIM) or saturated structured illumination microscopy (SSIM), all in one way or another addressing the problem of the limited spatial resolution of far-field optical microscopy. While SIM achieves a two-fold improvement in spatial resolution compared to conventional optical microscopy, STED, RESOLFT, PALM/STORM, or SSIM have all gone beyond, pushing the limits of optical image resolution to the nanometer scale. Consequently, all super-resolution techniques open new avenues of biomedical research. Because the field is so young, the potential capabilities of different super-resolution microscopy approaches have yet to be fully explored, and uncertainties remain when considering the best choice of methodology. Thus, even for experts, the road to the future is sometimes shrouded in mist. The super-resolution optical microscopy roadmap of Journal of Physics D: Applied Physics addresses this need for clarity. It provides guidance to the outstanding questions through a collection of short review articles from experts in the field, giving a thorough discussion on the concepts underlying super-resolution optical microscopy, the potential of different approaches, the importance of label optimization (such as reversible photoswitchable proteins) and applications in which these methods will have a significant impact.
Mark Bates, Christian Eggeling
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Optical nanoscopy: the road ahead
Stefan W Hell1,2 and Steffen J Sahl1
1 Max Planck Institute for Biophysical Chemistry, 37070 Göttingen, Germany
2 German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany
E-mail: shell@mpibpc.mpg.de and ssahl@mpibpc.mpg.de
Status.
The perception of what is possible in terms of spatial resolution in the optical microscope has dramatically changed over the last 25 years. It is worth reflecting for a moment on the observation by Louis Agassiz (1807–1873), displayed in the entrance area of the Nobel Museum in Stockholm:
'Every great scientific truth goes through three stages.
First, people say it conflicts with the Bible.
Next they say it had been discovered before.
Lastly they say they always believed it.'
Such may indeed be the case for many breakthroughs in natural sciences and technology. But several aspects of it undoubtedly hold true for the development of far-field optical (fluorescence) 'nanoscopy', also known as 'super-resolution' microscopy. The sceptics were numerous, as the foundations of the subject of optics and the works of eminent physicists of the 19th century—among them von Helmholtz, Lord Rayleigh and Ernst Abbe—weighed heavily in the initial years.
And yet the realization that a major leap in spatial resolution is possible by exploiting the spectral properties of the imaged molecules [1–4]—and not by fighting against the phenomenon of diffraction—clearly laid the foundation for what has been achieved. In little more than a decade, the early (1994/95) theoretical proposals of STED [1], GSD [2] and later (2003) RESOLFT [3] microscopy turned into powerful imaging strategies that have begun to unravel the inner details of cells and other transparent objects. Today, resolutions far exceed the old ones written in stone on Ernst Abbe's memorial in Jena. As a major paradigm shift, molecular states and the transitions between them have emerged as the enabling element and the key to radically overcoming the diffraction barrier in far-field optical imaging.
Meanwhile we have witnessed what has been dubbed by some observers 'the resolution revolution'. Far-field optical methods including STED, RESOLFT, PALM and STORM (the latter two are discussed by some of its inventors in the next contribution of this roadmap) are now routinely used in a growing number of labs to probe transparent matter at length scales a tiny fraction of the wavelength of the imaging light (compare figure 1, [5]). Membrane nanostructure (e.g. [6]), neuroscience (e.g. [7]), and systems with relevance to the study of diseases associated with protein aggregation, including Alzheimer's and Huntington's [8–10], are just a few examples where new insights have already been gained with 'super-resolution' fluorescence 'nanoscopy'. We could name many other topic areas such as visualizing the cell's nuclear machinery including chromosomal organization, or the molecular architecture of cell-cell interfaces, where new discoveries are currently awaiting us each month.
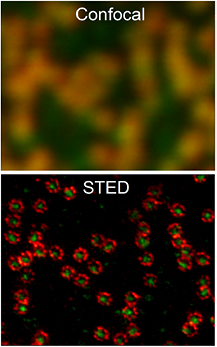
Figure 1. Nuclear pore complexes in an intact cell nucleus imaged by dual-color STED nanoscopy at ~20 nm spatial resolution (raw data is shown, for details of sample, see [5]). Image courtesy of Abberior Instruments GmbH.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
And yet, the field is still relatively young and there is a lot to do. We envision several important developments going forward, of which the main thrusts are clear: we wish to move more and more towards real-time, four-dimensional (4D) molecular analysis not only in cells, but tissue-like preparations or tissues themselves. In other words: three-dimensional (3D) in space, faster, and deeper. Already having moved far-field optical nanoscopy into experimental systems of the highest physiological relevance including the living mouse (figure 2, [11]) as a complete organism, the goal must be to enable the highest levels of resolution ultimately even under these challenging live-organism conditions.
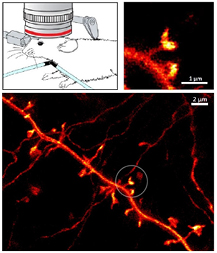
Figure 2. STED nanoscopy of the dendritic process in the molecular layer (somatosensory cortex) of a living mouse brain. Adapted with permission from [11], copyright 2012 The American Association for the Advancement of Science.
Download figure:
Standard image High-resolution imageThe present series of short articles by many of our leading colleagues gives insight into the current thinking of where the road ahead might lead us. Keep in mind that none of us actually knows, as the potential of a truly game-changing idea can only be guessed at. We are therefore fortunate to have them join us, detailing key advances as well as present obstacles of various aspects of methods development and application: Mark Bates and Xiaowei Zhuang discuss STORM, Rainer Heintzmann describes structured illumination microscopy (SIM), Martin Booth and Jörg Bewersdorf explain adaptive optics for super-resolution microscopy, Laurent Cognet and Brahim Lounis comment on applications to neuroscience, Simon Davis, Christian Eggeling and David Klenerman on those in immunological research. Alf Honigmann describes the prospects to dissect plasma membrane organization, Philip Tinnefeld shares with us insights into the calibration of super-resolution microscopes using DNA origami. Further topics are: live-cell super-resolution by RESOLFT (Ilaria Testa), switchable fluorescent proteins (Stefan Jakobs), nanobodies (Helge Ewers), in vivo super-resolution (Katrin Willig), correlative light and election microscopy (Gleb Shtengel and Harald Hess), advanced signal processing for STED / Gated STED (Giuseppe Vicidomini, Marco Castello and Alberto Diaspro) and approaches to improve fluorophore photostability (Thorben Cordes).
Advances in science and technology to meet challenges.
Resolution performance in fluorescence nanoscopy is now fundamentally related to the fluorophore's properties, notably its state-transfer or switching performance (kinetics, fatigue/longevity of the transfer, photostability and photon yield). The challenge of designing switchable fluorophores that hopefully will see further major improvements in these regards is a research topic of chemistry. On the optical side, improvements in detectors and lasers can be identified as major drivers of technical improvement and cost reduction, and thus ease of applicability.
Concluding remarks.
Coming back to the above observations about the course of discoveries, we have certainly witnessed that the global biological community is now beginning to embrace the new technology [12], and it is doing so rapidly. Could anyone have imagined one and a half decades back that multiple components of the nuclear pore complex (figure 1) would be rendered in an optical instrument as sharp as it is possible today? Probably not. Soon, it is likely that people will not want to see anything else anymore.
And it is evident from reviewing the literature that nanoscopy methods are beginning to enable numerous discoveries in molecular biology, neuroscience and beyond. Facilitating widespread access to these nanoscopes and associated expert advice is a key issue. Commercialization of the technologies will strongly contribute in this regard, helping to render fluorescence nanoscopy a new gold standard.
Super resolution microscopy by stochastic switching of single fluorescent molecules
Mark Bates1 and Xiaowei Zhuang2
1 Max Planck Institute for Biophysical Chemistry, 37070 Göttingen, Germany
2 Howard Hughes Medical Institute, Harvard University, Cambridge, USA
Status.
When 'on-off' switching was first observed in fluorescent molecules, it was not immediately obvious what an impact this would have on high-resolution fluorescence microscopy. Yet, this effect has emerged as a key concept in overcoming the 'diffraction limit' of optical resolution. For example, in STORM, a super-resolution imaging method reported in 2006, photo-induced fluorescence switching is used to temporally separate the spatially overlapping images of single molecules, allowing their positions to be determined with high precision and an image with sub-diffraction-limit resolution to be reconstructed from these molecular positions (figure 3(A)) [13]. The resolution of STORM is thus not limited by diffraction, but rather by the precision with which the molecular positions can be measured and, in some cases, also by the density of the localized molecules. Figures 3(B) and (C) show two-dimensional (2D) STORM images of in vitro assembled molecular structures as well as structures inside cells, resolved with a spatial resolution of ~20 nm, a 10-fold increase over diffraction limit [13]. Similar super-resolution methods based on photoactivation and localization of single molecules, PALM or Fluorescence Photoactivation Localization Microscopy (FPALM), were also published in 2006 [14, 15]. Variations of the switching mechanism, such as binding and dissociation of individual molecules, have also been used for super-resolution imaging [16].
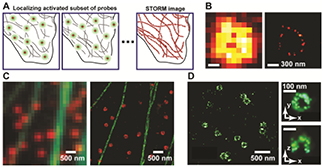
Figure 3. Super-resolution imaging by STORM. (A) At any time, only a sparse set of fluorophores decorating the sample are switched ON (green circles), and their positions are determined (crosses). Over time, many molecules have been localized, and their positions are plotted to create the STORM image. (B) Comparison of conventional (left) and STORM (right) images of a circular RecA filament. Adapted with permission from [13]. (C) Comparison of conventional (left) and two-color STORM (right) images of microtubules (green) and clathrin (red) in a cell. Adapted with permission from [17]. (D) (Left) XY-cross-section of a 3D STORM image of clathrin-coated pits in a cell. (Right) Magnified image of a single coated pit. Upper panel: an XY cross-section of the pit. Lower panel: an XZ cross-section of the pit. Adapted with permission from [18].
Download figure:
Standard image High-resolution imageSTORM was soon extended to multicolor and three-dimensional (3D) imaging [17, 18]. The rapid identification and development of new switchable organic dyes and fluorescent proteins (FPs) have not only enabled multicolor super-resolution imaging (figure 3(C)) but also improved the image resolution. 3D STORM was first achieved by the use of astigmatism to determine the 3D positions of individual molecules (figure 3(D)) [18]. Subsequently, a variety of additional 3D localization methods have been used for super-resolution imaging [19–24].
Advances in labeling, image analysis, and camera technology have allowed fast, time-resolved super-resolution imaging of live samples. Dependent on the configuration, a STORM microscope can achieve an image resolution of sub-10 nm, a time resolution of 30 ms, and the simultaneous detection of four or more spectrally distinct labels on a biological sample in certain cases, although these properties have not yet been simultaneously demonstrated on the same sample due to the trade-off between spatial and temporal resolution [25–30].
The improved resolution enabled by STORM, PALM and related methods has yielded fundamental insights into a variety of biological systems, as exemplified by the discovery of the periodic membrane skeleton structure in neurons (figure 4) [31], the elucidation of the molecular organization of chemical synapses in the brain [32], the structure of DNA at the end of the linear chromosome [33], and the architecture of the nuclear pore complex [34]. These examples illustrate the potential insights that can be gained when the spatial resolution of light microscopy is extended into a new regime: towards the scale of molecules and molecular complexes.

Figure 4. A novel periodic membrane skeleton structure in neurons revealed by STORM. (A) 3D STORM image of actin in axons of hippocampal neurons showing periodic ring-like structures evenly spaced along the axon. (B) STORM image of actin (green) and βII-spectrin (magenta) in an axon showing that the adjacent actin rings are connected by spectrin tetramers. Adapted with permission from [31].
Download figure:
Standard image High-resolution imageCurrent and future challenges.
While much progress has been made towards non-invasive, super-resolution imaging of biological specimens using single-molecule-based super-resolution methods, present technology still has limitations, which are the focus of current development. Here, we focus on three topics: (i) fluorescent labels, (ii) labeling methods, and (iii) spatial and temporal resolutions.
It is the photo-switching properties of the fluorescent label that ultimately determine what is attainable in terms of image resolution. The photon output (which determines localization precision), on-off switching contrast and duty cycle (which limits the localization density), photostability, and number of switching cycles are all important determinants of image quality [26]. The photon output of the most popular photo-switchable dyes is in the range of several thousand detected photons per switching cycle [26]. Recently developed ultra-bright photo-activatable dyes increased this number by 100–1000 fold, but their photoactivation efficiency is relatively low (~20–60%) [28]. The development of brighter photoswitchable dyes with high activation efficiency will allow the image resolution to be substantially improved. Another key characteristic is cell permeability. Most of the existing high-quality photoswitchable dyes are not cell permeable and cumbersome delivery methods, such as microinjection and electroporation, have to be used to deliver these molecules into living cells. The development of cell-permeable photoswitchable dyes with desirable photophysical properties would greatly benefit live-cell super-resolution imaging [35].
The labeling technologies that link fluorophores to their target molecules are also essential. Immunofluorescence labeling has the great advantage of probing endogenous proteins without the concern of genetic perturbation induced artifacts, however antibodies are bulky (~10 nm in size). The recently developed nanobody (~2 nm) technology could be a promising solution to counter this problem [36]. Enzymatic protein (e.g. SNAP and Halo) or small peptide tags allows dyes to be linked to proteins using genetic fusion approaches, which results in a relatively small label size (1–3 nm), and also allows live cell labeling and imaging [37].
Fluorescent proteins (FPs) offer an easier approach to labeling proteins in live cells. However, compared to the synthetic dyes, the photon output of photoactivatable FPs (PAFPs) is much smaller (~1000 photons or less). Moreover, the brightest PAFPs tend to occupy the same spectral region (red emission), while the blue/green PAFPs give even fewer photons (a few hundred), making multicolor imaging challenging with PAFPs. Hence, it is desirable to develop novel PAFPs with improved photon output over the entire visible spectral range [38]. In addition, the tendency of FPs to dimerize could also cause protein aggregation and artifacts in cellular structures. Finally, PAFPs tend to have a substantial dark fraction that leads to low signaling efficiency [39]. Hence, it is important to minimize the dimerization tendency and optimize the signaling efficiency of PAFPs. Finally, most live-cell labeling experiments use transiently transfected FP-fusion proteins, in which case the presence of unlabeled, endogenous proteins reduce the labeling efficiency. The recently developed CRISPR-based genome editing approach offers a promising route for achieving 100% labeling efficiency of proteins in cells.
In addition to the fluorescent labels, the optical detection system and analysis methods also play an important role in determining the localization precision and accuracy of fluorophores, which in turn impacts the image resolution. A plethora of high-precision 3D localization methods have been developed. However, at the molecular scale, localization accuracy may be compromised by fluorophore orientation effects, and methods combating this effect have begun to emerge [40, 41].
Recent advances in camera technology and data analysis have led to improved time resolution of single-molecule-based super-resolution imaging. The introduction of scientific complementary metal-oxide semiconductor (sCMOS) cameras has allowed near video-rate STORM imaging in some experiments [30]. New algorithms for fluorophore localization, in particular, those that are capable of localizing emitters with substantially overlapping images, promise to further increase the imaging speed (e.g. [42, 43]).
Aided by continuous technological advances, single-molecule-based super-resolution imaging has developed into a widely used and powerful method for visualizing previously unseen details in biology.
Structured illumination microscopy
Rainer Heintzmann1,2,3
1 Leibniz Institute of Photonic Technology, Jena, Germany
2 Institute of Physical Chemistry and Abbe Center of Photonics, Friedrich-Schiller University Jena, 07743 Jena, Germany
3 King's College London, Randall Division, London, UK
Status.
Although the term structured illumination has been used for diverse applications such as surface profiling, transmission phase imaging and optical sectioning or scattering and fluorescent samples, this section is limited to the application of structured illumination for super-resolution fluorescence microscopy.
Figure 5 shows one way of classifying various fluorescent super-resolution microscopy methods. In fluorescence structured illumination microscopy [44, 45] (SIM) the sample is illuminated with a typically dense distribution of lines and an image is formed on the camera for each illumination structure. SIM not only defines where a sample is illuminated but, due to the image formation, also obtains spatial information about the sample emission. All this information, where excited and where emitted, is equally useful and, when combined, leads to a resolution gain of a factor of two as compared to the limit spatial frequency of an imaging system as defined by Abbe's well known equation. STED typically operates with a rather open pinhole for maximum efficiency of signal detection. It thus bases its resolution increase only on the non-linearly coupled illumination profiles. However, SIM, saturated SIM (SSIM) or non-linear SIM [46] and RESOLFT [47] utilize the emission information. This is especially important since the illumination dosage and thus the resolution gain has to be kept low to preserve the living state of the sample.

Figure 5. Structured illumination in the context of other superresolution methods.
Download figure:
Standard image High-resolution imageIn addition to imaging the emission light at each projected pattern position, high-resolution SIM bears a second important hallmark: generating the illumination structure by coherent interference.
This allows very high spatial frequencies of the illumination structure up to Abbe's limit 1/d = 2NA/λ, with peak to peak distance d, the numerical aperture NA and the illumination wavelength λ, to be generated in the sample with 100% contrast.
For point-illumination processes as in confocal, STED or GSD microscopy, the illumination contrast diminishes with spatial frequency, reaching zero at Abbe's limit frequency. In SIM coherent illumination maintains the illumination contrast up to the limit. This enables SIM to efficiently mix a strong amount of high-resolution information from outside the frequency limit into the detection pass-band of the microscope.
In SIM an image is acquired at a series of illumination (phase) positions of the pattern and the images are processed to yield a high-resolution image. The reconstruction process essentially extracts (disentangles, unmixes) the overlapping down-modulated high-frequency information from the set of images and reassigns this information to the correct sample spatial frequencies [48].
Even though grating-like patterns can also be formed in an incoherent illumination process, e.g. by imprinting in a scan process with a focused laser beam, and high-resolution SIM images can be reconstructed [49], the full potential of SIM is only utilized in coherent illumination [50].
Current and future challenges.
A major disadvantage of the typically dense SIM illumination grating (2 or 3-beam interference) is the dominance of unwanted out-of-focus light in the detected images for thick densely labeled samples. Even though SIM processing can effectively remove this out-of-focus light, the final image quality still deteriorates due to the excessive noise generated by the Poisson statistics photons.
A second challenge of classical SIM is the resolution to remain limited to the factor of two. This can be overcome with non-linear SIM as outlined below.
Imaging speed is also an important consideration for SIM. It should be noted that single-beam illumination methods like confocal microscopy or STED are inherently limited in their rate of information generation, as only one photon can be obtained per fluorescence lifetime and molecule whilst the illumination spot is addressing this small sample area. For single particle situations, single-beam scanning can still yield impressive imaging rates [51]. This is possible, since for detecting a single molecule or small particle such as a 15 nm vesicle in front of a dark background, it suffices to collect very few photons per particle. The situation changes significantly in the case of densely labeled samples, unspecific background signal and large fields of view, where a good signal to noise ratio is required in each pixel to discern fine structural details. Thus especially for densely labeled samples the multiplex advantage of SIM should allow significantly higher imaging speeds at much larger fields of view. Another promising route to fast SIM acquisition of 3D image stacks is the combination of SIM with multi-plane imaging as developed by Gustafsson and Abrahamsson [52].
A further challenge to SIM is the image processing speed. Especially since the image acquisition speed is rapidly increasing it becomes more and more of a challenge to process this data quickly enough, ideally to allow online visualization of the acquired data.
A general challenge to microscopy and especially to high-resolution imaging is the presence of sample-induced aberrations. These therefore also need to be addressed in the SIM data reconstruction process or corrected for in the experimental data acquisition.
Advances in science and technology to meet challenges.
To address the out-of-focus problem of SIM there are essentially four approaches: (A) The periodic illumination structure can be made more sparse. This reduces the noise-contribution of the out-of-focus light but requires more phase-positions (scan positions) to be acquired with an associated loss in imaging speed. Even though several overlapping coherent beams can form 2D or 3D sparse periodic patterns [53], each new coherent illumination beam also contributes to the zero-frequency content which is solely responsible for the noise of uniform strength at all frequencies. Thus each additional beam reduces the in-focus signal-to-noise ratio of the highest frequency information. (B) The illumination geometry can be changed as in line-scanning SIM [54]. However, this slightly reduces the highest possible frequency and complicates the setup for the required rotation of the illumination structure and the orientation of the line-detection. (C) Coherent SIM illumination can be combined with the light-sheet geometry, either with the help of interfering Bessel beams7 or multiple interfering light-sheets [55]. A disadvantage of this approach is the uncommon sample mounting and the need for illumination access from the side of the sample. (D) Spot-light illumination geometry (figure 6), which is suggested here: two (or more) mutually interfering relatively large focal spots are overlapping in the sample to generate the finely striped illumination structure. This still allows very high angles of interference and thus still very high frequencies, yet due to the small illumination region in combination with the very high illumination angles, the out-of-focus contributions in the imaged region of interest are massively reduced. In addition, the polarization can be kept close to azimuthal guaranteeing close to 100% fringe contrast. The need for spot-light stepping (e.g. with a scanning and de-scanning galvanometric scanner) is an extra effort, but the loss in speed is at least partially compensated in practice by the reduced region of interest required in the detection process and thus the faster readout of the ROI on the camera.
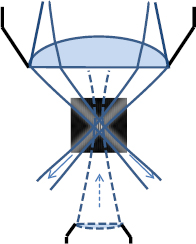
Figure 6. Spotlight SIM geometry as suggested in the text. The low-NA beam from below is optional and not included in the shown intensity structure.
Download figure:
Standard image High-resolution imageRegarding the resolution, non-linear SIM, closely related to RESOLFT, but different in some of the image processing aspects, can break the linear SIM resolution limit which is transferring twice the smallest frequency as set by Abbe's equation. However, to demonstrate non-linear SIM at 50 nm resolution in living samples, many cycles of illumination, photo-switching and detection need to be performed. A disadvantage of the choice of molecular switching for the required non-linearity is that the on/off switching of single molecules worsens the detections statistics. If an experiment is repeated multiple times the fluctuation between measurement is not governed by the Poisson statistics according to the detected number of photons but rather dominated by the Poisson statistics of the on/off switching of the few molecules contributing to a pixel. A high label density and multiple switching cycles as well as better processing software can counteract this photon-bunching-like molecular switching statistics introduced into the measurement process. Some of this will undoubtedly be addressed in future SIM image processing work.
SIM imaging speed has continuously been improving now reaching rates of several hundred raw-data frames per second. This is mostly due to current advances in camera and spatial light modulator technology. In terms of image processing, recent developments include the use of graphics processing units (GPUs) to speed up the process, but a convenient and fast general purpose SIM algorithm still has to be developed.
With respect to sample-induced aberrations, novel data processing strategies ('blind-SIM' reconstruction) are under development, which utilize the redundancy in the SIM data to reconstruct and correct for aberrations.
Concluding remarks.
High resolution structured illumination microscopy for fluorescence microscopy has since its development [44, 45] gained a lot of interest in biological applications as it is exposing the sample very effectively causing a minimal amount of photo-damage. Especially with the recent strategies [50, 52–54] to reduce disturbing out-of-focus light, SIM promises to become a preferred choice for long-time super-resolution fluorescence imaging of living samples in routine applications.
The prospects of adaptive optics for super-resolution microscopy
Martin J Booth1,2 and Joerg Bewersdorf3,4
1 Centre for Neural Circuits and Behaviour, University of Oxford, Oxford, UK
2 Department of Engineering Science, University of Oxford, Oxford, UK
3 Department of Cell Biology, Yale University School of Medicine, New Haven, Conneticut, USA
4 United States Department of Biomedical Engineering, Yale University, New Haven, Conneticut, USA
Status.
Adaptive optics (AO) has the potential to transform the abilities of super-resolution microscopes. Spatial variations in the refractive index of a specimen cause wavefront aberrations that lead to a reduction in image contrast and resolution. Using a dynamically reconfigurable optical element, such as a deformable mirror (DM) or liquid crystal spatial light modulator (SLM), it is possible to correct aberrations, restoring operation of the microscope close to the diffraction limit. All specimens exhibit aberrations at some level. Depth-dependent spherical aberration is introduced by a mismatch in refractive index between the objective lens immersion medium and specimen and/or by a non-optimum coverglass thickness. The structures of tissues, cells and organelles create further, more complex aberrations. Even high-specification microscope systems have residual aberrations that can affect image quality.
AO for conventional microscopes has been developed over the last decade and more and has shown clearly the benefits of aberration correction in extending high resolution imaging to thicker specimens [56]. The application of AO to super-resolution microscopy is however in its infancy. All super-resolution methods, whether stimulated emission depletion (STED), single molecule switching (SMS), such as PALM or STORM, or structured illumination microscopy (SIM), place higher demands on specimen preparation, optics and/or data processing than conventional microscopy. It is not surprising therefore that the tolerance of these microscopes to aberrations is tighter.
The first appearance of AO in this field was in STED microscopy, where depletion beam phase masks were implemented using SLMs [57, 58]. Later developments showed how such SLMs could be used to compensate specimen aberrations in a feedback correction loop (figure 7) [59]. AO has also been introduced to SMS microscopy, where DMs correct for aberrations in the imaging path (figure 8) [60]. A similar configuration has been shown for SIM [61]. This work so far has provided a sound basis for future developments, but there are still several challenges to meet before AO becomes widespread in super-resolution microscopes. However, the adaptive, self-correcting nature of this technology promises to transform what are currently sensitive instruments into more robust and easy-to-use imaging tools.

Figure 7. Aberration correction in 3D STED microscopy showing 200 nm beads imaged through 25 μm of tissue: confocal (left), STED (middle) and STED with aberration correction (right). Reproduced with permission from [59], copyright 2012 The Optical Society.
Download figure:
Standard image High-resolution image
Figure 8. 3D SMS images of microtubules before and after aberration correction. Reproduced with permission from [60], copyright 2015 The Optical Society.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
Aberrations affect each of the super-resolution techniques (STED, SMS and SIM) differently due to the considerably different image formation process in each case. This leads to varied challenges in the ways AO is implemented.
In STED microscopy, the critical aspect is the formation of the focus of the additional STED laser, which ultimately determines the efficiency and resolution. This focus ideally requires a point of 'zero' intensity surrounded by a bright ring. Even a small increase of the minimum intensity by aberrations leads to a strong reduction in detected fluorescence. In 2D STED, the zero intensity is fairly robust to aberrations. In 3D STED, however, even a small distortion can lead to a serious loss of signal [59].
The optics of an SMS microscope resemble those of a conventional widefield microscope. The main negative impact of aberrations occurs in the data processing stage, where the images of the individual emitters are compared with a model point spread function (PSF). As the model PSF is usually aberration-free, there is a mismatch between the data and the model due to any aberrations that are present. This leads to a lower probability that each emitter image meets the acceptance criteria and hence to fewer emitters contributing to the final image. Modifying the criteria to increase the number of acceptable fits leads to a reduction in average localization precision. It has also been shown that localization accuracy can be detrimentally affected by aberrations [60]. Correction is best performed using a DM in the imaging path, as the mirror-based device works well with the broadband, randomly polarized fluorescence emission.
SIM relies upon the projection of high spatial frequency sinusoidal patterns into the specimen and the imaging of the resulting fluorescence. It is therefore the reproduction of this high spatial frequency that is the most important aspect of the SIM system and, correspondingly, the aspect that is most critically affected by aberrations. It has been shown in non-super-resolution SIM, where structured illumination was used for optical sectioning, that certain aberration modes affect the imaging of the illumination spatial frequencies much more than other modes [62] and this affects the design and operation of AO correction. In principle, correction of illumination and detection paths could be implemented using a single DM placed in a common path.
Advances in science and technology to meet challenges.
It is already known that specimen-induced aberrations are significant enough to degrade super-resolution images even at depths of a few micrometres [59–61]. As aberrations change across specimens, it is vitally important to be able to measure and correct aberrations in a feedback system. Correction based upon specimen models, such as a uniform but mismatched refractive index, can only provide a partial solution. Efficient image-based aberration measurement—or sensorless AO—has proven successful in this area; direct wavefront sensing, as deployed in other microscopes [63] may well have further application in super-resolution methods. In all cases, an acceptable balance between speed and accuracy of correction and specimen exposure will need to be found.
Super-resolution methods, where the aberration tolerances are tighter than conventional microscopes, may also be more susceptible to the effects of aberrations that vary across the image field. Such field-dependent aberrations could be compensated in STED microscopy using faster correction elements that reconfigure during scanning. Certainly, the development of kilohertz rate, continuous phase, liquid crystal SLMs would be beneficial here. In imaging systems (SMS, SIM), the field dependence could be solved using multiple conjugate AO, where several correction elements in different optical planes compensate for complex specimen structures. A reduction in cost of DMs would clearly be of benefit in making this a practical proposition. New DM designs that are better tailored to the demands of microscopy—for example, the correction of spherical aberration in high numerical aperture systems—would also be valuable.
There is further scope for SLMs to be used for control of polarization, which plays an important role, in different ways, in all super-resolution microscopes. For example, in STED microscopy, the quality of the depletion beam is highly dependent upon the polarization state, which can be affected by system and specimen effects. Correction of these polarization aberrations could form part of more advanced adaptive systems. The SLM can also be used to provide combined 2D and 3D STED foci superimposed using polarization multiplexing [64].
In all super-resolution microscopes, the increasing complexity of aberrations as the focus moves deeper may necessitate the use of multiple adaptive elements to provide large-amplitude complex wavefront correction. One could, for example use a DM with a large deflection range, but a small number of actuators—for compensation of low order (e.g. spherical) aberrations—in tandem with a smaller deflection range, more complex device, to compensate higher order aberrations.
Concluding remarks.
Now that super-resolution imaging is established as a valuable tool for biological sciences, the next level of development will be to extend its application to a wider range of scientific questions. In one sense, this means that the microscopes should be capable of working with a wider range of specimens. Using AO to access thicker specimens will permit more use of super-resolution in tissue applications, with significant potential benefit in developmental biology, neuroscience and other areas. Another advantage of the wider use of AO will be increased usability of super-resolution systems, as AO provides the opportunity for self-calibration and auto-alignment of systems [65], thus reducing the need for in-house technical support. Technological advances such as these will facilitate the transformation of these methods into commonplace laboratory tools.
Correlative super-resolution optical and electron microscopy roadmap
Gleb Shtengel and Harald Hess
Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA, USA
Status.
Correlative light and electron microscopy (CLEM) is attractive because it exploits two microscopy techniques that give very different and very complementary information. Electron microscopy (EM) provides structural information often necessary for proper interpretation of light microscopy (LM) data. Alternatively, LM provides protein specific information which is harder to get with EM, but does not generally provide ultrastructural details. Therefore, by combining LM and EM, one achieves protein specific localization in the context of a global structure.
When LM is performed on a live sample first, doing subsequent EM imaging allows linking the dynamics of the biological organism to its ultra-structure.
In regular CLEM, the LM resolution is restricted by optical diffraction to ~200–300 nm, often limiting CLEM to illustration and identification applications.
Super-resolution optical microscopy may have a resolution of 10–20 nm, providing a much closer match to the resolution of EM (0.1–10 nm) and thus allowing for correlative imaging on a comparable resolution scale.
Correlative super-resolution optical and electron microscopy (CSREM) was first demonstrated as a combination of PALM and transmission electron microscopy (TEM) performed consecutively on Tokuyasu cryosections. Other CSREM modalities had since been realized. PALM and SEM as well as STED and SEM had been demonstrated in plastic-embedded sections [66]. PALM and SEM had also been performed on thin Tokuyasu cryosections [67]. Interferometric PALM (iPALM) had been combined with focused ion-beam (FIB)-SEM to allow for 3D CSREM in thick Tokuyasu cryosections [68] (figure 9). iPALM and Pt replica TEM [69] as well as dSTORM and SEM [70] were combined for studies of membrane-bound structures. New OsO4—resistant version of fluorescent protein mEos allowed for CSREM in plasticized sections of cells fixed with OsO4, critical for better ultrastructure preservation [71] (figure 10). The preservation of fluorescence and antigenicity of LR-white or HM20 acrylic embedded samples offers another path to CSREM. Finally, performing LM and EM at cryogenic temperatures is very appealing as it allows to avoid chemical fixation (or perform it in frozen sate) and eliminates a potentially disruptive step of dehydration [72].
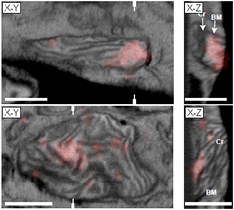
Figure 9. X–Y (left row) and X–Z (right) slices through correlated 3D data sets generated by iPALM (red, mEos-TFAM, colocalized with mitochondrial DNA) and FIB-SEM. Reproduced with permission from [67]. Labels denote the mitochondrial boundary membrane (BM) and cristae (Cr). White hatch marks indicate the locations of the X–Z sections. Scale bars: 500 nm.
Download figure:
Standard image High-resolution image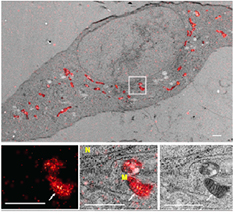
Figure 10. Correlative PALM and TEM images of GMA section of 3T3 cells expressing mitochondrially targeted mEos4a. Reproduced with permission from [71]. Top, whole cell. Bottom, PALM, merge and TEM images of boxed area. Arrows indicate a mitochondrial cristae fold. Labels indicate nucleus (N) and mitochondrion (M). Scale bars, 1 μm.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
Several important challenges must be addressed in order to achieve useful CSREM. These are not so much in the instrumentation but rather in the protocols and dealing with the unique labelling constraints that each biological sample imposes.
The protocols allowing for high resolutions of each individual technique have to be developed and adjusted to be compatible with each other. The fixation and staining protocols optimized for LM and EM are very different and often mutually exclusive. Strong fixatives, such as gluteraldehyde, OsO4 and uranyl acetate are commonly used in order to preserve the cellular ultrastructure for high fidelity EM imaging. These strong fixatives adversely affect fluorescence of optical labels and also degrade antigenicity, resulting in poor labeling density and specificity.
The individual imaging modalities must provide the critical information expected from them (protein specific imaging with good labeling density and specificity for FM and good ultrastructure preservation for EM).
Sample handling protocols need to be developed, which do not result in sample deformation and degradation during preparation, imaging, and transfer from aqueous medium of LM into vacuum environment of EM.
In order for CSREM to be truly quantitative, a registration algorithm must be developed, which allows not only to register LM and EM data sets, but also to quantify the precision of this registration, since the uncertainty of a correlative imaging technique is a quadrature sum of all contributing uncertainties [68]. For this purpose we had used Au nanoparticles that can be embedded into the substrate or sample itself. The Au nanoparticles can be designed to have photoluminescence over the entire visible spectrum. They are single dipole emitters which do not bleach, and are also electron dense, making them excellent fiducial markers for multiple color LM and EM image registration. Vertical registration in the third dimension remains a challenge since the dehydration step between LM and EM imaging may result in sample shrinkage.
Advances in science and technology to meet challenges.
In order to overcome the above challenges, many aspects of CSREM need to be further improved.
One of the most critical areas is improvement in compatibility of sample preparation protocols for fluorescent and electron microscopies. We think that for 3D CSREM plasticized sections are most promising. But the choice of high quality (high density and high specificity) fluorescent labeling strategies compatible with OsO4 fixation and resin embedding (particularly epoxy resins, such as EPON or Durcopan) are extremely limited. There are two alternatives, both need to be explored:
- Development of the fluorescent labeling strategies that are compatible with EM sample preparation protocols, particularly with resin embedding. This includes more fluorescent label colors and engineering fluorescent proteins for such CSREM compatibility as well as adapting caged dyes into biological systems and into a CSREM protocol.
- Investigating alternative EM preparation protocols allowing for better preservation of fluorescence and/or antigenicity.
CSREM will also benefit from improvement of both parts—super-resolution optical and electron microscopy. We had concentrated on PALM/STORM as a super-resolution method. It would be beneficial to further investigate other modalities, such as STED and SIM, particularly non-linear SIM, as it offers higher optical resolution.
Finally, cryogenic CSREM looks very promising. Possibility of imaging the biological sample in its near native state (having not undergone chemical fixation, embedding, etc.) is very appealing.
Concluding remarks.
Recent advances in super-resolution light microscopy allow for optical imaging with resolution approaching that of electron microscopy. This, in turn, allows for correlative light and electron microscopy which combines protein specific and ultructructural information on biological samples.
Microscopy reference samples produces by DNA nanotechnology
Philip Tinnefeld
Institute for Physical & Theoretical Chemistry, Braunschweig Integrated Centre of Systems Biology (BRICS), Laboratory for Emerging Nanometrology (LENA), Braunschweig University of Technology, Braunschweig 38106, Germany
Status.
The spatial resolution of new superresolving fluorescence microscopes has traditionally been demonstrated with fluorescent beads or cytoskeletal filaments in fixed cells. Resolution is, however, defined as the ability to distinguish two objects and is not fully characterized by the point-spread-function of a fluorescent bead or the localization precision of a single molecule. Fluorescent beads also have the dyes included in a polymer with an unnatural environment with different refractive index and altered photophysical properties [73]. Alternatively, cytoskeletal filaments are one-dimensional (1D) structures and distances between two filaments arbitrarily occur in cells. The distance between filaments used to demonstrate resolution cannot be reproducibly fabricated. In addition, small distances between filaments occur stochastically and no statistics to also exclude the influence of noise (which can lead to larger or smaller apparently resolved distances) can be obtained. Filament labeling requires labels of substantial size and the labeling density is not rigorously defined. Other natural structures such as nuclear pore complexes are well reproduced in size and stoichiometry but labeling is more difficult and the achievable geometries are limited [74]. Despite some efforts (see [75, 76] and references therein) there has not been a sample, which allows simple comparison of different methods or to check every component required for a successful super-resolution measurement. These components do not only imply the optical alignment of the microscope but also include the imaging buffers, the optimal laser powers and several other factors. Is there a possibility for a simple quick test of the microscope on a daily basis as well as for a quantitative, robust and reproducible characterization of its resolving power including all the complexities of emerging fluorescence imaging modalities?
In recent years, we have introduced DNA origami nanorulers as reference samples for fluorescence microscopy [77]. Structures obtained from DNA nanotechnology have several unique properties which cannot be met by any other technology: (i) structures ranging from nanometers up to micrometers can be constructed with high yield by self-assembly; (ii) these structures are easily modified incorporating fluorescent dyes or other orthogonal chemistry at defined positions with subnanometer accuracy. This allows, for example, creating patterns of dye molecules with defined geometry and a defined number of fluorescent dye molecules. (iii) After production, DNA nanostructures are fairly robust and samples can be stable over months or even years.
Figure 11 shows the principle of DNA origami, a very important DNA nanotechnology method [78]. In DNA origami, a long scaffold strand is folded into defined structures which have been designed with the aid of a computer program. Overall, a ~2.6 μm long DNA strand can be folded into almost any desired shape.

Figure 11. Scheme of the formation of DNA origami. A long scaffold stand is folded into the desired conformation by short staple strands. Dyes are incorporated using labelled staple strands.
Download figure:
Standard image High-resolution imageFor applications with super-resolution microscopy (SR), a variety of DNA origami nanorulers has been presented [77]. Starting from resolving two dye molecules at a distance of 88 nm on a DNA origami rectangle, the range of nanorulers was quickly expanded [79]. Meanwhile DNA origami nanorulers are available in a variety of colors and intermark distances. 27 nm–90 nm nanorulers are preferred for STED microscopy (figure 12(a)), 120–160 nm are typical for SIM (figure 12(b)), and 350 nm can be readily resolved by conventional diffraction limited microscopes [73]. Super-resolution techniques relying on the successive localization of single molecules have special requirements such as switching off of the largest fraction of molecules. Rulers in the range of 30–90 nm have been made for dSTORM where switching is achieved by a thiol-containing oxygen depleted buffer (figure 12(c)) [73].

Figure 12. Examples of DNA origami nanorulers: (a) 71 nm STED, (b) 160 nm SIM, (c) 94 nm dSTORM, (d) 80 nm triple-mark two-color DNA PAINT. Insets show sketches of the respective nanoruler.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
A wide range of DNA origami rulers are conceivable that can be used for demonstrating the performance of a microscopy technique in terms of sensitivity and resolution. Two marks or lines are designed at specific distances to resolve two objects according to a resolution criterion. Custom-production can create almost any distance in the range of 3–360 nm. Research groups have started using DNA origami nanorulers to demonstrate the potential of their techniques including counting of molecules [80], 2D and 3D super-resolution [81, 82], and dye performance [83]. The nanorulers are also useful as benchmark samples in microscope production, demonstration and microscopy training. Especially beginners in super-resolution microscopy find it difficult to achieve required labeling densities, to apply the optimal buffer conditions and to use the right laser intensities and camera integration times. Without reference samples, it is fairly difficult to evaluate the quality of an image especially for 3D samples. Single-molecule localization based SR additionally requires well-defined buffer conditions which are not stable over time. As an alternative localization based technique, DNA PAINT nanorulers have emerged as the reference sample of choice because they only require a single laser wavelength and they are less prone to bleaching and buffer conditions [84]. In DNA PAINT, the apparent blinking of molecules is induced by transient binding of a short labeled oligonucleotide to a docking strand protruding from the DNA origami. By adapting the length of the double-stranded region and the oligonucleotide concentration, the switching kinetics are totally controlled and a maximum number of photons can be extracted enabling a resolution of 6 nm or better [85].
Researchers have also started using DNA origami nanorulers as a decision helper before purchasing a microscope by comparing systems from different companies with nanorulers. Still, a large number of research articles would strongly benefit if researchers demonstrated their abilities with samples that make measurements comparable (e.g when for demonstrating dyes with improved properties and quantum yield [35, 86], self-blinking dyes [87], or new fluorescent proteins and for all research that is claiming quantitative resolution e.g from localization precision or from widths of point-spread functions).
The information from DNA origami reference samples can provide more than resolution and sensitivity. Using 3D nanorulers, the refractive index mismatch correction factor that experimentally might deviate from theoretical consideration can be determined [88], and chromatic aberrations could be ultimately corrected e.g. by using DNA PAINT with different-colored imager strands that bind to the same docking strand on the DNA nanostructure (figure 12(d)). In the future, 3D chromatic aberrations should also become amenable for correction. Another big advancement will be in situ reference structures that are added to the biological specimen similar to fiducial markers and allow for direct corrections and referencing e.g. of molecular brightness and bleaching steps for determination of stoichiometries. DNA origami nanorulers will also be realized with the important class of fluorescent proteins and biological environment could be more realistically reproduced with dye labeled proteins on DNA origami. A main challenge in many of these applications is the provision of affordable reference samples of sufficient amount, robustness and labeling homogeneity.
Advances in science and technology to meet challenges.
Fluorescent beads are common reference samples. They are robust but they also bleach, contain dyes in unnatural environment and cannot easily be adjusted to specific needs. In contrast, DNA nanostructures are modular, flexible, well-defined and can be equipped with further functional groups for specific immobilization. If researchers manage to achieve a similar degree of robustness and accessibility, DNA reference samples will become a key technology for the training, spreading and improvement of emerging microscopy techniques.
Concluding remarks.
Fluorescence microscopy has tremendously evolved but our abilities to quantify the progress has lagged behind. DNA nanotechnology allows designing probably the largest man-made objects with control down to the atomic level. Equipping these structures with fluorescent dyes gives us new handles to address, test and develop fluorescence microscopy with reference samples that are able to face the increased complexity, resolution and finesse that is characteristic for the impressive innovation pace of the field.
Super-resolution microscopy to dissect plasma membrane organization
Alf Honigmann
Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany
Status.
The plasma membrane (PM) is the interface between the outside environment and the internal cytoplasm of the cell. Many important cellular functions are localized at this interface zone for example signaling and sensing, selective transport of matter, cell adhesion or electrical action potential propagation. The specific functions of the PM in principle result from the spatiotemporal organization of proteins and lipids into distinct functional complexes. Despite the success of lipidomics, proteomics and glycodomics in identifying the molecular components of the PM we still lack the connection between composition, structure and biological function of the PM. Our simple models do not explain why cells need thousands of different lipid species for functioning. The question is how lipids interact with proteins to self-organize into functional structures. Without understanding the fundamental principles of membrane organization we certainly miss an important layer in the regulation of cellular processes. To reveal the principles of bio-membrane organization new approaches and methods are required, that provide the spatiotemporal resolution and sensitivity to capture interactions of lipids and proteins directly in living cells.
Current and future challenges.
It is generally agreed that the simple model of the PM as a 2D passive fluid of lipids with embedded proteins has to be substituted by a more complex model which takes self-organization of different lipid and protein species into account as well as the active remodeling of the membrane by different cellular structures and processes (for example the cytoskeleton, cell adhesion and exo-/ endocytosis) (figure 13). Over the years several membrane models have been proposed, the most prominent models of the last 20 years being the lipid raft [89] and the membrane skeleton fence model [90]. The first postulates dynamic nano-domains, which are formed by favorable interactions of sphingolipids, cholesterol and specific glycoproteins, while the latter proposes actin enclosed membrane compartments. Both are suggested to be important for localizing and stabilizing signaling complexes. However, due to technical challenges, experimental underpinning of either model is insufficient.

Figure 13. Model of plasma membrane organization. (1) Cholesterol and glyco-lipid enriched rafts. (2) Cell cortex membrane interactions (picket fence). (3) Clusters of poly-anionic lipids on the inner PM-leaflet. (4) Palmitoylated proteins on the inner PM-leaflet. (5) Site of endocytosis. (6) Cell adhesion receptors. (7) GPI-anchored proteins on the outer PM-leaflet. (8) Membrane turn-over by vesicular transport. (9) Extracellular matrix.
Download figure:
Standard image High-resolution imageWith the ability to detect and follow the dynamics of fluorescently labelled lipids and proteins (single molecule tracking and fluorescence correlation spectroscopy) in the PM both models have been revisited. Most membrane proteins were found to be indeed confined to clusters or compartments. Unfortunately, direct experiments on lipid dynamics or lipid-protein interactions are still the exception and the reported results so far provide no clear picture [91]. The technical challenge is to resolve very transient structures on the macro-molecular scale, which requires live cell compatible imaging with high temporal as well as spatial resolution. Super-resolution microscopy methods in general address this problem. However, these techniques require labelling of proteins and lipids with bright and photo stable fluorescent dyes. Since already minor modifications of lipids have a large impact on interactions in the membrane, fluorescent labelling of lipids is delicate and often results in biased membrane interactions. Thus, a key challenge will be developing labelling strategies, which make functional lipid probes accessible to super-resolution microscopy methods.
Advances in science and technology to meet challenges.
Up to now super-resolution imaging of protein and in rare cases also lipid distribution in the plasma membrane by STED and PALM/STORM has revealed that in many cases proteins are arranged in nanoscale clusters [92–94]. Unfortunately, most super-resolution imaging is done on chemically fixed cells using antibody staining, which likely perturbs membrane organization. However, new labeling approaches have been developed such as Snap-, Clip- or Halo-tags or site specific click chemistry that in combination with cell permeable organic dyes allow studying protein distribution and dynamics in live cell membranes with high resolution. Unfortunately, fluorescence labeling of lipids for super-resolution microscopy is still problematic and requires distinct strategies depending on the type of interaction that wants to be investigated [95]. Another challenge is that analyzing the distribution of proteins and lipids from 2D super-resolution images can be significantly biased by membrane ruffling, sites of endocytosis or any other 3D structure which is projected onto a 2D image. Therefore without 3D information the ability to dissect the organization of the PM is very limited.
Studying membrane organization with super-resolution microscopy in living cells requires high temporal resolution in addition to high spatial resolution (sub-millisecond for lipid dynamics). Since STED imaging is a raster-scanning method it can be sped up simply by decreasing the field of view or by parallelization, which is technically more challenging. In PALM/STORM the single molecule photon yield and the switching kinetics in combination with the frame rate of the camera are limiting the acquisition speed. Recent improvements have pushed the time resolution from minutes to video rate (30 Hz), which allowed to follow slowly moving membrane structures such as caveolae in living cells [30]. For the characterization of fast lipid dynamics at the nanoscale the combination of STED with fluorescence correlation spectroscopy (FCS) has proven to be advantageous. STED-FCS combines the high spatial resolution of STED with the statistical analysis of photon fluctuations caused by the dynamics of molecules in the detection spot. This combination enables characterizing anomalous lipid diffusion heterogeneities with very high spatio-temporal resolution. Advancements of this technique allow mapping diffusion dynamics along a micrometer long line (scanning STED-FCS) in contrast to the previous single point measurements [95, 96]. So far, STED-FCS measurements of live cell membranes did not indicate the presence of traditional lipid phase separation driven domains (i.e. lipid rafts) in the outer leaflet of the PM in undifferentiated cultured cells, even though a cytoskeleton and cholesterol dependence of diffusion dynamics had been shown for some lipid analogues before. In any case, it remains to be shown to what extent the local probe mobility extracted by STED-FCS or single-molecule tracking actually reflects weak and transient interactions of lipids and proteins. Clearly, additional strategies are required to reveal these interactions.
Technical improvements like parallelization (multiple foci and detectors) with fast electro-optical beam scanning or advanced light-sheet based methods [55] may allow acquiring full field of view image stacks with a temporal resolution suitable to capture diffusion dynamics and lipid protein interactions with an illumination dose that minimizes phototoxic effects. For direct observations of interactions between lipids as well as lipids and proteins multicolor acquisition schemes are required, which impose higher instrumental complexity and raise the bar for quantitative (≈100% efficiency) labeling strategies.
Certainly, a comprehensive understanding of membranes on the cellular level has to include membrane organization of internal organelles and the metabolic lipid pathway. Yet again, to make this step new strategies for labeling such as organelle specific lipid delivery are required and the dense packing of internal membranes calls for isotropic sub 20 nm spatial resolution. A first step towards characterization of internal membranes could be the development of new lipid order probes (to replace laurdan) that are suitable for super-resolution imaging.
Concluding remarks.
Applying super-resolution microscopy to reveal the fundamental principles of plasma membrane organization is a promising direction, with the potential to map lipid protein interactions in live cell membranes with unprecedented detail. However, technical improvements like increasing acquisition speed and sensitivity are required, and more importantly new fluorescent probes have to be developed that are more photostable, less phototoxic and compatible with live cell protein and lipid labelling.
Photoswitchable fluorescent proteins for nanoscopy
Stefan Jakobs1,2
1 Max Planck Institute for Biophysical Chemistry, 37070 Göttingen, Germany
2 University Medical Center of Göttingen, Department of Neurology, Göttingen, Germany
Status.
Diffraction-unlimited far-field super-resolution fluorescence (nanoscopy) methods typically rely on transiently transferring fluorophores between two states [97]. The insight, that it is no longer the quality of the objective lens but the properties of the fluorophore that ultimately determine the attainable resolution in super-resolution microscopy, spurred an amazing quest to generate fluorophores tailored for super-resolution microscopy.
For live cell imaging, the most widely used labels are fluorescent proteins (FPs) that are structurally similar to the green fluorescent protein (GFP). Indeed, the discovery of GFP from the jellyfish Aequorea victoria has revolutionized cell biology [98]. FPs are soluble small compact proteins of ~27 kDa. Fused to a host protein, they may be used as molecular light bulbs to highlight the localization and movement of the host protein. All common FPs exhibit a GFP-like fold, namely a beta-barrel enclosing an alpha-helix containing the autocatalytically formed chromophore (figure 14). Except for one oxygen molecule, maturation does not require any co-factors. The enclosure of the chromophore in the beta-barrel allows for a very rich (protein-)chemistry that determines the photophysical properties of the FP.

Figure 14. The GFP. (A) Structure of GFP shown in a top and a side view (PDB 2Y0G). (B) Comparison of the size of GFP (green) to a microtubule (red). Depicted is a cross-section through a single microtubule (PDB 3J2U).
Download figure:
Standard image High-resolution imageDedicated protein engineering by several research groups has led to a large collection of FPs exhibiting various colors and other properties. Notably, FPs with different switching properties were created (figure 15). Photoswitchable FPs (also named phototransformable fluorescent proteins; PTFPs) may be classified into different categories. (1) Photoactivatable fluorescent proteins (PAFPs) are irreversibly photoconverted from a weakly or non-fluorescent state to a fluorescent state; (2) photoconvertible fluorescent proteins (PCFPs) undergo a photoinduced change of their color; and (3) reversibly switchable fluorescent proteins (RSFPs) may be repeatedly photoswitched between a fluorescent on- and a non-fluorescent off-state. This is not a strict categorization, since also photoswitchable FPs have been engineered that combine properties of the different categories. In addition to irreversible phototransformations or reversible switching between long-lived metastable states, presumably all FPs may be transferred into (4) short-lived dark-states, a phenomenon known as 'blinking'. From these short-lived dark-states, which often are still ill-defined, they relax thermally back in the fluorescent state, a process that may be accelerated by light irradiation, depending on the dark-state. All of the various switching mechanisms have been utilized for super-resolution microscopy by some means or other.

Figure 15. Various photoswitching modes.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
Switchable FPs that are utilized for live-cell super-resolution microscopy have to fulfill a number of requirements, some of which may even be mutually contradictory. Obviously, switchable FPs must match the best non-switchable FPs (i.e. they must exhibit a high quantum yield, a high extinction coefficient, low photobleaching, a fast maturation at 37 °C, must be monomeric, etc). As detailed elsewhere [3], the current super-resolution approaches may be divided into two broad categories. The first class comprises scanning approaches that rely on a patterned illumination to spatially control the fluorescence behavior of the molecules, such that molecules that are closer than the diffraction-limit are not emitting simultaneously, thereby achieving sub-diffraction limit resolution. Prominent examples are STED and RESOLFT microscopy. The second class uses various (photoinduced) mechanisms to stochastically activate individual molecules within a diffraction-limited region at different times. Images with sub-diffraction resolution are then reconstructed from the determined positions of individual fluorophores. Prominent approaches of this class are PALM, STORM, GSDIM, and dSTORM.
As a scanning method, RESOLFT nanoscopy (as the closely related non-linear structured illumination microscopy) requires RSFPs, and the resolution attainable is largely determined by the number of switching cycles, the switching contrast and the signal-to-noise ratio. Hence for this method, RSFPs exhibiting low bleaching, high resistance against switching fatigue and a high contrast between the on- and the off-state are needed. Compared to the first RSFPs available, tremendous progress has been made by protein engineering. For example, the GFP-derived RSFP rsEGFP2 can undergo several thousand switching cycles at high speed before photodestruction [99]. The archetypical RSFP Dronpa [100] would undergo less than 10 cycles at the same light intensities. Still, several ten thousand or even several hundred thousand switching cycles would be desirable to enable massively prolonged live cell RESOLFT super-resolution microscopy. Stochastic imaging approaches typically rely on PAFPs or PCFPs. Here, the localization accuracy of a single emitter depends primarily on the number of photons detected from the FP while it is in the on-state. Hence in the case of the stochastic super-resolution methods, high photon yields in combination with short on-times are desired. Whereas RSFPs outperform current organic dyes with respect to the number of possible switching cycles, current PAFPs or PCFPs generally emit less photons in one burst than dyes.
A major and largely unmet challenge is the generation of (photoswitchable) FPs with an excitation maximum above 600 nm [101]. Such FPs are highly desirable, because red light is scattered less and many tissues are relatively transparent to light between 600 nm and 1000 nm wavelength. Hence photoswitchable FPs that can be excited by far-red light would be highly useful as genetically encoded markers for non-invasive deep-tissue super-resolution imaging.
Advances in science and technology to meet challenges.
Most current approaches to further optimize fluorescent proteins rely on random mutagenesis and screening rather than targeted approaches. Indeed, screening approaches mimicking evolutionary concepts proved to be tremendously powerful in the quest to identify better fluorescent proteins. Even photoswitchable proteins with entirely novel properties emerged from these approaches [102]. However, screening approaches may miss new properties that potentially could be introduced into FPs if the implementation of the new property would be very improbable requiring the exchange of many amino acids.
The lack of targeted approaches to generate entirely new or improved photoswitchable FPs is partly rooted in insufficient predictive power of current molecular dynamics simulations and on lacking detailed insights into the actual switching mechanisms. Most crystallographic studies on photoswitchable FPs concentrated on the relaxed on- and off-states, but provided only limited insight into the dynamical changes occurring during switching [102, 103]. Likewise, we are only beginning to understand the bleaching pathways in fluorescent proteins [104]. Advances in ultra-fast spectroscopy and structure determination, notably in time-resolved serial crystallography using x-ray free electron laser (XFELs) [105] are expected to help in closing this gap, which in turn, combined with improved protein modeling, may facilitate more targeted approaches in protein design.
Concluding remarks.
Currently, only GFP-like FPs are known to autocatalytically form a chromophore. However, several naturally occurring and artificially designed fluorescent proteins have been described that rely on exogenously supplied chromophores [106]. Such proteins might be developed into attractive alternatives to FPs. Nonetheless, the GFP-like scaffold is presumably far from being fully exploited, yet. Ever more advanced mutagenesis and screening strategies, especially when combined with detailed spectroscopic and structural data dynamically describing the switching mechanism and the bleaching behavior, are likely to provide further improved or even entirely novel fluorescent proteins for years to come. The development of new labels will also spur new developments in the technical implementations of super-resolution microscopy, so that an end to the exciting developments in this field is not in sight.
Drop down the light: RESOLFT microscopy in living cells and tissues
Ilaria Testa
Royal Institute of Technology, SciLifeLab, Stockholm, Sweden
Status.
The ability to discern individual organelles, molecular complexes and structures in the interior of a living biological system and to follow their fate in real time is an open and exciting challenge. A suitable method to meet this challenge needs to satisfy multiple requisites: (1) to be minimally invasive (2) to distinguish molecules of interest in the context of the biological system (3) to observe molecules at a relevant length scale.
RESOLFT [3, 107–109] super-resolution microscopy represents a powerful tool to decipher spatio-temporal information coded in the life sciences by (1) minimizing the illumination light intensities (2) employing genetically encoded markers (3) improving the spatial resolution of conventional fluorescence microscopy down to the nanoscale. In RESOLFT nanoscopy the spatial resolution is improved by taking advantage of 'long-lived' (μs ms) molecular states, which are populated/depopulated with light intensities in the range of W cm−2 to kW cm−2. Such precious states are found in reversible switchable fluorescent proteins (rsFPs) so far. The available rsFPs for RESOLFT were generated by introducing a few mutations in the sequence of well-known fluorescent proteins such as Dronpa, EGFP, YFP or Cherry. rsFPs can be light driven in different molecular states which inhibit or permit their ability to fluorescence if exposed to visible light. These states are associated with cis-trans [109] or hydrated-dehydrated [105] molecular configurations. For RESOLFT to work these states need to be reversibly populated or depopulated multiple times. The improvements in spatial resolution is in fact achieved by saturating the OFF switching transition using a light pattern featuring one or more intensity minima or 'zeros' (ring or line shaped, figures 16(b)–(d). The role of this light beam is to transiently silence the emission of rsFPs by switching them to the long lived OFF state. Once switched off, the rsFPs cannot be excited anymore and remain dark. Only fluorophores residing in the direct vicinity of the zero-intensity minimum of the RESOLFT light pattern focus can effectively remain into the fluorescent ON state and hence contribute to the fluorescence signal.
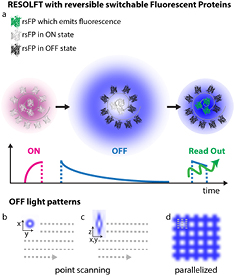
Figure 16. RESOLFT pulse scheme. The typical pulse scheme applied for RESOLFT imaging of the Dronpa and rsEGFP family is depicted in (a). Each step is performed with different colours and duration depending on the absorption spectra of both states of a chosen rsFP. In (b)–(d) are illustrated the light patterns currently implemented in point scanning and parallelized RESOLFT microscopy to improve the spatial resolution along the focal plane (b) or in 3D (c) or in a parallelized fashion (d).
Download figure:
Standard image High-resolution imageImportantly, the kinetics of the switching to the OFF state determines the final resolution. Current point-scanning RESOLFT microscopy equipped with a ring shaped light pattern (figures 16(a) and (b)) reach a lateral resolution down to 30–40 nm [102, 110].
The axial resolution can be improved as well down to 70–90 nm by adding an additional light pattern (figure 16(c)) [110].
This system is comparably cheap and easy to use since it requires only a few microwatts of continuous wave lasers for sample illumination (switching and excitation). Multi-channel variants employing spectral and lifetime information were also successfully realized enabling colocalization studies at the nanoscale [111, 112]. This microscope was also upgraded for brain tissue imaging using a glycerin objective lens to match the refractive index of the tissue and additional lenses to correct for spherical aberrations. Neuronal proteins were resolved up to 50 μm deep in the brain slice unraveling different dendritic spine morphologies (figure 17, panel (a)) [110]. Their spontaneous activities were recorded in different time windows with a record of more than an hour observation time (>140 frames acquired from the exact same region in the sample), which is a promising result to meet the challenge of long-term imaging.

Figure 17. (a) RESOLFT nanoscopy in brain tissues unravels actin fine structures in dendrites and dendritic spines previously hidden, as highlighted by the comparison with the confocal image. (b) Parallelized RESOLFT imaging of a subresolved keratin network of a whole cell (120 μm × 100 μm) recorded in 3 s. Scale bar 1 μm in panel (a) and 10 μm in panel (b). Reproduced with permission from [47], Copyright 2013 Nature Publishing Group.
Download figure:
Standard image High-resolution imageCurrent and future challenges.
A point-scanning implementation of the RESOLFT concept intrinsically compromises the temporal resolution of the technique, especially in large fields of view where many pixels are recorded. This caveat can be overcome by implementing parallelized illumination modes to switch and excite the rsFPs. Periodic line patterns rotated at different angles [113], as routinely done in structured illumination microscopy, or superimposed at 90 degrees [113] (light pattern illustrated in figure 16(d)) can be successfully implemented to speed up the acquisition of large fields of view in RESOLFT microscopy. In fact, pioneering studies demonstrated super resolved recordings of a living cell in a field of view of about 100 μm × 100 μm within a few seconds (figure 17, panel (b)).
A second factor that influences the temporal resolution is the time that a certain rsFP requires to undergo a complete switching cycle. At the moment the fastest switching kinetics in rsFPs are found in the so-called 'negative switchers', i.e. proteins that, after a certain number of excitation cycles followed by fluorescence emission, switch to the dark state. So the 'OFF switching' needed to improve the spatial resolution and the 'fluorescent excitation' needed to generate an image are closely coupled. Consequently, faster switching kinetics will lead to the detection of only fewer photons, thus dictating a trade off between contrast and time resolution. Fortunately novel switching mechanisms are found in protein types such as Dreiklang where the switching to the dark state and the fluorescence emission are initiated at different wavelengths and are therefore 'uncoupled'. However, the variants with uncoupled switching available nowadays are still relatively slow (>10 ms) and not efficient (few 100 cycles) compared to the negative switchers (us, >1000 cycles) impeding the full potential in fast recording and spatial resolution.
Advances in science and technology to meet challenges.
Compared to other super-resolution approaches, RESOLFT nanoscopy is quite a young technique. In fact, even if the principle has been around for quite some years the winning combination of optics and switching ability to resolve living cells was reached only a few years ago. Since then the technique rapidly improved showing its potential for tissue imaging, time lapse recording in living cells also with multicolor detection and faster recording modes. Also novel probes [114] with optimized switching kinetics and expression in living cells become available.
The future will see a more extensive application of the technique in various biological systems from different types of cells to embryos and more complex organisms such as mice and rats. To do so, development of new optical schemes as well as novel bright reversible switchable probes and labelling strategies will be necessary.
Point scanning RESOLFT microscopy has potential for imaging in higher penetration depth for example by taking advantage of adaptive optics for wave front correction. Also, multiphoton RESOLFT variants will improve the penetration ability and will further minimize photo toxicity by shifting the illumination to the less energetic part of the spectrum. On the other hand, parallelized illumination schemes need to improve the axial spatial resolution as well as the imaging ability in thick samples. Illumination modalities based on two or more objectives as in light sheet or 4pi microscopy will be a future step in this development.
Either with point scanning or parallelized recording, RESOLFT nanoscopy will benefit from bright reversible switchable probes which can switch quickly multiple times and still be bright and photostable when left in the ON state. Molecular mechanisms, which allow optical decoupling of switching and fluorescent excitation, are the most promising ones [105]. Such features will enable faster recording at higher image contrast and spatial resolution compared to previous results. Also, the application of RESOLFT nanoscopy to biologically relevant questions will benefit from advanced labeling strategies to substitute protein overexpression. So far, live cell super-resolution microscopy studies relied on the overexpression of a host protein fused to a fluorescent protein. CRISPR-mediated endogenous protein tagging in human cells preserves native protein expression levels [115]. Such genome-edited 'knockin' cells will facilitate quantitative and dynamic studies based on RESOLFT live cell imaging.
Concluding remarks.
RESOLFT nanoscopy is a super-resolution technique advisable for live cell imaging due to the minimal light doses required. This feature is also a technical advantage for its simple implementation as less powerful, routinely used visible lasers are required.
The gentle illumination allowed long-term recording of neuronal proteins and their dynamics in cells and tissues at the nanoscale. Cellular processes such as cytoskeleton dynamics and ER rearrangements could be detected with sub-second time resolution.
Furthermore, the implementation of parallelized illumination schemes based on the RESOLFT concept or nonlinear structured illumination enabled fast recording of a whole cell surface at high spatial resolution (<100 nm).
Parallelized illumination schemes combined with light sheet microscopy and applied to switch and read-out new brighter photostable reversible switchers will push the spatio-temporal resolution of RESOLFT nanoscopy to a new level opening up its application to biologically relevant questions.
Super-resolution microscopy in neurosciences: zoom in on synapses
Laurent Cognet and Brahim Lounis
University of Bordeaux, Institut d'Optique & CNRS, Talence, France
Status.
The remarkably efficient functioning of the brain largely mirrors its multiscale complexity. In particular, it became clear since the emergence of modern neuroscience that basic investigations of brain organization at the subcellular scales are key for the understanding of brain processes. Since then, our knowledge of the cellular mechanisms involved in neuronal communication and their evolution during life has literally exploded. Most of our comprehension comes from the discovery of molecules, genes and signaling cascades that play central roles in the maintenance and plasticity of the neuronal communication. Notably, the identification of the synapse has attracted much attention. It contains key molecules required to induce plastic changes and mediates a large fraction of fast neuronal communication and long-term adaptations. The dimensions of brain synapses are, however, small at the submicron scale and therefore they cannot be resolved with conventional optical microscopes. Electron microscopy has been providing ultra-structural information of synapse architectures and to some extent, knowledge about the content and local molecular densities in these structures. However, this knowledge is out-of-reach in living cells and in intact tissues with this imaging modality. In this context, light microscopy constitutes the imaging modality of choice to study synapses in live cells. Continuous developments and refinements of optical imaging techniques delivered an impressive amount of information about synaptic molecule organizations at the nanoscale. A first decisive achievement came from the possibility to detect single molecules in living cells. It not only allows localizing molecules with nanometer accuracies, far below the optical resolution, but also provides the ability to probe the intimate variety of molecule dynamic environments from nano- to micro-scale levels. In the early 2000s the presence of mobile receptors for neurotransmitters was revealed for the first time [117, 118] in the postsynaptic membrane of dissociated neuronal cultures and the role of neurotransmitter receptor diffusion in fast synaptic transmission was demonstrated [119]. The advent of several super-resolution methods that followed these early achievements raised great expectations in neurosciences by providing optical images of neuronal structures with unprecedented resolutions.
Current and future challenges.
Super-resolution microscopy methods mostly rely on the control of the number of emitting molecules in specific imaging volumes. Two major types of techniques found major applications in neurosciences: those based on STED where highly localized fluorescence emission volumes are optically shaped and those based on single-molecule localizations (PALM, (d)STORM or (u)PAINT) [120]. Single-molecule localization based methods have proven to be powerful to study nanoscale molecular organizations such as that of postsynaptic receptors (figure 18) and scaffold proteins (e.g. [32, 116, 121, 122]) but also that of actin and actin binding proteins in axons [31]. On the other hand, STED microscopy allows visualizing dendritic spine shapes in living neurons with remarkable resolution and revealed that spine neck plasticity regulates compartmentalization of synapses [123] (figure 19) and further deciphered the dynamic organization of actin.
Figure 18. High-density single-molecule trajectories (top) and super-resolution imaging (bottom) of endogenous glutamate receptors measured on a live neuron with the uPAINT method. The circle highlights the presence of a synapse. Reproduced with permission from [116], copyright 2010 Elsevier. Scale bar = 500 nm.
Download figure:
Standard image High-resolution image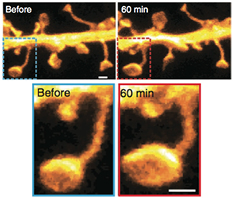
Figure 19. Effect of long term potentiation on the morphology of synaptic spines and heads revealed by STED microscopy (bottom panels are zoomed in versions of the top ones). Reproduced with permission from [123], copyright 2014 Nature Publishing Group. Scale bar = 500 nm.
Download figure:
Standard image High-resolution imageAs exemplified in the aforementioned achievements, imaging synaptic structures and their molecular contents with nanometer resolutions already constitute a breakthrough for the understanding of synaptic functioning. Because these demonstrations have predominantly been performed on dissociated neuronal cultures or thin fixed brain slices, one of the main challenges will be to transfer such cutting edge technologies to more integrated and ultimately intact living samples. This will allow major fields of neurosciences, such as development, aging and neurodegenerative disorders, to benefit from the unprecedented degree of detail provided by these microscopies.
Advances in science and technology to meet challenges
The possibility to perform super-resolution imaging in intact living brain tissues to address major neurosciences questions is conditioned to several requirements. Because both light scattering and absorption limit the penetration depth in thick biological samples in the visible domain, super-resolution imaging in tissues is only just emerging. Scattering is particularly detrimental for STED microscopy, since the realisation of a zero intensity region for depletion is mandatory. Two-photon microscopy, which uses near-infrared laser excitations, is a common strategy to excite molecules deep in brain tissues. However, due to the high intensities required, photobleaching rates are drastically enhanced and single molecule based super-resolution with two-photon excitation remains difficult. To fully exploit the biological transparency window, near-infrared probes with favourable photophysics still need to be developed, as current red-shifted dyes are not strong emitters.
Another key development will consist of achieving fast wide field super-resolved imaging at rates compatible with the inherent movements of living samples in 3D. Current super-resolution methods are indeed restricted to rather small imaging areas (sub-millimetric), which is constitutive to the novel opportunity to obtain images at superior resolutions. Indeed, by increasing the image resolution typically by a factor 10–50 per dimension, the size of a 3D image will be increased by a factor 103 up to ~105. This imposes constraints on the imaging speed as well as data handling. A promising strategy would consist of performing multi-scale imaging where only 'relevant' brain sub-areas are imaged at the nanometer scales while the other regions are imaged at lower resolutions. The definition of imaging 'relevance' within the brain might depend on time and/or the physio-pathological local state of the sample as well as the nature of the information provided by the low resolution imaging modality. In this context, correlative imaging with other modalities, which do not have to be limited to optical techniques, e.g. ultrasound imaging or magnetic resonance imaging, are interesting routes. Combining multiple super-resolution approaches can also provide brand novel opportunities. Finally, a full understanding of the brain function cannot be obtained solely by images. Manipulation of the brain physio-pathological state will be needed using methodologies that are compatible with super-resolution imaging. Optogenetic methods provide promising tools in this context.
Concluding remarks.
For widespread application of super-resolution imaging in neuroscience, the main challenge will be to link subcellular information gathered at the molecular scale (e.g. about synaptic processes) to the global organ function obtained at macroscopic scales, in a well-defined functional state. To this aim, a multidisciplinary effort will be needed where physicist, chemists, computer scientist, neurophysiologists and neuropathologists will have to work together. The task is vertiginous, but is at the level of the complexity of the brain.
Nanobodies for high-resolution imaging
Helge Ewers
Freie Universität Berlin, Berlin, Germany
Status.
In the early nineties, a new class of antibodies assembled merely from a homodimer of heavy chains was discovered in camelids [124]. Like in previously known heavy chains, camelid heavy chain hypervariable domains contain four framework regions (FRs) and three highly variable complimentary determining regions (CDRs). In camelid heavy chains, the CDRs are located to loops emanating from the beta fold formed by the FRs and all contribute to antigen binding. Since these camelid IgG epitope-binding domains are formed from a single domain within a polypeptide chain, rather than from the combined hypervariable domains of a heavy and a light chain, the potential for the engineering and production of recombinant binders from these antibodies was immediately apparent. The epitope binding VHH domains are of ~15 kDa molecular weight and about 2 by 3 nm in size compared to the 150 kDa and 10 nm of regular IgG antibodies and are thus called nanobodies.
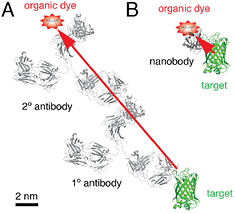
Figure 20. Nanobodies as efficient probes for the accurate delivery of fluorescent dyes. (A) In traditional immunostaining, a specific primary antibody and a dye-coupled secondary antibody are combined for the detection of molecules in cells. The relatively large size of the antibodies and the frequent use of polyclonal antibodies that may recognize a number of different epitopes, result in significant error in the localization of the target molecule, when the dye is imaged. (B) When an organic dye is directly coupled to a nanobody, it is delivered into closest proximity of the target structure. Structures are to scale and taken from www.pdb.org.
Download figure:
Standard image High-resolution imageNanobodies profit from better solubility, higher stability in solution and far more efficient expression than conventional antibodies and engineered epitope binding domains of IgG antibodies, so called scFv fragments. The generation of antigen-specific nanobodies is well-established for immune, non-immune or semisynthetic libraries from camelids such as llamas using phage, yeast or ribosome display. The small size of nanobodies leads to significantly better access to antigens in cells and to higher tissue permeability, which renders nanobodies highly investigated as therapeutic agents against cancer and as probes for medical imaging. In this context, nanobodies also profit from a lower plasma half-life since they are excreted via the kidneys. Taken together, these advantages have been recognized as very valuable by the pharmaceutical industry and several nanobodies are being developed as drugs or imaging reagents.
In basic biological research nanobodies against the green fluorescent protein have been employed successfully as binders for immunopurification, but especially as probes for fluorescence microscopy. Anti-GFP nanobodies coupled to green fluorophores allow for imaging modes that require very photostable labeling such as structured illumination microscopy [125] by enhancing fluorescence in the GFP channel. On the other hand, anti-GFP nanobodies coupled to very bright dyes suitable for single molecule localization microscopy have provided a method for very accurate imaging of target structures in cells and in consequence to unparalleled resolution of cellular structures [36]. In this way, the technique has for example allowed for resolution of the spatial organization of an important multiprotein complex within the nuclear pore [126].
Current and future challenges.
While the use of nanobodies against GFP is a useful solution for a large number of proteins and yields high resolution compared to antibody-mediated labeling, it always precludes the successful endogenous or exogenous expression of functional GFP-tagged molecules in cells. As cell biologists know, not all proteins can be readily tagged with GFP while retaining their function and in some systems, transient overexpression is not a straight-forward approach. Furthermore, overexpression is naturally non-quantitative as well. Thus more nanobodies directed against native proteins rather than tags need to be developed. Unfortunately at this time nanobodies are available only against a small amount of target proteins commercially, even less nanobodies are thoroughly tested and freely available in the community.
The final resolution in single molecule localization microscopy depends on the labeling density [14]. However, for very dense structures like a mitotic spindle or the postsynaptic density of synapses, where tens of thousands of molecules are distributed within few diffraction-limited voxels, the duty cycle of the best dyes [127] or the contrast of photoactivatable fluorescent proteins at present pose a limit to the number of high precision localizations that can be yielded regularly from an experiment. This is an important problem to be solved by novel labeling technology.
Advances in science and technology to meet challenges.
A significant neglected problem in current experimental research is the use of thousands of different, often polyclonal antibodies. For many targets, tens of commercial antibodies are available, often several polyclonal antibodies, i.e. complex mixtures of varied affinity and specificity. Such polyclonal antibodies are produced in different species, adding to the uncertainty of antibody properties and specificity and making reproducibility of results between antibodies used and thus between laboratories and publications all but impossible. Furthermore, hundreds of millions are wasted each year on 'bad' antibodies [128]. A highly desirable aim is thus the generation of clearly characterized standard binders for each protein, ideally with a solved structure of the binding interface and clearly defined kinetics and specificity. In addition they should be cheap to manufacture, stable and contain the capability to be developed into a humanized therapeutic reagent. Nanobodies bring many of these properties to the table and it is highly desirable that more nanobodies will be developed, characterized and made available to the community. Such an endeavor would be supported by the fact that recombinant libraries containing the entire repertoire of amino-acid combinations possible in the CDRs of camelid heavy chains are now available and new strategies for the highly productive generation and characterization of nanobody binders have been developed [129].
The challenge posed by the limited contrast of photoswitchable probes or the limited duty-cycle of organic dyes, which limits the final resolution of very dense 3D structures, may be overcome by approaches based on the transient binding of bright organic dyes to labels with high specificity. Such methods like points accumulation for imaging in nanoscale topography PAINT [16] combined with DNA-mediated specific binding and clearly defined and multitarget binding [130] should be developed further based on the combination of DNA-oligomers with nanobody binders for high density, high resolution imaging. As an alternative, new approach to the generation of small, chemically defined binders may come from the development of so-called bicyclic peptides [131]. Aptamers have been proposed as specific imaging reagents, but their use is still not widespread.
Concluding remarks.
In conclusion, nanobodies are very useful reagents even beyond the traditional use as probes. What is needed now is the development and thorough characterization of nanobodies against more cellular targets, ideally against all expressed proteins and other cellular molecules. The result will be a portfolio of accessible, well-defined and efficient labeling reagents for high-resolution imaging.
Super-resolution imaging of early signaling events at the leukocyte cell-surface
Simon J Davis1, Christian Eggeling1 and David Klenerman2
1 MRC Human Immunology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, Headley Way, Oxford, OX3 9DS, UK
2 Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK
Status.
The immune system is particularly dependent on the ensemble behaviour of proteins at cell–cell interfaces and one of the best places to study cellular signaling processes. The emerging view of leukocyte signaling is that it is controlled by the local redistribution and interactions of a relatively large set of cell surface proteins that starts (within seconds) at a scale likely to be below the resolution of confocal or deconvolution fluorescence microscopy [132, 133], is followed within minutes by the reorganization of receptors into meso-scale (sub μm-scale) protein assemblies called microclusters (figure 21(A)), and after minutes to hours culminates in the formation of μm-scale structures called 'immunological synapses' (figure 21(B)) [133–135]. Given the scale of these organizational changes, it seems clear that super-resolution imaging will have a very large impact on our understanding of immune signaling. The major immediate areas of enquiry concern the resting organization or 'ground-state' of the leukocyte cell surface, and the early changes linked to incipient receptor signaling that precede the meso- and larger-scale changes. Super-resolution imaging and/or spectroscopy are particularly well suited to following the molecular events leading to lymphocyte receptor triggering and signaling, and will inevitably provide new biophysical insights into the selectivity and sensitivity of these processes.
Figure 21. Structures formed by leukocytes. (A) TIRF image of T-cell receptor (TCR, green) and ZAP70 kinase (red) microclusters formed by T-cells interacting (1 min) with a supported lipid bilayer containing antigen. Scale bar 5 μm (from [135]). (B) Several μm-large confocal images of TCR (pink) and adhesion protein LFA-1 (blue) in T cells interacting (15 min, upper; 30 min, lower) with 'antigen-presenting cells', depicting the immunological synapse >1 μm in diameter (lower). Reproduced with permission from [142], Copyright 2002 American Association for the Advancement of Science.
Download figure:
Standard image High-resolution imageThis does not mean that the analyses will be straightforward. There are, for example, already conflicting observations about the resting state of the leukocyte cell surface that need to be resolved. Whereas PALM/STORM experiments have suggested that antigen receptors, the proteins that initiate adaptive immune responses, are preclustered into 'protein islands' on resting (i.e. non-activated) cells [136, 137], other single-molecule approaches suggest that these types of receptors are apparently freely diffusing and functionally monovalent [138–140]. Significant challenges exist in designing and performing imaging experiments on leukocytes, which extend from how we handle the cells in the course of experiments and control, e.g. the initiation of signaling processes, through to how we label the proteins that we want to follow. These issues and possible strategies to address them are discussed in this article.
Current and future challenges.
There are four main classes of problems that can be identified in performing super-resolution imaging of the signaling responses of leukocytes. These are associated with labelling the proteins of interest, controlling the triggering of the immune response so that imaging can be performed at the correct time, imaging cell–cell contacts and with the necessary time resolution and, lastly, getting good statistics.
Advances in science and technology to meet challenges.
There are generic problems with labelling the proteins of interest and ensuring and confirming that any labelling strategy maintains biological function. The fraction of labelled proteins is one issue, particularly when the goal is to determine protein oligomerisation states. Recently developed strategies for tagging proteins, e.g. with Halo- or Snap-tags are good at preserving protein function, but the levels of labelling of cytoplasmically-expressed tags in our experience can be as low as 10–30%. It is also often important that the total number of proteins on the cell surface should not be significantly altered, since this may lead to artificial association. In general this means that it is preferable to label endogenous proteins rather than transiently transfect cells and overexpress the protein of interest. The constraint here is that intracellular proteins cannot be easily accessed for labelling in intact cells, restricting labelling to the extracellular regions of cell-surface proteins. On the other hand reducing the expression level of labelled endogenous proteins may be useful to both slow down signaling and reduce the resolution required for imaging. To study the interaction between proteins it is necessary to label more than one protein and this requires two well-separated fluorophores for STED, PALM/STORM or SIM. Lastly, since formation of a signaling complex will result in downstream signaling i.e. calcium responses, actin rearrangements and NFκB production, it is often important to be able to simultaneously detect signaling in order to relate the structures observed on the cell surface directly to the resulting cellular response. This may require the use of a non-fluorescence-based method to measure signaling such as electrophysiology or the capability to switch between or add imaging modes (such as observation of fluorescent calcium indicators or labelled actin).
Sampling is another related problem for higher resolution STORM/PALM imaging since a gain in precision is unlikely to be useful if there is not also an increase in the density of the labels in experiments. Sampling can be improved by higher labelling efficiency, using smaller labels (because of steric hindrance) and via the better expression, folding and activation efficiency of the fluorescent proteins. Still, overexpression of proteins should be avoided due to potential biasing effects on their function, e.g. artificial clustering.
Since the triggering of the immune response can be fast, e.g. within 6 s [141], it is important to firstly ensure that the cell is not triggered before the imaging starts. Ideally, signaling needs to be initiated at a defined time in order to capture the earliest events and to reduce issues with cellular photobleaching and photodamage resulting from prolonged exposure to laser radiation. Because lymphocytes have to respond to surfaces, e.g. other cells or microbes 'presenting' stimulating ligands (i.e. antigens), it is very likely that their physiology will be sensitive to interactions also with artificial surfaces such as the microscope cover glass. Current TIRF imaging has the potential problem of surface-induced protein re-organization, as well as being capable of only studying parts of cell (e.g. the basal membrane). It would be better to study also the apical surface or the entire cell, and also to develop general new methods to avoid surface-induced triggering. One approach is to build artificial cell surfaces on, e.g. lipid bilayers. Historically, out of necessity, these have been relatively simple systems comprising 2–3 proteins that can be produced in large amounts inserted into a one-component lipid bilayer, but many more or fundamentally different proteins and lipids may have to be incorporated into the bilayers in order to reproduce appropriate physiological responses. Ideally it is best to be able to control the initiation of signaling by the fast addition of soluble ligands in the case of the innate response or bringing one cell in a controlled way to an activating surface or to another cell to study early signaling. One attractive approach for controlling the timing of signaling initiation is to use photo-activatable ligands of the receptors of interest [141].
For studying the resting lymphocyte cell surface, it may be necessary to eschew TIRF imaging completely and suspend cells away from the coverslip if it turns out that contacts with, e.g. glass surfaces perturb the underlying biology. This may require careful control of the embedding media to slow down mobility of cells in suspension. Because so much of lymphocyte behaviour is dependent on specific cell–cell interactions, the goal in any case has to be to image cell–cell conjugates in 3D, which needs setups such as AFM tips, micropipettes or optical tweezers for positioning of the cells, bearing in mind that these manipulations might also induce cellular changes. On the other hand, imaging above the coverslip reduces the numerical aperture of the objective and thus the efficiency of collecting photons. Also, the fluorescence photons may have to pass through the cell leading to aberrations and deterioration of the signal, calling for correction using adaptive optics.
However, the greatest obstacle to obtaining a detailed understanding of signaling in lymphocytes is to obtain images of sufficient time and spatial resolution during the six seconds it takes for a response to start, particularly since it is still unclear what minimum spatial resolution will be required to understand the biology. In PALM/STORM, the imaging speed is limited by the frame rate of the camera (fortunately continuously improving) and ultimately by the photo-physics of the fluorophore. The total number of photons detected from a single fluorophore per frame and the rate with which they are emitted determine the spatial and temporal resolution, respectively. Unfortunately, these are restricted by the saturation of the fluorophore's electronic transitions as well as by the population of dark states, both presently intense topics of current research. The requirement for scanning the beam on the other hand slows down the image acquisition in STED and SIM, deteriorating with increasing spatial resolution. This means there is usually a compromise between image resolution and speed. A remedy is sacrificing large field of views, employing improved scanning routines by parallelization or use of faster scanners, and combination with techniques such as fluorescence-correlation-spectroscopy [96, 143]. Also, recent light-sheet based microscopy approaches [144, 145] have demonstrated considerable potential, especially for studying the entire cell in 3D, but the spatial resolution of these methods should ideally be improved, e.g. by combination with SIM and photoswitchable labels, and the interpretation of data requires extensive image analysis (in contrast to direct scanning approaches such as wide-field, confocal or STED microscopy). Phototoxic effects by the illumination light will probably not play a role when imaging for seconds only. However, long observation times of up to hours for investigating for example the formation of the immunological synapse require imaging with low illumination doses, such as light-sheet based approaches [144, 145].
It is further necessary to be able to image a large number of cells to get good statistics and remove any bias in the selection of the cells analysed. This is harder at high resolution, which is largely limited to imaging one or a few cells simultaneously and will require methods to both control triggering and automate image acquisition and analysis.
Concluding remarks.
The main challenges that currently exist are to firstly find the best method to solve each of the generic problems discussed above individually and then to integrate these methods together to allow a large amount of data to be taken and analysed. This is likely not only to produce significant advances in understanding the molecular basis of signaling during the adaptive immune responses of leukocytes but also be highly applicable to innate immune responses, particularly the triggering of Toll-like receptors and other pattern-recognition receptors. In the longer term, given the general nature of the problems, these methods could then be straightforwardly applied to a wide range of other cellular signaling processes triggered either by cell–cell contacts or by soluble ligands. This will allow the full potential of super-resolution microscopy to be exploited to study the molecular basis of signaling at the cell surface in both normal and diseased cells.
In vivo super-resolution microscopy
Katrin I Willig
Center for Nanoscale Microscopy and Molecular Physiology of the Brain Göttingen, Germany
Status.
Most of the super-resolution microscopy applications in biology study cultured cells grown outside of their natural environment, i.e. in vitro. However, synapses, for example, which are the most basic functional units for information processing and memory formation in the brain, are best studied in tissue of brain slices or, ideally, in the living animal. Moreover, in developmental biology it is indispensable to work with model organisms such as C. Elegans, zebrafish, or mice. These organisms are to some extent transparent for visible light and a wide range of genetically modified organisms with modifications of genes modelling diseases or expressing genes of interest fused to a fluorescent protein are available. Although the term in vivo is often used for living cells as well I will focus here on super-resolution of whole, living organisms. Imaging of living tissue is a challenge for all light microscopy techniques due to scattering of light within the tissue, aberrations caused by a mismatch in refractive index, and light absorption. From all super-resolution techniques, targeted switching techniques, such as STED or RESOLFT, are more suitable for tissue imaging than stochastic switching techniques (such as PALM or STORM) because of their sectioning capability and speed. Imaging of actin in dendritic spines in organotypic and acute brain slices was performed using STED and RESOLFT microscopy at a penetration depth of up to 120 μm [115, 146]. Of these two, only STED microscopy was utilized for in vivo imaging in living organisms; cytosolic expression of GFP in a neurosecretory-motor neuron of a living roundworm Caenorhabditis elegans [147] was imaged as well as cytosolic expression of YFP in neuronal structures in the somatosensory cortex of a transgenic mouse [11]. Moreover, in vivo STED microscopy was used to study actin, a part of the cytoskeleton required for the stability and mobility of cells, in the dendritic protrusions of a living, anaesthetized mouse (figure 22) [148] and the abnormal spine growth after increased expression of a membrane-bound isoform of neuregulin-1 [149]. Thus far, PALM microscopy has been applied in vivo to image bacteria and zebrafish embryos, namely FtsZ, a cell division protein which forms a 'z-ring' during cell division in living bacteria Caulobacter crescentus [150] and caveolin-1 membrane domains in living zebrafish embryos, 24 h of age [151]. For completeness, light sheet based microscopy has gained high impact as an in vivo technique especially in developmental biology because of its high speed. By combining the light sheet technique with PALM or structured illumination super-resolution images can also be acquired [152].
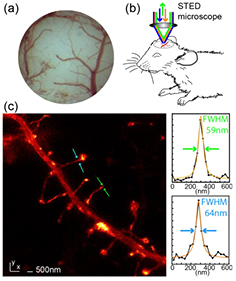
Figure 22. In vivo STED microscopy of neuronal structures. (a) Clear view of the visual cortex through an optical window. (b) Upright STED microscope for imaging of the cortex of the anaesthetized mouse. (c) Actin filaments in dendritic spines and dendritic arborization recorded with STED microscopy. Maximum intensity projection of raw data. The line profiles (right) of the marked areas show a full-width at half-maximum (FWHM) of ~60 nm indicating a resolution <60 nm. Adapted with permission from [148].
Download figure:
Standard image High-resolution imageThe need for in vivo measurements in neuroscience or developmental biology is undisputable and in vivo super-resolution could enable scientists to unravel fundamental working principles of the brain or development of the brain in context of the operating organism, although in vivo applications of super-resolution are still in its infancy.
Current and future challenges.
Like all applications of super-resolution microscopy to cultured cells, in vivo super-resolution microscopy depends critically on the method of tagging specific proteins and the photo-stability and brightness of the fluorophore. Overexpression of fusion proteins leads to artifacts and novel tags which optimally label the endogenous proteins without disturbing their function have yet to be developed. Challenges specific for in vivo super-resolution microscopy are discussed focusing on STED microscopy of the brain of mice, the only mammals imaged with in vivo super-resolution up to now:
Penetration depth: although the penetration depth in C. Elegans or zebrafish embryos is sufficient when using STED microscopy, it is still a challenge to penetrate deeper than the superficial layer 1 of about 50 μm thickness in the adult mouse cortex. Layer 1 is of interest when studying synaptic connections though cell bodies are at a depth still unattainable using STED microscopy. Deeper cortical layers have different synaptic input than layer 1 connections and are therefore of high scientific interest as well as even deeper brain regions such as the hippocampus.
Photo-toxicity: as for all fluorescence microscopy techniques photo-toxicity is an issue for in vivo STED microscopy. Non-linear photo-toxic effects are reduced by choosing long STED pulses of >200 ps and the photon dose is kept low by short pixel dwell-times of a few μs.
Chronic experiments: repeated imaging over days or months is of interest e.g. to study synaptogenesis, the process of synapse formation which contributes to learning and memory. The challenge is to obtain the clear optical view over this period which normally blurs due to healing processes.
Image 'artifacts' caused by vital functions: vital functions like breathing and pressure pulse in the blood vessels cause motion of the inspected tissue. This motion is of high relevance if microscopy is performed at the nanometer scale. To overcome these problems, details should be chosen as small as it is worthy for super-resolution to increase the framerate to image ideally within one heartbeat.
Awake animals: brain function studied in anaesthetized mice might be influenced by the anesthesia. To avoid this influence and to be able to study e.g. visual perception, experiments are often conducted in awake mice. This results in general in a degradation of imaging quality due to motion artifacts, either due to breathing or the movement of the entire, awake animal.
Advances in science and technology to meet challenges.
Labeling: in vivo STED microscopy would benefit greatly from improved red-emitting labels for two reasons: firstly, typically green or yellow emitting fluorescent proteins (GFP, YFP) are excited at 440–500 nm and depleted at 560–600 nm. In this range the absorption of proteins and hemoglobin is high, which might lead to phototoxic effects. Secondly, a higher wavelength would improve the penetration depth. Unfortunately, the quantum yield of red-emitting fluorescent proteins is still very low and needs to be improved.
Penetration depth: the penetration depth of all light microscopy techniques is limited by scattering of the light, aberrations of the penetrating light, and absorption [153]. While absorption is relatively low and can be neglected, scattering is the limiting factor at penetration depths >100 μm. Aberrations are distortions of the wave form, which limit the super-resolution capability already at depths of a few micrometers when focusing with oil immersion objectives, the standard high numerical aperture objectives for super-resolution in tissue. Aberrations can, in principle, be compensated by the use of adaptive optics [65] or to some extent by using the correction collar of the objective [148]. Adaptive optics is still not routinely used for in vivo imaging because it is costly and difficult to apply.
Low light levels with RESOLFT microscopy: RESOLFT is an alternative method for STED microscopy in vivo because it uses low light levels but also allows sectioning capability [115]. Until now, the scanning speed of RESOLFT is slower making it difficult to image fast processes in vivo, but the development of faster reversible switching fluorescent proteins is ongoing.
Chronic experiments: in principle, repeated imaging with in vivo super-resolution microscopy over days, by letting the mouse wake up and anaesthetizing it again, is possible and could be adapted. The challenge is to develop a brain implant allowing perfect imaging capability over the duration of the chronic experiment. Typically the opening in the scull starts to close below the glass window leading to image degradation and therefore a quick loss of super-resolution.
Motion artefacts: Super-resolution recording need to be synchronized with the vital functions to avoid artefacts caused by breathing and pressure pulse in vessels or by developing post processing routines of the in vivo recordings.
Concluding remarks.
In vivo super-resolution microscopy is a powerful tool for imaging simple organisms up to small mammals. However, applications should concentrate on the key characteristic of super-resolution, which is imaging of features not accessible using standard in vivo microscopy. This means that details of the cells and their reaction to peripheral stimuli should be imaged rather than whole cells. One could also think of in vivo super-resolution in different parts of the body of mammals which are accessible, e.g. muscle, liver, skin etc. However, all these organs or organ systems are accessible by cultures allowing us to mimic the in vivo situation for these organs in vitro. The central nervous system is different: its single reactions may of course be observed and estimated independently, however, every reaction has to be estimated in conjunction with the signals from both, inside and outside the intact organism. Therefore, studying sub-cellular processes with super-resolution in vivo could play a substantial role in the study of the living brain.
Acknowledgment.
I thank H Steffens and M Simard for proofreading of the manuscript.
Advance image deconvolution in STED microscopy
Giuseppe Vicidomini, Marco Castello and Alberto Diaspro
Nanoscopy, Nanophysics, Istituto Italiano di Tecnologia, Via Morego, 30, 16163, Genoa, Italy
Status.
Image deconvolution is a computational method that mitigates the distortions introduced by an optical system [154]. In 1983, Agard and Sedat first applied deconvolution to fluorescence wide-field microscopy to obtain digital optical-sectioning [156]. With the advent of physical optical-sectioning techniques, such as confocal and multi-photon microscopy, deconvolution has been partially put on a shelf. From a signal theory perspective, the diffraction limit imposes a maximum spatial frequency kc (cut-off frequency) that can be transmitted by the conventional microscope. Indeed, features of the sample at a distance below the diffraction limit, thus represented by frequencies above kc, cannot be distinguished through the microscope. Despite this limit, when prior information about the sample and/or the image formation process is introduced into the deconvolution problem, the restored image can contain features with frequencies above kc, namely, the deconvolution can potentially overcome the diffraction barrier. However, the integrity of these frequencies depends on the accuracy of the prior information [154]. In other words, the high frequencies 'generated' by deconvolution (and not transmitted by the microscope) could be linked to artifacts and not to real features. The skepticism regarding the ability of deconvolution to alone overcome the diffraction barrier has so far limited its dissemination as a general approach and relegated its application mainly to wide-field microscopy. The combination of image deconvolution and wide-field microscopy represents an important tool for all such applications that need high temporal resolution but not the ultimate spatial resolution.
In stark contrast with conventional microscopy, the stimulated-emission-depletion (STED) microscope [1, 157] is a band-unlimited system, i.e. all the frequencies of the sample can be transmitted to the image. As with all current super-resolved microscopy techniques, STED microscopy resolves features closer than the diffraction limit by registering their signal sequentially in space and time [158]. To this end, the fluorescence molecules defining these features are driven by an excitation beam into the (bright) excited-state, meanwhile, a second beam, the so called STED beam, transfers the excited molecules located in a well defined region back to the (dark) ground state. As a result, only the residual excited molecules contribute to the signal. Because of the transition of the molecule between the excited- and ground-state its probability of contributing to the signal is proportional to Iexc × exp(−ISTED/Is), where: Iexc and ISTED are the intensity of the excitation and STED beam; Is is the STED beam intensity at which the fluorescence probability reduces to 1/e and is a property of the molecule. The nonlinear dependence of the fluorescence probability (with the STED beam intensity) helps demonstrate the band-unlimited nature of STED microscopy. In real space the resolution of a microscope is defined by the point-spread function (PSF), in frequency space it is defined by the optical transfer function (OTF), describing the strength with which the sample's frequencies are transferred to the image (figures 23(a) and (b)). The OTF and the PSF are linked through the Fourier transformation. The radial PSF(r) of a (non-confocal) scanning STED microscope is proportional to Iexc(r) × exp(−ISTED(r)/Is). If we expand the exponential as a Taylor series the PSF(r) reads as Iexc(r) ×  . Because of diffraction Iexc(r) and ISTED(r) do not contain frequencies above kexc/STED = 2NA/λexc/STED (where NA is the numerical aperture of the objective lens and λexc/STED the wavelength of the excitation/STED beam), but the m-th term of the series has a support of mkSTED. Because the product in the space domain corresponds to a convolution in the frequency domain, the m-th function power has m-times the support of the basic function. Hence, at least theoretically, the STED microscope's OTF has infinite support.
. Because of diffraction Iexc(r) and ISTED(r) do not contain frequencies above kexc/STED = 2NA/λexc/STED (where NA is the numerical aperture of the objective lens and λexc/STED the wavelength of the excitation/STED beam), but the m-th term of the series has a support of mkSTED. Because the product in the space domain corresponds to a convolution in the frequency domain, the m-th function power has m-times the support of the basic function. Hence, at least theoretically, the STED microscope's OTF has infinite support.

Figure 23. PSFs (a) and OTFs (b) for different intensities of the STED beam (ζ = ISTED/Is) and for different value for the time-gating (Tg as multiple of the fluorophore's excited-state lifetime τ, ζ = 5). Insets show the normalized counterpart. (c) Comparison between raw CW-STED (left), gated CW-STED (middle) and deconvolved (multi-image deconvolution. Reproduced with permission from [155], copyright 2014 AIP Publishing LLC) gated CW-STED (right) images of immunolabelled microtubules. Scale bar 1 μm.
Download figure:
Standard image High-resolution imageAs a matter of fact, the job of deconvolution on STED images becomes simpler, because, at least theoretically, the microscope transmits the sample's high frequencies into the image.
Current and future challenges.
In STED microscopy the resolution is no longer limited by diffraction but instead by the noise. In particular, the resolution limit is given by the highest frequency whose strength is above the noise level.
The support of the OTF is unlimited, but the strength of the high frequencies fast reduces (each term of the Taylor series increases the support of the OTF by a factor kSTED, but at the same time the factorial m! in the denominator reduces the strength). Notably, increasing the intensity of the STED beam or similarly decreasing the saturation intensity Is boost the high frequencies (figure 23(b)) [159]. The first approach increases the potential photodamage effects; the second approach requires the engineering of new fluorescent molecules with long excited-state lifetime and/or high-stimulated emission cross-section. On the other hand the noise level can be reduced by enlarging the acquisition time, but at the expense of photodamage and temporal resolution. For these reasons the implementation of deconvolution methods with the ability to recover the sample's high frequencies partially 'hidden' by the noise represents an important challenge towards an efficient, fast and low-invasive STED microscope.
Deconvolution uses information describing how the microscope produces the image (forward model) as the basis of a mathematical inversion that reconstructs the fluorophore's map that defines the sample. State-of-the-art deconvolution methods are based on statistical formulation of the forward model and use maximum-likelihood or maximum a posteriori approaches to derive the relative algorithms [154]. These formulations allow the inclusion of statistical properties of the data into the problem: most of the deconvolution algorithms assume Gaussian noise dominating the images and only a few assume Poisson noise. Because of the low fluorescence counts rates of STED microscopy, a mixture of photon counting noise (Poisson) and other detector's noise (dark-count, read-out, afterpulsing, etc) corrupts the images. Surely, new algorithms based on noise mixture can better recover the sample's high frequencies.
Another key element for the success of deconvolution is the estimation of the PSF (or the OTF). Indeed, the precise knowledge of the OTF allows assigning its original strength to each sample's frequency. In contrast to conventional microscopy the PSF of a STED microscope does not depend only on the optical parameters of the system, but on the fluorophore and sample conditions as well. Thus an estimation of the PSF cannot be obtained by measuring calibration sub-resolved particles, but has to be extracted directly from the image. Blind deconvolution algorithms meet these requests, but increase the complexity of the problem. A reduction of complexity is obtained through semi-blind deconvolution. These methods estimate the parameters of a PSF's analytical model together with the fluorophores's map defining the sample. Up to now no blind or semi-blind deconvolution algorithms have been tested or developed for STED microscopy.
Advances in science and technology to meet challenges.
In the absence of noise, image deconvolution can theoretically extend the resolution of a STED microscope to infinity, also in the case of limited STED beam intensity. Practically, improving the signal-to-noise ratio (SNR) helps to operate at substantiallly lower STED beam intensity. Thus, advances in detector technology, which reduce noise and/or increase quantum-yield and count-rate directly translate into a resolution improvement. Similar enhancements can be obtained through advances in optical components, which increase the fluorescent collection efficiency of the microscope, and through developments of fluorescence molecules with high quantum-yield and high-photostability to persist to the relative higher intensity of the STED beam.
An important feature of STED microscopy is its ability to instantaneously produce sub-diffraction images, thus avoiding delays in the pipeline analysis. To preserve this ability, when STED microscopy is combined with deconvolution, it is necessary to implement real-time deconvolution algorithms. Graphics process unit (GPU) based implementations are recently moving in this direction [160], however for a large data set the computational time grows rapidly and new parallelization strategies need to be explored.
Concluding remarks.
The STED microscope theoretically offers unlimited resolution. Typically, its resolution is tuned by increasing the intensity of the STED beam, which potentially increases the photodamage effects. Image deconvolution offers an important alternative. Deconvolution has the ability to 'recover' from the noise the sample's high frequencies that are normally boosted only by increasing the STED beam intensity.
At this stage it is important to compare deconvolution with the so-called gated continuous-wave (CW) STED implementation [161]. This approach improves the effective resolution of a CW-STED microscope without increasing the STED intensity and similarly to deconvolution acts on the frequency space. In particular, it suppresses the low frequencies of the image, but in contrast with deconvolution it does not 'recover' high frequencies (figure 23(b)) [162]. Furthermore, the time-gate inherently reduces the SNR of the image, thus the high frequencies can be lost under the noise level. For these reasons, time-gating is not an alternative to deconvolution, but they can be synergically combined (figure 23(c)) [155].
All the efforts above will be in vain if the resulting deconvolution algorithms cannot be refined to the point where they can be widely and routinely used by biologists who are not experts in image processing. Thus a fundamental aspect for a wide dissemination of deconvolution in STED microscopy is to derive full and user-friendly software packages.
The limiting factor: fluorescent probes for optical super-resolution microscopy
Thorben Cordes
University of Groningen, Groningen, The Netherlands
Status.
The resolution limit of optical microscopes, i.e. their inability to resolve structural details smaller than half the wavelength of light, is based on physical phenomena that blur the final image [4]. In 2014 the 'Nobel Prize for Chemistry' was awarded for the development of super-resolved microscopy, which allows to overcome this limitation in far-field fluorescence microscopy. But what is the chemical aspect of optical nanoscopy?
While the main use of super-resolution microscopy is of an interdisciplinary nature, we find its key link to chemistry in the way the diffraction barrier can be broken: by preparation of fluorescent labels in different molecular states [163]. The optical resolution can be enhanced by restriction of emission to an area much smaller than the point-spread function (PSF) via spatial fluorescence confinement e.g. off-switching of the fluorescence during scanning, figure 24(a). Only close to the actual position of the emitter, the fluorescence is 'on'. The resolution is given by pixel size (scanning steps) and is limited by the number of possible excitation cycles C (figure 24(a)/(b)). The requirement for this on-off switching by e.g. stimulated emission depletion, STED [4], goes in hand with the demand for high photostability [163]. A second approach is video-imaging of single isolated fluorescent labels (STORM, PALM, PAINT) [4, 163, 164]. Resolution beyond the diffraction limit is achieved by temporal separation of the labels via on-off switching of emission (i.e. again preparation of molecular states with discernable emission properties, figure 24(c)) and localization of the fluorophore position via centroid fitting. The achievable resolution is influenced by photostability since the number of detected photons P during the 'on' period determine fitting accuracy [4, 163].

Figure 24. The resolution limit of optical super-resolution imaging is determined by the properties of the fluorescence label. (a) In scanning-based approaches, a stable emitter undergoes C excitation cycles that result in the emission of at least one photon ('on'). Here, a limitation is photobleaching. (b) Idealized resolution in % of the original FWHM for 2D imaging. Reproduced with permission from [163]. (c) In single-molecule based approaches, the number of emitted photons P per on cycle determines resolution.
Download figure:
Standard image High-resolution imageThe key element for optical super-resolution is hence control of the emission signal (stable versus blinking) as well as photostability. The second factor quantitatively limits the achievable resolution to a fraction of the full width of half-maximum (FWHM) of the original PSF (figure 24(b)). I here discuss strategies to engineer fluorescent labels for super-resolution microscopy by (i) molecular design and (ii) use of strategies to improve photophysical properties externally [163, 164].
Current and future challenges.
Fluorescent labels face a strict assessment procedure before they find use in (life-science) applications. The accepted standard quality criteria are high values of absorption cross section, fluorescence quantum yield, chemical stability, water solubility, low environmental sensitivity and finally the flexibility to link them to various biomolecular targets [163, 164]. It is needless to say that the (partially) opposing requirements face researchers with an impossible challenge to design the perfect fluorophore—only the concrete application determines which of the properties are essential.
These requirements are further accompanied by damage pathways that either irreversibly or transiently diminish fluorescent signals via intersystem crossing (rate kisc), redox or chemical reactions (rate kredox, kbleach, figure 25(a)) [163, 164]. The major problem with the formation of triplet- or radical-states is their high reactivity that results in photobleaching via various pathways (figure 25(a)). Additionally, their lifetime (μs ms) is sufficient to reduce the fluorescence count rate and to induce blinking in single-molecule traces. The final complication is molecular oxygen, which depletes the triplet-state effectively via triplet-triplet annihilation, but generates singlet oxygen during quenching. This reactive oxygen species is not only toxic for biological systems but also damages fluorophores. If possible, i.e. under in vitro conditions, oxygen is excluded from experiments and remains one of the most ambiguous players when trying to optimize photophysical properties of fluorophores.
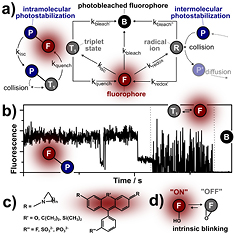
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 25. (a) Damage pathways and photostabilization strategies of fluorophores. (b) Typical fluorescent time trace of a single self-healing fluorophore with stable emission (F-P), triplet blinking (T1 ↔ F) and bleaching (B). Reproduced with permission from [167]. (c) Structural modifications of fluorophores that lead to improved properties or (d) increased functionality.
Download figure:
Standard image High-resolution image{kind=link}
Advances in science and technology to meet challenges.
An improvement of fluorophore properties is typically achieved via two distinct strategies: (i) structural modifications of the core to impose desirable properties or (ii) indirect support of the fluorophore via photostabilizers.
The general structural design of fluorophores aims at high absorbance cross sections, high fluorescence quantum yield, and small rates for intersystem crossing and internal conversion e.g. via exclusion of heavy atoms (see [163] and references cited therein). Typical fluorophores are rhodamines that consist of a conjugated ring system with different heteroatoms (N, C, or Si for R and R' [35, 165]) and substituents accessible for modification, figure 25(c). Using four-membered azetidines instead of the standard N,N-dimethylamino substituents for R allows to completely block fast non-radiative deactivation pathways via twisted internal charge transfer (TICT) states. This approach improves fluorescence quantum yield with little penalty on the extinction coefficients (>20-fold increase for a coumarine derivative) [35] even under live-cell conditions [86]. Especially silicon- and carborhodamines (R' = C(CH3)2, Si(CH3)2, figure 25(c)) show superb chemical stability in these challenging surroundings [35, 86, 165]. Another frequently used strategy uses fluorination or sulfonation (R'' = F,  ,
,  ) that modifies net charge (→water solubility) and increases chemical stability [165]. Recently, molecular design of silicon-based rhodamines allowed to add a functionality that is of great interest for single-molecule-based super-resolution: intrinsic blinking. The reversible switching between non-fluorescent and fluorescent states with on- and off-times tailored for STORM-type microscopy could be realized via an intramolecular nucleophilic addition (figure 25(d)) [88].
) that modifies net charge (→water solubility) and increases chemical stability [165]. Recently, molecular design of silicon-based rhodamines allowed to add a functionality that is of great interest for single-molecule-based super-resolution: intrinsic blinking. The reversible switching between non-fluorescent and fluorescent states with on- and off-times tailored for STORM-type microscopy could be realized via an intramolecular nucleophilic addition (figure 25(d)) [88].
Besides a molecular design, photostabilizers such as triplet-state quenchers or redox-active compounds offer a viable route to suppress potential damage pathways (figure 25(a)) [163, 164]. The required collisions between the reactive fluorophore state and the photostabilizer can be achieved via covalent linkage (intramolecular [166–168]) or diffusion (intermolecular [163, 164]), figure 25(a). Both strategies suppress signal blinking and photobleaching via a collision-based mechanism (figure 25(a)) [168]. The recent revival of self-healing fluorophores, which were originally pioneered by the Lüttke-lab [166] in the 1980s, shows, however great promise. Strikingly, a single photostabilizer can fully remove triplet-blinking (figure 25(a)/(b)) and at the same time increase photostability. A typical fluorescent time trace of a self-healing fluorophore shows stable emission with active triplet-quenching (figure 25(b), F-P), subsequent deactivation of the photostabilizer with then dominant triplet-blinking (figure 25(b), F ↔ T1) and final photobleaching (figure 25(b), B). More work is needed to make self-healing fluorophores fully competitive with established strategies since the latter (photostabilizers as a buffer-additive) are still more effective [168]. The lower photostability of self-healing fluorophores is, however, partially compensated by the fact that self-healing is often the only viable option for live-cell imaging and for assays where the addition of photostabilizer to the buffer remains ineffective or cannot be tolerated by the biological system. Furthermore, no general synthesis concepts are available to link the fluorophore, photostabilizer and biomolecular target in an arbitrary fashion. This limits the choice of fluorophores usable in self-healing to certain structural classes and their properties (charge, redox potential, colour)—so far only cyanine fluorophores are used for self-healing [167, 168].
Concluding remarks.
The structural design as well as the available photostabilization strategies of organic fluorophores have been optimized to a very large extent and offer superb fluorophore performance to perform optical super-resolution microscopy. There is, however, space for improvement under live-cell conditions (e.g. for RNA stains, fluorescent proteins etc.) where oxygen and the heterogeneous chemical surroundings of the cells cause unpredictable behavior of fluorophores. Another goal should be to combine functionality of a fluorophore (e.g. intrinsic blinking) with high photostability via self-healing or to achieve substantially higher brightness and photostability than is accessible with single chromophroes via use of plasmonic effects or multi-chromophore systems. In that respect nanodiamonds or collectively blinking nanoparticles provide interesting alternatives for future use in optical super-resolution microscopy.