


Abstract
The formation of carbon–heteroatm bond is the key step of synthesis of numerous organic compounds, including socially important products such as pharmaceuticals, crop protection agents and organic functional materials. These reactions proceed most efficiently when catalyzed by compounds of transition metals, first of all palladium. However, this approach has considerable drawbacks, in particular, high cost and toxicity of transition metal compounds and harsh reaction conditions required in some cases, resulting in limited functional group tolerance. This review describes the recent advances in the development of methodology of transition metal-free carbon–heteroatom bond-forming cross-coupling. It is shown that single-electron transfer and homolytic bond cleavage result in the generation of highly reactive radical and/or radical ion intermediates, enable the formation of new carbon–heteroatom bonds. These intermediates are generated using either visible light or electricity as energy sources or simple organic compounds acting as electron donors. Methods for carbon–heteroatom bond formation based on radical reactions proceeding under mild conditions and in the presence of labile functional groups are considered. The key mechanistic aspects of the reactions are highlighted. The review mainly covers the original publications of the current decade.
The bibliography includes 302 references.
Export citation and abstract BibTeX RIS
| D.I.Bugaenko. PhD in Chemistry, Researcher at the Division of Organic Chemistry, Department of Chemistry, Moscow State University. |
| E-mail: bugad357@yandex.ru |
| A.V.Karchava. PhD in Chemistry, Assistant Professor at the same Division. |
| E-mail: karchava@org.chem.msu.ru |
| Current research interests of the authors: chemistry of heterocyclic compounds, new synthetic methods, non-catalytic methods for C–heteroatom bond formation. |
| M.A.Yurovskaya. Doctor of Chemical Sciences, Professor, Leading Researcher at the same Division. |
1. Introduction
Cross-coupling reactions resulting in the formation of carbon – heteroatom bonds are among the key tools of organic chemistry used for the synthesis and functionalization of pharmaceuticals, organic functional materials and other practically useful organic compounds both in laboratory practice and on an industrial scale. 1, 2
Traditionally, these reactions are catalyzed by palladium, copper, nickel and some other transition metal compounds. Although catalytic transformations are characterized by high efficiency and reliability, they also suffer from certain drawbacks. 3 The first one is high and continuously growing cost of transition metals and their compounds, which leads to increase in the prime cost of the products synthesized with these catalysts by 5 – 20% every year. 4 For example, the price of palladium troy ounce increased from ∼200 to >1500 US dollars in the period from 2005 to 2019. 4 In addition, some reactions require the use of expensive ligands, which may be even more expensive than transition metals.
One more important problem is the presence of transition metal trace impurities in the products of synthesis, because they have a pronounced toxicity and can promote decomposition and/or isomerization of organic compounds. 5 In addition, there are cases where impurities have caused considerable changes in the electric properties of organic compounds and in the performance of functional devices based on them 6 and were responsible for the erroneous results in medicinal chemistry studies. 7 When transition metal-catalyzed reactions are used for the synthesis of pharmaceuticals or for the design of functional materials for organic electronics, especially thorough purification of the products from transition metal traces is required; this is associated with significant time and material expenses. 8–10 Transition metal-catalyzed reactions also have a number of other drawbacks: as a rule, they proceed at elevated temperatures 11 and are sensitive to steric factors. 12 Furthermore, the possibility of activation of not only one carbon – halogen bond in polyhalo-substituted substrates brings about the problem of selectivity. 13, 14
In recent years, the synthetic community has made attempts to find efficient alternatives to catalytic processes, which resulted in the development of transition metal-free cross-coupling strategy. The most significant progress was noted for three main types of these transformations induced by visible light, electric current or alkali metal bases. Their key advantages are mild conditions and high selectivity in the activation of chemical bonds. This allows the use of more diverse functionally substituted substrates than in conventional catalytic cross-coupling reactions.
In the absence of transition metals, the alternative activation of chemical bonds in organic molecules is attained via single-electron transfer (SET) and, in some cases, via homolytic bond cleavage, which can be induced, for example, by visible light irradiation of organic compounds. The SET processes can be initiated by the energy of light (for visible light this is usually mediated by photocatalysts, see Section 2) or by electric current (see Section 3). In addition, organic electron donors can be introduced into the reactions to induce SET processes (see Section 4). Single-electron transfer processes involving organic molecules give rise to organic radicals, radical ion species or electron donor – acceptor complexes (EDA). All of these intermediates usually have high reactivity, which facilitates the formation of new bonds.
Upon the reaction with a photocatalyst (PC) in the electronically excited state (PC*), substrate (S) can be either reduced (Fig. 1, cycle A) or oxidized (cycle B). In this case, the starting molecule is activated as a result of photo-induced electron transfer.
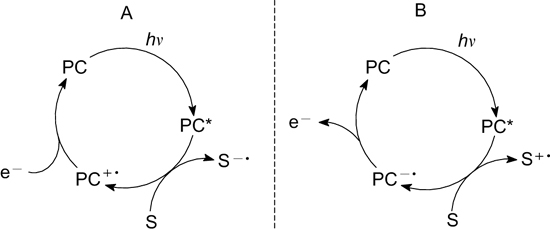
Figure 1. Substrate activation upon photoinduced electron transfer via substrate reduction or oxidation.
Download figure:
Standard imageSimilarly, the substrate can be activated by electric current upon anodic oxidation (pathway C) or cathodic reduction (pathway D) (Fig. 2).
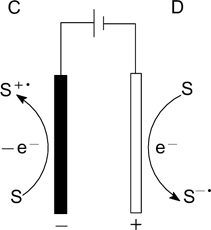
Figure 2. Substrate activation by electric current at the anode or cathode.
Download figure:
Standard imageThe formation of carbon – heteroatom bonds upon the substrate activation by the single-electron transfer mechanism can occur along three pathways (Fig. 3). Pathway a is typical of carbon substrates containing π-electron systems (compounds with carbon–carbon double bonds or aromatic derivatives) and possessing low oxidation potentials. In this case, the carbon substrate (CH) first undergoes photoinduced or anodic oxidation to be converted to the corresponding radical cation (CH+•), which then reacts with nucleophilic heteroatomic substrate (X) to give a C–X bond. Alternatively, heteroatomic substrate X can undergo single-electron oxidation, thus giving the corresponding radical cation (X+•), which reacts as an electrophile with electron-rich carbon substrate (pathway b). Transformations of the third type (pathway c) are inherent in carbon substrates containing an electron-withdrawing leaving group — a halogen atom. In this case the carbon substrate (CY) is first reduced to the corresponding radical anion (CY−•), which eliminates the leaving group (Y−) to give the C. radical. This radical reacts with the heteroatomic substrate to give a carbon – heteroatom bond. Figure 3 shows simplified transformation schemes in which the SET activation of organic molecules results in the formation of carbon – heteroatom bonds. In some cases, these schemes can be supplemented by the steps of formation and transformations of electron donor – acceptor complexes, redox transformations involving photocatalysts and intermediates, and hydrogen atom elimination or addition.
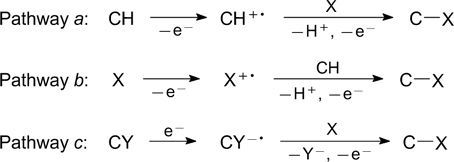
Figure 3. Transformations to form C–X bonds upon substrate activation via single-electron transfer.
Download figure:
Standard imageThe intensive development of the strategy of cross-coupling free from organometallic catalysts was reflected in a number of analytical reviews. Among them, note the review by Sun and Shi 3 published in 2014 and covering the literature up to 2012. This review considers, in particular, reactions that have no analogues among transition metal-catalyzed processes. Various types of photoinduced 15 and electrochemically induced 16 processes are also subjects of short review publications. Some reviews analyze the photochemically and electrochemically driven formation of carbon – nitrogen 17 – 19 and carbon – sulfur 20 bonds. In addition, there are reviews 21, 22 that give examples of reactions induced by alkali metal bases, but the reported data are limited to analysis of carbon – carbon bond-forming reactions.
The present review considers the key achievements of the last decade in the development of transition metal-free cross-coupling reactions that result in the formation of carbon – heteroatom bonds. Since this review is mainly addressed to synthetic chemists, we arranged the material according to the method used to activate organic compounds (visible light, electrical current and organic electron donors) and according to the type of carbon – heteroatom bond formed (nitrogen, sulfur, phosphorus and oxygen). Each part of the review discusses the formation of carbon–heteroatom bonds, first, as a result of functionalization of C–H bonds at sp3- and sp2-hybridized carbon atoms and, at the end, as a result of displacement of leaving groups. Without claiming to be exhaustive, we tried to demonstrate the most significant results of this type and the extensive possibilities of this strategy for the synthesis of structurally diverse organic compounds. For most of the considered reactions, the mechanisms of substrate activation and formation of carbon – heteroatom bonds are discussed and examples of compounds synthesized in this way are given.
2. Photoinduced reactions
In recent years, particular attention has been attracted by organic reactions induced by visible light, which represent an efficient and robust methodology of organic synthesis. The key step of these reactions is the formation of reactive radical type intermediates, which provide route to new products inaccessible by thermal activation of the substrates. 23
2.1. Formation of C–N bond
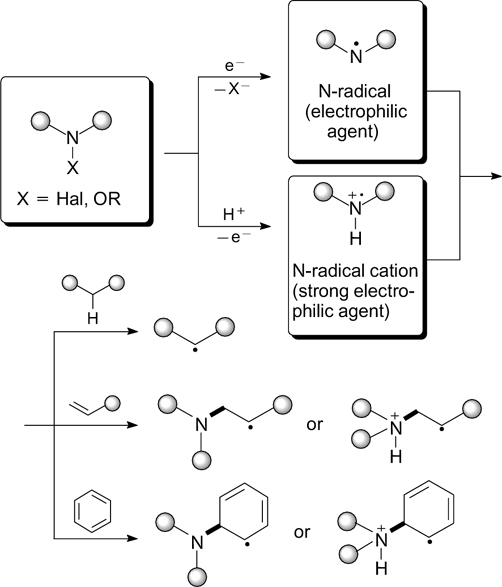
The C–N bond formation reactions induced by visible light are accomplished by generation of N-centred free radicals upon activation of amines, amides, hydrazones, and their N-derivatives. The reactivity of N-radicals depends on the type of hybridization of the nitrogen atom and the nature of substituents. 24 N-Centred radicals have been rarely used in organic synthesis for a long time because of the absence of selective methods for their generation under mild conditions. The development of photoredox-catalyzed reactions driven by visible light and based on single-electron transfer and energy transfer processes 25 crucially changed the situation in this field. In recent decades, significant progress has been attained in the development of photoredox generation methods of N-centred radicals and radical cations from diverse precursors containing labile N–O, N–N and N–halogen bonds and from compounds containing N–H bonds. In most of these reactions carried out to date, transition metal (iridium, palladium, ruthenium) complexes are used as photoredox catalysts. 26 – 28 Meanwhile, there are known examples of generation of N-radicals in the absence of transition metal catalysts, which form the subject of this part of the review.
N-Centred radicals are either converted to C-centred radicals upon hydrogen atom transfer (HAT) or add to electron-rich systems — compounds with multiple bonds or aromatic rings (Scheme 1).
Scheme 1
Download figure:
Among the former type reactions, mention should be made of cross-dehydrogenative benzylic C(sp3)–H amination using 9,10-dicyanoanthracene (DCA) as an organic photoredox catalyst and N-methoxyamides as precursors of amidyl radicals (Scheme 2). 29
Scheme 2
Download figure:
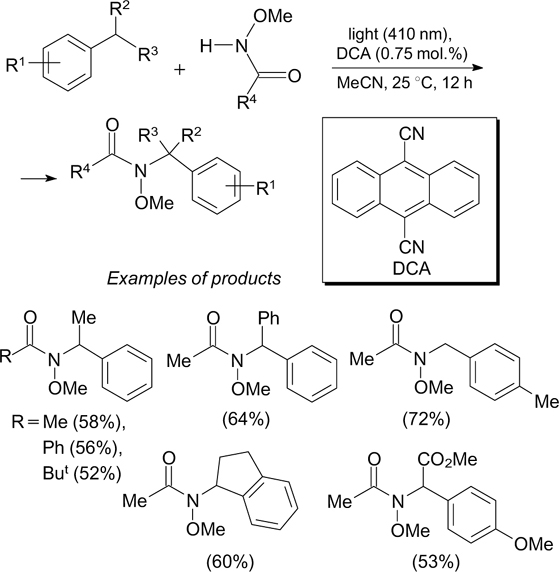
The putative reaction mechanism includes the step of formation of amidyl radical 1, stabilized by the captodative effect 30 (simultaneous presence of electron-donating and electron-withdrawing substituents at the radical centre), as a result of single-electron oxidation of N-methoxyamide with 9,10- dicyanoanthracene in the singlet excited state (DCA*) (SET process, Scheme 3). The hydrogen atom transfer from the benzylic substrate to the N-centred radical (HAT process) leads to a stable benzyl radical, which is then oxidized by DCA* to the corresponding benzyl cation. This cation reacts with N-methoxyamide to give the amination product.
Scheme 3
Download figure:

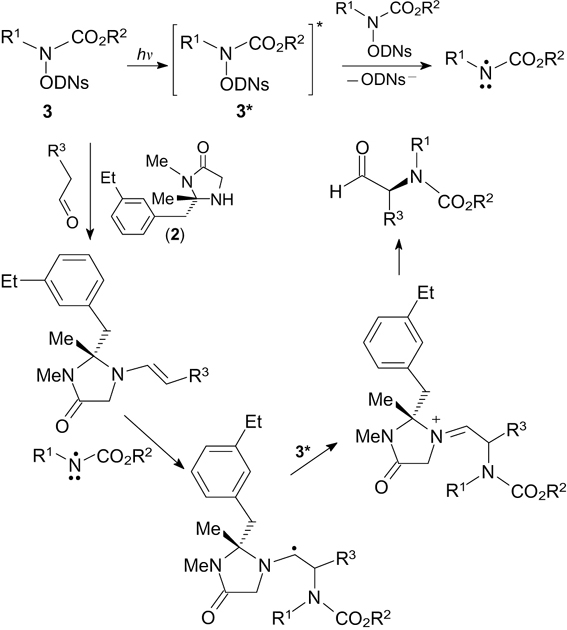
The asymmetric α-amination of aldehydes is based on the reaction of electrophilic N-radicals with nucleophilic enamines, which are obtained in situ from aliphatic aldehydes and chiral imidazolidinone 2 (Scheme 4). 31 In this case, the initial N-(2,4-dinitrobenzenesulfonyloxy)carbamates 3 serve as precursors of N-radicals.
Scheme 4
Download figure:

The radicals are generated without a photoredox catalyst upon the single-electron reduction of the substrate in the electronically excited state (3*) with the starting molecule 3. The addition of N-radicals to enamines affords C-radicals, which undergo SET oxidation involving compound 3* and are thus converted to the corresponding cations (Scheme 5).
Scheme 5
Download figure:
A related generation of amidyl radicals upon irradiation of N-(2,4-dinitrophenyloxy)amides with a compact luminescent lamp provides the basis for the synthesis of pyrrolidinones. 32 Radical 4 formed upon 5-exo-dig-cyclization is converted to the corresponding pyrrolidine derivative via hydrogen atom transfer from cyclohexa-1,4-diene (Scheme 6).
Scheme 6
Download figure:

Similarly, visible light-induced homolytic N–O bond cleavage in O-aryloximes and the subsequent 5-exo-trig-cyclization of the resulting N-radicals 5 provides the synthesis of five-membered N-heterocycles (see Scheme 6). 33
Depending on the aryl group in O-aryloximes, the N–O bond activation giving rise to iminyl radicals 5 may be performed either in the presence of organic photoredox catalyst eosin Y (EY) or via the intermediate formation of the electron donor–acceptor complex with Et3N (not shown in Scheme 6; see Ref. 33).
The amidyl radicals generated upon green light irradiation of N-(2,4-dinitrophenyloxy)amides, which contain no multiple bonds, undergo intermolecular reactions with electron-rich aromatic compounds (substituted pyrroles, indoles, furans, thiophenes, naphthalenes, azulenes, etc.) in the presence of 2 mol.% eosin Y to give amination products. 34
The photoinduced intramolecular amination of C(sp3)–H bonds is also used to synthesize six-membered fused heterocyclic systems. The visible light-induced intramolecular formation of C–N bonds in benzamides in the presence of 1,1'-binaphthyl-2,2'-phosphoric acid (BPA) and pinacol borane yields fused heterocycles. This method was used to prepare the natural compounds tryptanthrin and rutaecarpine and related products (Scheme 7). 35
Scheme 7
Download figure:

This reaction proceeds via photoinduced isomerization of amides to short-lived photoisomers 6 stabilized by intermolecular hydrogen bonds with the (R)-BPA molecule. As a result of SET processes, photoisomers 6 are converted first to N-radicals 7 and then to iminium ions 8 (Scheme 8).
Scheme 8
Download figure:

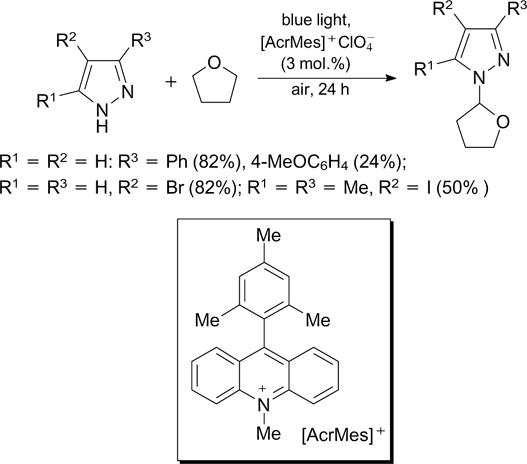
Ethers can serve as substrates for the initiated photochemical amination of the C(sp3)–H bonds, which was demonstrated in relation to THF.
36
In this case, the reaction involves 9-mesityl-10-methylacridinium perchlorate  as the photoredox catalyst (Scheme 9). In the electronically excited state, the latter compound
as the photoredox catalyst (Scheme 9). In the electronically excited state, the latter compound  , which is a potent oxidant, transforms azole molecules to the corresponding N-radicals. This reaction also involves air oxygen, which oxidizes the reduced form of the photoredox catalyst and promotes generation of C-radicals from THF. Pyrazoles and 1,2,3-triazoles can act as aminating agents.
, which is a potent oxidant, transforms azole molecules to the corresponding N-radicals. This reaction also involves air oxygen, which oxidizes the reduced form of the photoredox catalyst and promotes generation of C-radicals from THF. Pyrazoles and 1,2,3-triazoles can act as aminating agents.
Scheme 9
Download figure:
The acridinium photoredox catalyst  and oxygen as the terminal oxidant were used to generate hydrazonyl radicals from N-tosylhydrazones of β,γ-unsaturated ketones.
37
and oxygen as the terminal oxidant were used to generate hydrazonyl radicals from N-tosylhydrazones of β,γ-unsaturated ketones.
37
An alternative approach to the formation of N-centred radicals is based on the application of iodine(III). In this case, the highly reactive N-radicals are generated as a result of photoinduced homolysis of the N–I bond. An example of this type is the intramolecular oxidative amination of tosylamides, giving rise to saturated nitrogen-containing heterocycles (Scheme 10). 38
Scheme 10
Download figure:
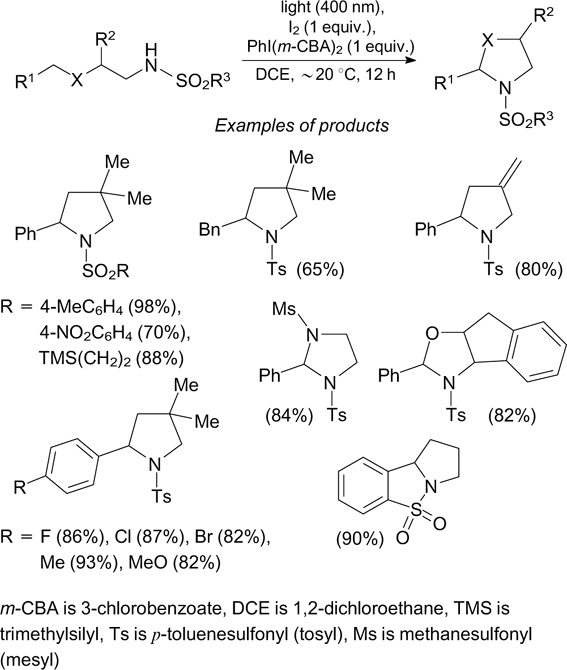
The putative mechanism of cyclization includes the initial reaction of molecular iodine with hypervalent iodine(III) compound to give I(m-CPB), which is the true catalyst of the process. The subsequent reaction of this compound with the substrate yields key intermediate 9, containing an N–I bond and functioning as a photosensitizer. The irradiation of intermediate 9 affords N-radical 10, which undergoes 1,5-transfer of hydrogen and thus isomerizes to C-radical 11. The elimination of a iodine atom from intermediate 9 under the action of this radical initiates a radical chain reaction (Scheme 11).
Scheme 11
Download figure:

A synthetic strategy that makes it possible to avoid the drastic conditions of the Hofmann – Löffler reaction (cyclization of N-haloamines on heating in a strong acid
39
) is based on 1,5-cyclization of N-alkyltosylamines to give substituted pyrrolidines catalyzed by the  anion (Scheme 12).
40
This anion is generated upon oxidation of the iodide anion under the action of PhI(OAc)2. In this case, the amidyl radical is also formed upon the homolytic cleavage of the N–I bond in the intermediate N-iodotosylamide induced by irradiation of the reaction mixture with a compact fluorescent lamp (CFL).
anion (Scheme 12).
40
This anion is generated upon oxidation of the iodide anion under the action of PhI(OAc)2. In this case, the amidyl radical is also formed upon the homolytic cleavage of the N–I bond in the intermediate N-iodotosylamide induced by irradiation of the reaction mixture with a compact fluorescent lamp (CFL).
Scheme 12
Download figure:
The I2–PhI(m-CBA)2 system was used for photoinduced C–H amination of arenes. 41 The considered methods for the generation of compounds with a N–I bond are based on the use of iodine compounds and are accompanied by the formation of aryl iodides as by-products. This circumstance makes these methods uneconomical and somewhat cumbersome, since it is necessary to separate the target products from undesirable compounds formed in equimolar amounts.
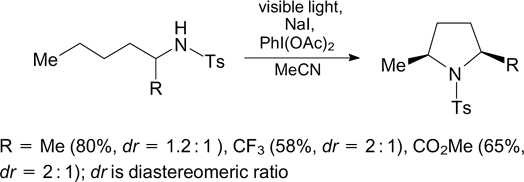
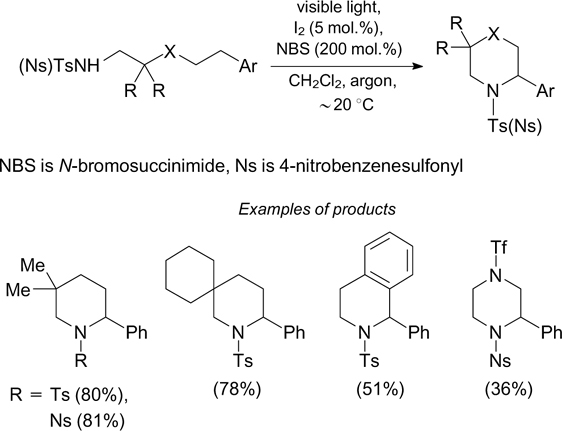
The cyclization of N-alkyltosylamides to give substituted pyrrolidines can also occur in the presence of N-iodosuccinimide for generation of photolabile compounds containing N–I bonds. 42 Using 2,4,6-triphenylpyrylium tetrafluoroborate as a photoredox catalyst for the oxidation of molecular iodine with air oxygen, it is possible to do without oxidants based on hypervalent iodine compounds. 43 A process of this type applied to N-alkylsulfonylamides containing an aryl substituent in position 5 leads to selective formation of substituted piperidines (Scheme 13). 44
Scheme 13
Download figure:
The generation of amidyl radicals upon the visible light-induced homolytic cleavage of the N–I bond is the key step of the benzylic C–H amination of aromatic hydrocarbons in the presence of chloramine T and molecular iodine. 45
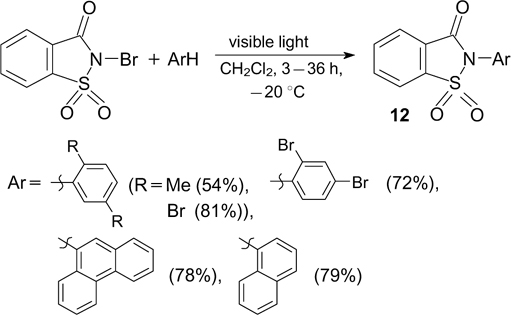
An alternative method for the generation of imidyl radicals comprises visible light irradiation of N-bromosaccharins. 46, 47 The use of these radicals underlies the imidation of (hetero)arenes to give N-arylsaccharins 12 (Scheme 14). 46 This method is applicable to a wide range of (hetero)arenes containing electron-donating and weak electron-withdrawing (for example, halogen atoms) substituents. A similar generation of amidyl radicals takes place in the case of substituted N-bromobenzamides, 48 although the synthetic use of the latter is not associated with the preparation of nitrogen compounds.
Scheme 14
Download figure:
In the synthesis of an extensive series (58 examples) of azine-substituted tertiary amines 13, the photoinduced generation of N-radical cations by single-electron oxidation of primary and secondary amines is followed by their coupling with the radical anions formed from the corresponding cyanoazines. 49 3,5-Dicyano-2,4,6-tris(diphenylamino)fluorobenzene (3DPAFIPN) serves as the photoredox catalyst (Scheme 15).
Scheme 15
Download figure:
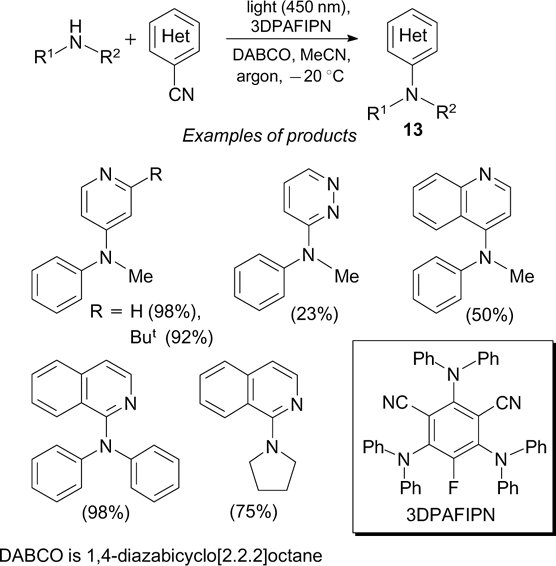
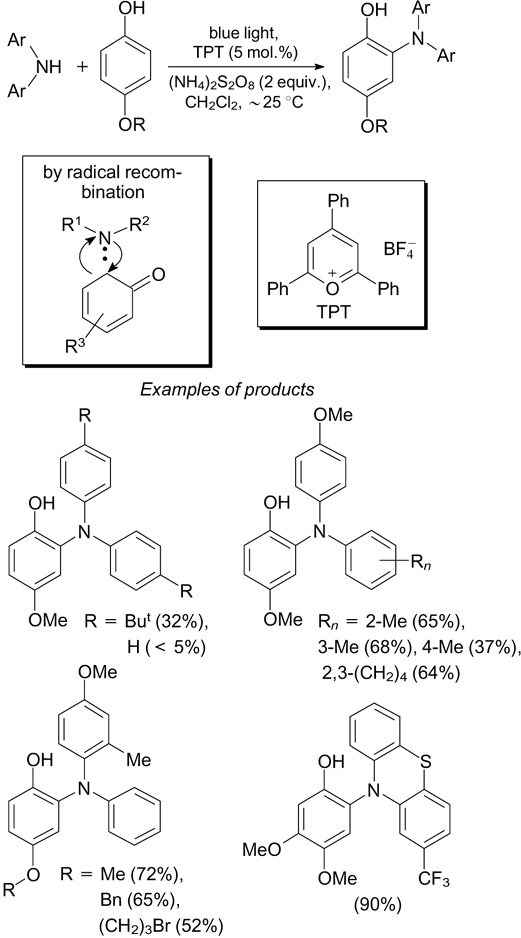
The cross-dehydrogenative amination of 4-alkoxyphenols with diarylamines on exposure to blue light takes place in the presence of 2,4,6-triphenylpyrylium (TPT) tetrafluoroborate as the photocatalyst and ammonium persulfate as the oxidant (Scheme 16). 50 The mechanism of this reaction is based on radical coupling (shown in a frame in the Scheme).
Scheme 16
Download figure:
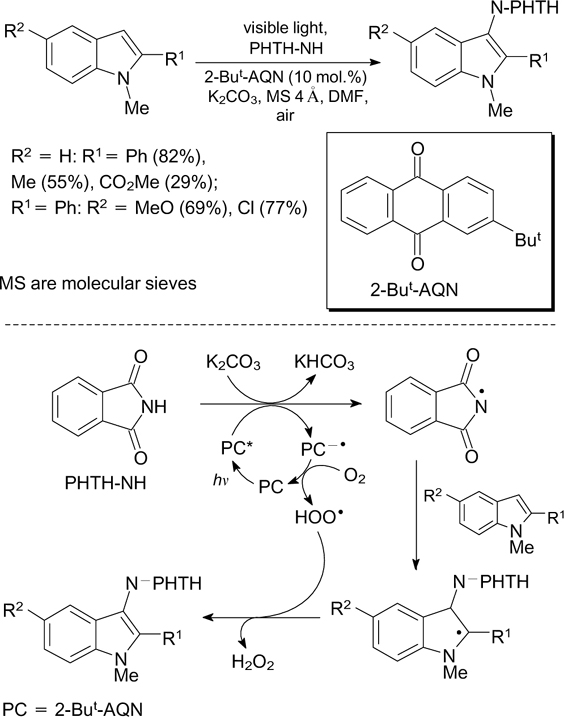
The generation of the imidyl radical from phthalimide is the key step of the synthesis of phthalimide (PHTH-NH) derivatives of indole and some other five-membered heterocyclic compounds. The photoinduced dehydrogenative coupling of substrates with phthalimide takes place when the reaction mixture is irradiated by CFL in the presence of a base and 2-tert-butylanthraquinone (2-But-AQN) as the photoredox catalyst (Scheme 17). 51
Scheme 17
Download figure:
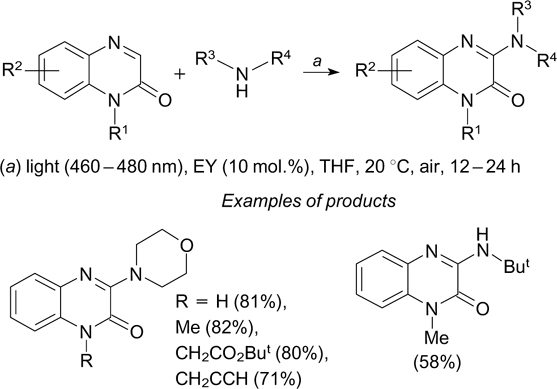
A similar mechanism of formation of the C–N bond is implemented in the amination of quinaxolin-2(1H)-ones with secondary amines induced by visible light at a 460 – 480 nm wavelength in the presence of eosin Y as the photocatalyst (1 – 5 mol.%) (Scheme 18). 52
Scheme 18
Download figure:
One more example of amination of an aromatic system is provided by regioselective cyclization of 2-styrylquinolines, resulting in quinolino[1,2-a]quinolinium salts 14, potential biologically active compounds (Scheme 19). 53
Scheme 19
Download figure:

The visible-light induced dehydrogenative coupling of electron-deficient alkenes with azoles catalyzed by the rose bengal (RB) dye affords N-vinylazoles. In this case, the reaction mechanism includes the intermediate formation of azolyl N-radical. First, the single-electron oxidation of azole with the electronically excited photoredox catalyst yields the corresponding radical cation, which is then deprotonated to the N-radical (Scheme 20). 54
Scheme 20
Download figure:

The acridinium photoredox catalysts 15a,b were used to generate alkylaminium radical cations upon oxidation of the corresponding primary aromatic alkylamines in an oxygen atmosphere under irradiation with light at 455 nm. The N-radical cations generated in this way were used for direct C–H amination of aromatic compounds; amino acid ester hydrochlorides were also used as aminating agents in this case. 55 When optically pure chiral amines were used, their enantiomeric purity was retained. Although this method is fairly selective for the synthesis of monoamination products, the regioselectivity of C–H amination in the absence of pronounced steric restrictions remains moderate (Scheme 21; the value indicated in parentheses after the product yield is the ratio of ortho- and para-isomers). Meanwhile, the amination of heteroarenes is virtually regioselective under the same conditions.
Scheme 21
Download figure:
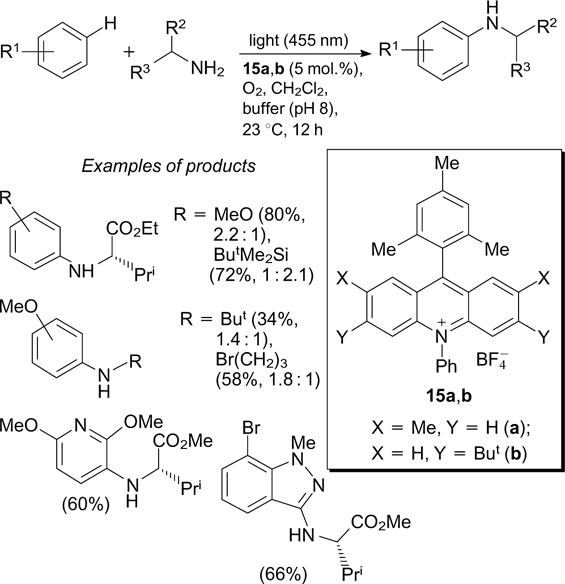
A possible mechanism of C–H amination of aromatic compounds proposed by the authors includes the initial oxidation of the primary amine to the corresponding radical cation with the acridinium photoredox catalyst in the electronically excited state. The addition of the N-radical cation to the aromatic molecule results in the formation of cyclohexadienyl radical, which aromatizes under the action of oxygen (Scheme 22). However, since some aromatic compounds with electron-donating substituents have oxidation potentials close to the oxidation potentials of amines, an alternative mechanism is also possible. The mechanism includes the initial visible light-induced oxidation of aromatic substrate to give the C-radical cation, which then reacts with N-nucleophile (see Scheme 22).
Scheme 22
Download figure:
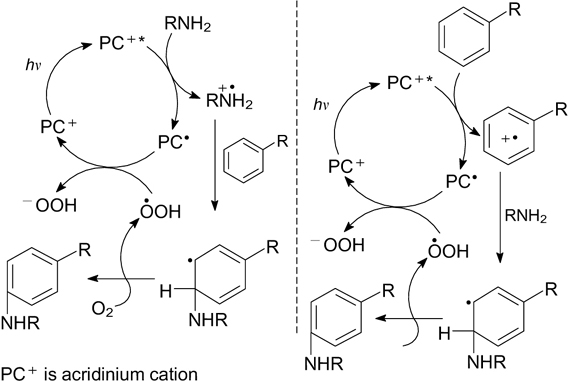
Acridinium and xanthylium salts were employed as photoredox catalysts in the visible light-induced N-arylation of primary amines and azoles with fluoro- and methoxybenzenes via formal nucleophilic substitution with replacement of F atom 56 and MeO group. 57, 58 In both cases, the probable mechanism implies the generation of C-radical cations (similar to that shown in Scheme 22), which then react with N-nucleophilic reagents. Unlike conventional aromatic nucleophilic substitution SNAr, visible light-induced photocatalytic reactions proceed under mild conditions and in the absence of strong electron-withdrawing substituents in the substrate molecule.
The decarboxylative coupling of NH-azoles with redoxactive N-acyloxyphthalimides 59, 60 in the presence of N-phenylbenzo[b]phenothiazine (PHT) as a photoredox catalyst and collidinium tetrafluoroborate on blue light irradiation leads to the synthesis of azole derivatives containing a tertiary alkyl substituent at the nitrogen atom (Scheme 23). 61 The key intermediates of the reaction are alkyl radicals, formed as a result of photochemically induced fragmentation of N-acyloxyphthalimides. The collidinium salt is necessary to suppress the side elimination reaction involving phthalimide anion as the base.
Scheme 23
Download figure:

2.2. Formation of C–S bond
The photocatalytic reactions that give C–S bond in the presence of either photoactive transition metal complexes or organic photoredox catalysts are addressed in detail in a review 62 published in 2018. Another review 63 that appeared the same year considers the photocatalyzed formation of sulfones and sulfoxides. In this part of the present review, we mention only the most important reactions of this type described earlier and consider in detail the data obtained after 2018.
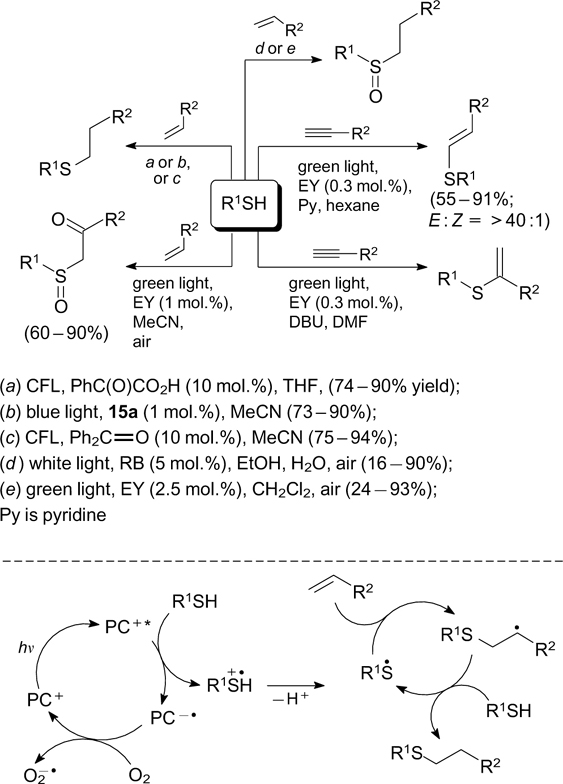
The visible light initiation was used to prepare the products of addition of aliphatic and aromatic thiols to multiple bonds (Scheme 24). Several photocatalysts were proposed for these reactions, including eosin Y, 64, 65 phenyl-glyoxalic acid, 66 benzophenone, 67 1,2-diphenylethane-1,2-dione, 68 9-mesityl-10-methylacridinium salts 69 – 71 and the acridine orange dye, which acts simultaneously as a Lewis base. 72 Although the reaction mechanisms vary to some extent depending on the conditions and the nature of the photocatalyst, the key step is the single-electron oxidation of the starting thiol to the corresponding radical cation, which is converted to thiyl radical upon deprotonation. 73 The addition of S-radicals to alkenes or alkynes affords the corresponding C-centred radicals, which participate in the subsequent generation of S-radicals. It is noteworthy that the selectivity of addition of thiols to terminal alkynes depends on the base used. In the case of pyridine, the major reaction product is E-alkene formed upon the anti-Markovnikov addition, 65 whereas the use of 1,8-diazabicyclo[5.4.0]undec-1-ene (DBU) results in the formation of the Markovnikov addition product. 74 The reactions carried out in the presence of oxygen gave products of oxidative addition of thiols to alkenes (see Scheme 24). 64, 75, 76 The photoredox-catalyzed formation of C–S bonds induced by visible light was used for the synthesis of benzothiophenes 69 and for direct C–H sulfenylation of indoles, 77 quinoxalinones 78 and 4-hydroxycoumarins. 79
Scheme 24
Download figure:
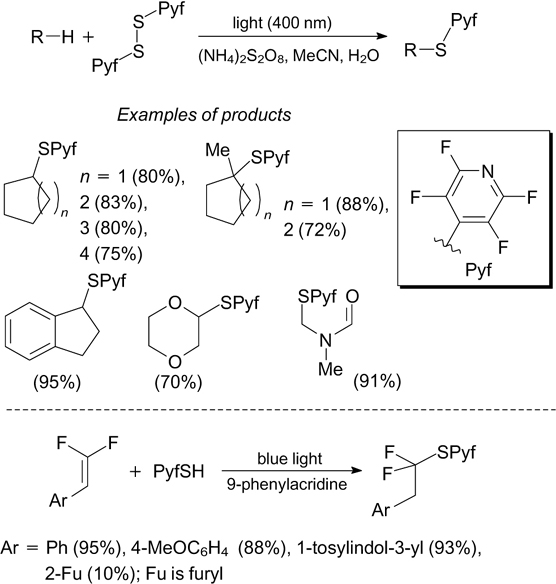
The C–H sulfenylation of alkanes and cycloalkanes was accomplished on treatment with bis(2,3,5,6-tetrafluoropyridin-4-yl) disulfide under irradiation with light at 400 nm in the presence of an oxidant. The reaction was found to be highly selective to tertiary C–H bonds of alkanes. The presumed mechanism of sulfide formation includes the generation of alkyl radical upon hydrogen atom transfer (HAT) from an alkane molecule to the thiyl radical, formed upon the photolytic dissociation of the disulfide (Scheme 25). 80 The thiyl radical can be obtained by blue light irradiation of the complex of 4-sulfanyl-2,3,5,6-tetrafluoropyridine with 9-phenylacridine via photo-induced proton coupled electron transfer (PCET). The thiyl radical generated in this way was used for the addition to gem-difluorostyrenes (see Scheme 25). 81
Scheme 25
Download figure:
One of the synthetic routes to benzothiazoles is based on cyclization of N-arylthiobenzamides upon C–H functionalization. First, thioamide or its deprotonated form is oxidized to give S-radical. The addition of the thiyl radical to the aromatic ring of the aniline moiety results in generation of a cyclohexadienyl radical, which aromatizes via hydrogen atom elimination. Although most transformations of this type are based on the use of ruthenium complexes as photoredox catalysts, catalytic systems that allow transition metal-free synthesis of benzothiazoles have been proposed in recent years. Reactions have been carried out in the presence of the following systems: riboflavin – K2S2O8 – blue light, 82 (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) – light of CFL, 83 carbon nitride – air oxygen – red light, 84 eosin Y – diisopropylethylamine (DIPEA) – green light. 85
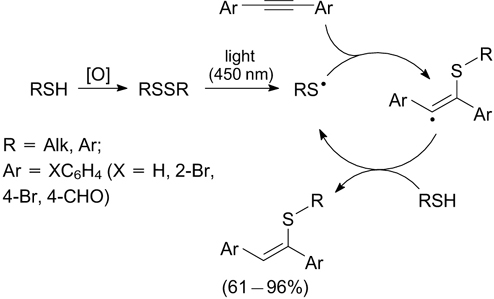
Blue light irradiation at 450 nm in the absence of a catalyst stimulates the addition of alkane- and arenethiols to diarylacetylenes to give 1,2-diaryl-1-alkyl(aryl)ethylenes as mixtures of cis- and trans-isomers. According to the mechanism proposed by the authors, the reaction starts with the oxidative coupling of thiols to give the corresponding disulfides, which dissociate under irradiation via homolytic cleavage of the S–S bond to give S-radicals, which initiate a chain radical reaction (Scheme 26). 86
Scheme 26
Download figure:
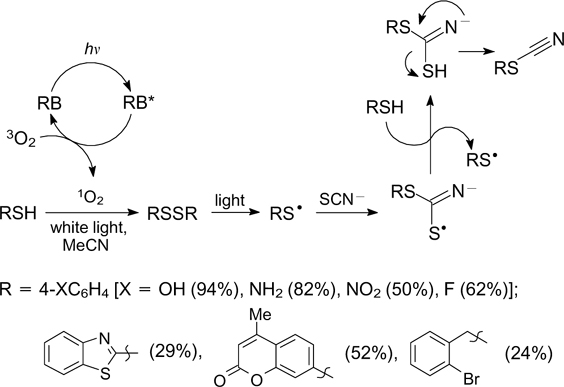
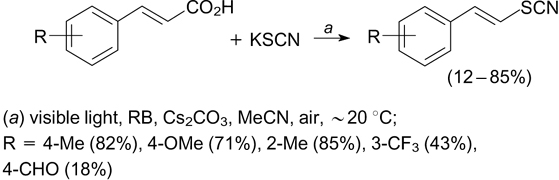
The addition of S-radicals generated from thiols to the thiocyanate anion under irradiation with white light in the presence of 1 mol.% rose bengal affords organic isothiocyanates. The mechanism of this reaction includes the initial generation of singlet oxygen1O2, which acts the oxidant for thiol and thus converts it to disulfide. Under irradiation, the latter dissociates into S-radicals, which add to the thiocyanate anion. Unlike other methods for the synthesis of organic isocyanates, this method is applicable to aromatic substrates containing substituents with different electronic properties and can be implemented under mild conditions requiring no toxic cyanide ion (Scheme 27). 87
Scheme 27
Download figure:
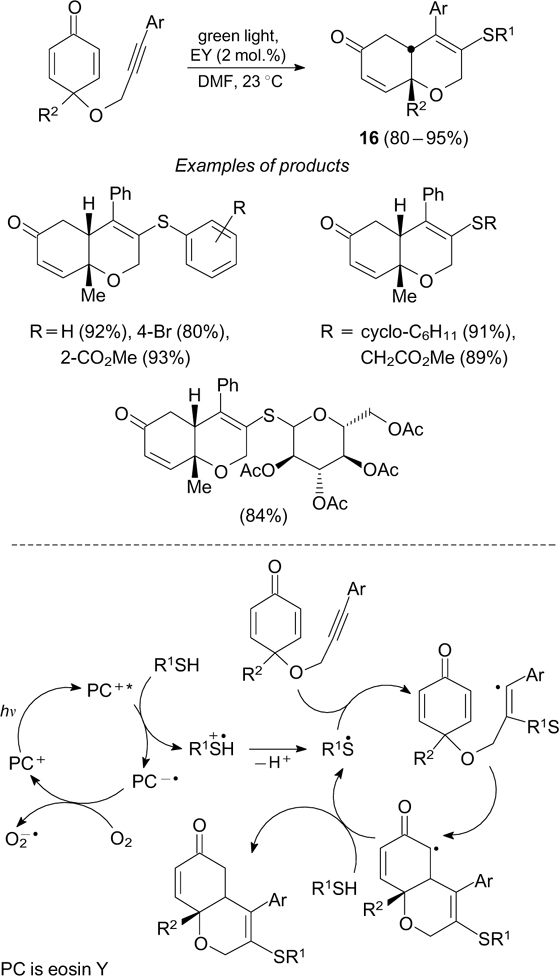
The addition of thiyl radicals to multiple bonds is the initial step of a number of cascade radical reactions that finally furnish complex molecules. An example is the synthesis of dihydrochromenones 16 by the sulfenylation – annelation cascade reaction (Scheme 28). 88
Scheme 28
Download figure:
Similar addition reactions to multiple bonds induced by visible light are also characteristic of thiocyanate anions (Scheme 29). These reactions begin with single-electron oxidation to give a thiocyanyl radical, which then adds to carbon – carbon double bonds of styrenes 89 and electron-rich aromatic systems — indoles, 90 imidazopyridines 91 and indolizines. 92 The visible light-induced addition of N≡CS radical to 2-hydroxyaryl β-dialkylaminovinyl ketones in the presence of rose bengal furnishes 3-thiocyanochromenes. 93 In all of the above examples, reactions proceed under aerobic conditions, which facilitates regeneration of photoredox catalysts. When styrenes are used as the substrates, oxygen serves as the oxidant for the benzyl radical that is formed upon the addition of the S-radical to the C=C bond, and 5-(hetero)aryl-2-imino-1,3-oxathiolanes are isolated in the final step (see Scheme 29). 89
Scheme 29
Download figure:

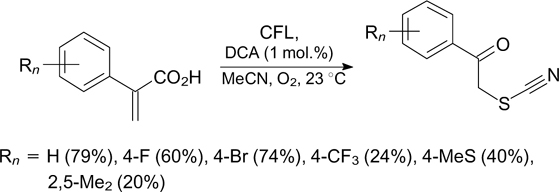
A similar oxidative transformation was observed upon the addition of the N≡CS radical generated from the thiocyanate anion to α-arylacrylic acids under irradiation with a fluorescent lamp in the presence of 9,10-dicyanoanthracene in an oxygen atmosphere. 94 In this case, the C–S bond formation followed by decarboxylation and oxidation of the benzylic position afford α-thiocyanato ketones (Scheme 30).
Scheme 30
Download figure:
One more type of visible light-induced transformation involving thiocyanate anion takes place for cinnamic acids. The synthesis of vinyl thiocyanates is based on decarboxylative cross-coupling of cinnamic acids with potassium thiocyanate taking place upon irradiation with CFL in the presence of rose bengal as the photocatalyst under aerobic conditions (Scheme 31). 95 In the first step, the electronically excited dye molecule (RB*) oxidizes the thiocyanate anion to the corresponding radical, which adds to cinnamic acid thus giving a benzyl radical. The latter is oxidized with air oxygen to the benzyl cation, which is decarboxylated to yield vinyl thiocyanate. Air oxygen is also responsible for regeneration of the photoredox catalyst.
Scheme 31
Download figure:
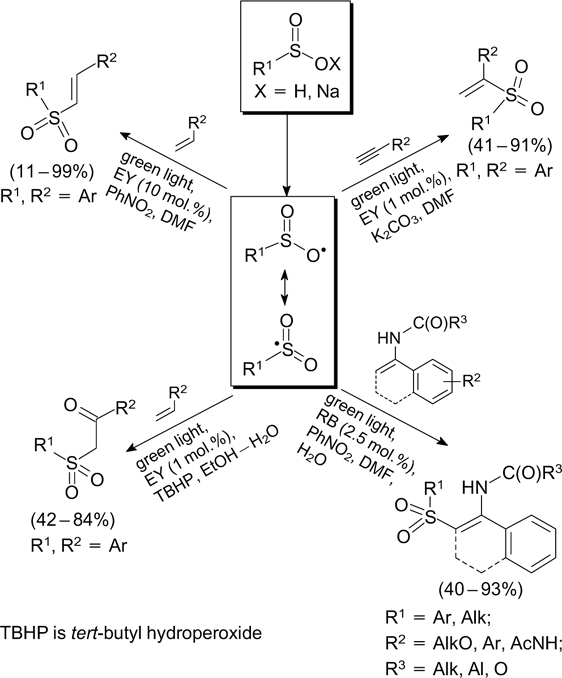
The generation of S-radicals via visible light-induced reactions of sulfinic acids and their salts followed by addition of the radicals to alkenes and alkynes underlies the synthesis of β-keto sulfones 96 and anti-Markovnikov 97 and Markovnikov isomers of vinyl sulfones 98 and β-acetaminovinyl sulfones (Scheme 32). 99
Scheme 32
Download figure:
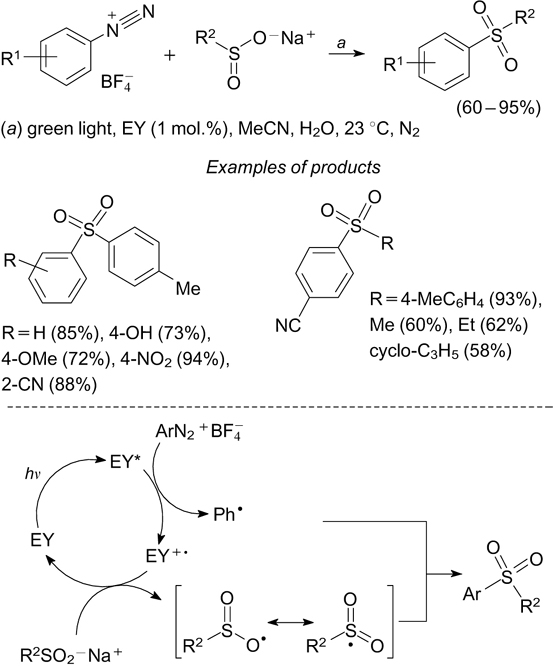
Alternative routes to the generation of arenesulfonyl radicals include the addition of preliminarily generated aryl radicals to sulfur(IV) oxides. In turn, aryl radicals can be generated from arenediazonium and diaryliodonium salts. 25
One of the synthetic routes to aryl alkyl sulfones and diaryl sulfones is based on the reaction of arenediazonium tetrafluoroborates with sodium aryl- and alkylsulfinates in the presence of eosin Y (1 mol.%) under irradiation with green light at 535 nm.
100
According to the mechanism postulated by the authors, eosin Y is initially transferred to the excited state (EY*) upon photon absorption, while the excited state can act as a single-electron reducing agent for the arenediazonium cation. The electron transfer from EY* results in degradation of the arenediazonium cation to give an aryl radical and a nitrogen molecule and in the formation of the EY+• radical cation. The EY+• radical cation generated in this way serves as an oxidant for the arylsulfinate anion, which is thus converted to the  radical. The recombination of Ar• and
radical. The recombination of Ar• and  radicals gives rise to sulfones (Scheme 33). There are examples of C–S bond formation upon coupling of sulfonyl radicals with stabilized tertiary alkyl radicals to give sulfones.
101
radicals gives rise to sulfones (Scheme 33). There are examples of C–S bond formation upon coupling of sulfonyl radicals with stabilized tertiary alkyl radicals to give sulfones.
101
Scheme 33
Download figure:
The generation of aryl radicals upon the photoinduced decomposition of diaryliodonium salts involving eosin Y and the subsequent reactions of the radicals with organic thiosulfates were used for the synthesis of aryl sulfides and aryl sulfoxides. The reaction selectivity was determined by the chosen conditions: the reaction carried out in an inert atmosphere under white light irradiation gave aryl sulfides, whereas green light irradiation in air promoted the formation of sulfoxides. 102
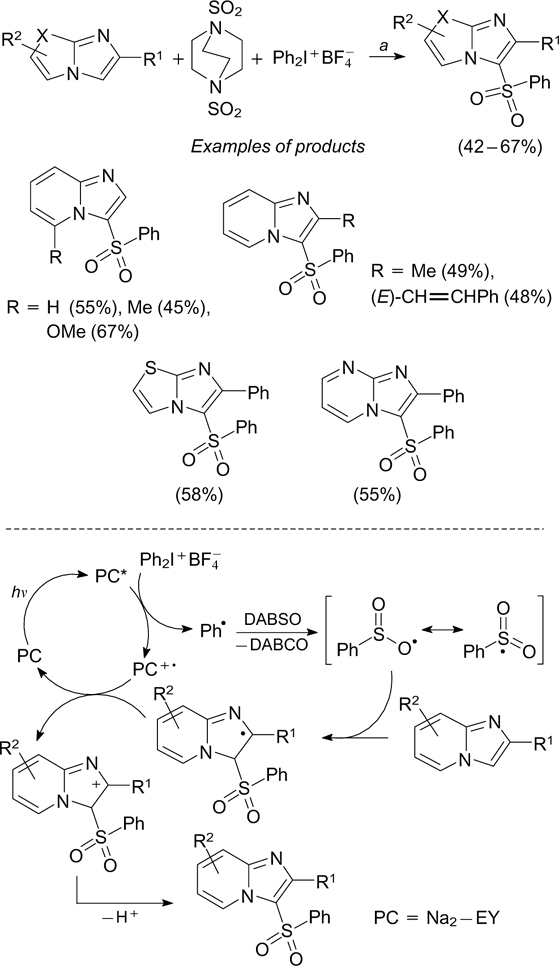
In recent years, DABSO, the complex of 1,4-diazabicyclo[2.2.2]octane and sulfur(IV) oxide, has become a popular reagent in the practical organic synthesis as a stable, easily handled substitute for toxic sulfur dioxide gas. 103 – 105 In particular, the three-component reaction of diphenyliodonium salt, DABSO and substituted imidazopyridines induced by green light irradiation in the presence of the disodium salt of eosin Y (Na2-EY) results in the formation of 3-sulfonylimidazopyridines in satisfactory yields (Scheme 34). 106
Scheme 34
Download figure:
One more alternative to sulfur dioxide used in organic synthesis is sodium or potassium pyrosulfite M2S2O5 (M = K, Na). 107 For example, photoinduced three-component reaction of arenediazonium tetrafluoroborate, thiourea and Na2S2O5 in MeCN under irradiation with CFL in the presence of rhodamine G results in the formation of S-aryl thiosulfonates in good yields. According to the mechanism postulated by the authors, the photoinduced decomposition of the diazonium salt leads to generation of an aryl radical, which is converted to arenesulfonyl radical upon the addition to sulfur dioxide. In parallel, the arenediazonium cation reacts with thiourea to give S-aryl isothiouronium salt, a source of the arenethiolate anion. The oxidation of this anion with a photoredox catalyst in the excited state gives rise to an arylthienyl radical, which recombines with the arenesulfonyl radical to give S-aryl thiosulfonate. 108 A related process is implemented when diaryliodonium salts, N,N-disubstituted hydrazines and K2S2O5 are allowed to react under irradiation with blue light in the presence of perylene derivative as a photosensitizer, which results in the formation of sulfonyl hydrazides. 109
There are examples of cascade radical reactions involving arenesulfonyl radicals, which are generated upon the addition of aryl radicals to a SO2 molecule. For example, 3-arenesulfonylquinolines were obtained by the green light-induced reaction of diaryliodonium salts, DABSO and N-arylpropargylamines in the presence of eosin Y. 110 Related cascade reactions that give C–S bond in the key step were used to prepare arylsulfonylcoumarins 111 and arylsulfonyl oxindoles. 112
The reaction of 3-trifluoromethylphenyl esters of arenesulfonic acids with styrenes under blue light irradiation at 427 nm in N,N-dimethylacetamide (DMA) in the presence of Cs2CO3 furnishes trans-vinyl sulfones. 113 According to the proposed mechanism, the arenesulfonyl radical generated from arenesulfonate is the key intermediate in the formation of the C–S bond. In the first stage, the electron donor – acceptor complex is formed from arenesulfonate and a solvent molecule. The photoexcitation of this complex initiates a single-electron transfer from the DMA molecule to arenesulfonate, thus inducing S–O bond cleavage and giving an arenesulfonyl radical. The addition of this radical to styrenes to give benzylic radical and the subsequent elimination of the hydrogen atom yield vinyl sulfones (Scheme 35). The reactions of arenesulfonic acid esters with vinylheteroaryls proceed in a similar way.
Scheme 35
Download figure:
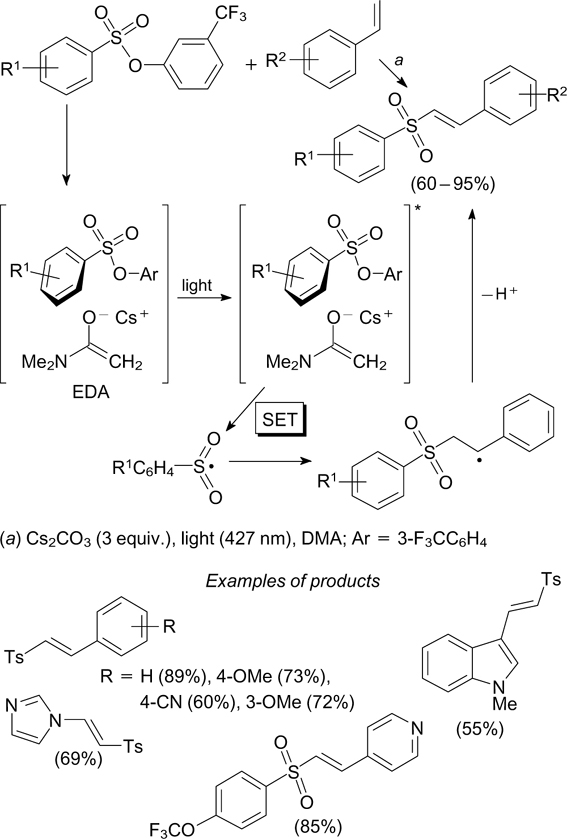
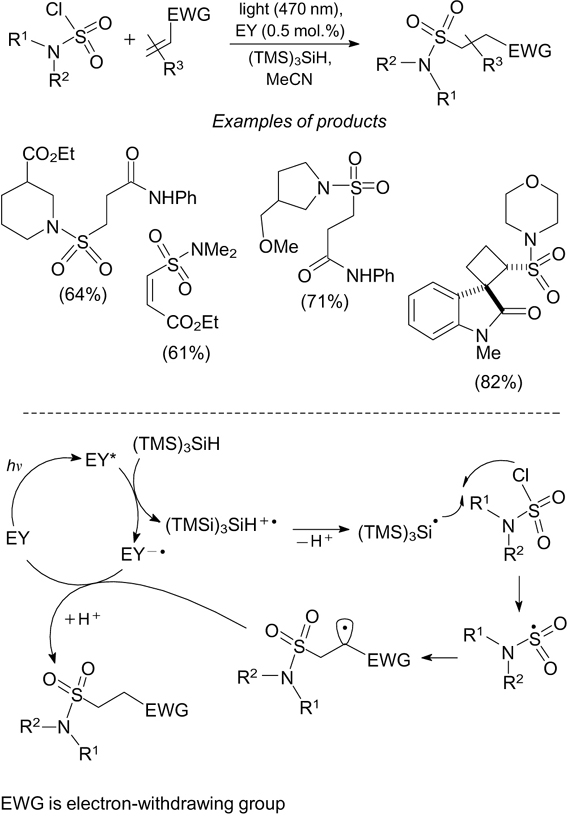
A method for activation of sulfamoyl chlorides to form the corresponding sulfamoyl radicals is based on elimination of a chlorine atom from the substrate under the action of tris(trimethylsilyl)silyl radical. 114 The latter radical is generated by photoinduced oxidation of tris(trimethylsilyl)-silane followed by deprotonation in the presence of eosin Y on exposure to blue light at 470 nm. The sulfamoyl radicals generated in this way were used in the addition reactions to compounds with multiple bonds to obtain sulfonamides (Scheme 36).
Scheme 36
Download figure:
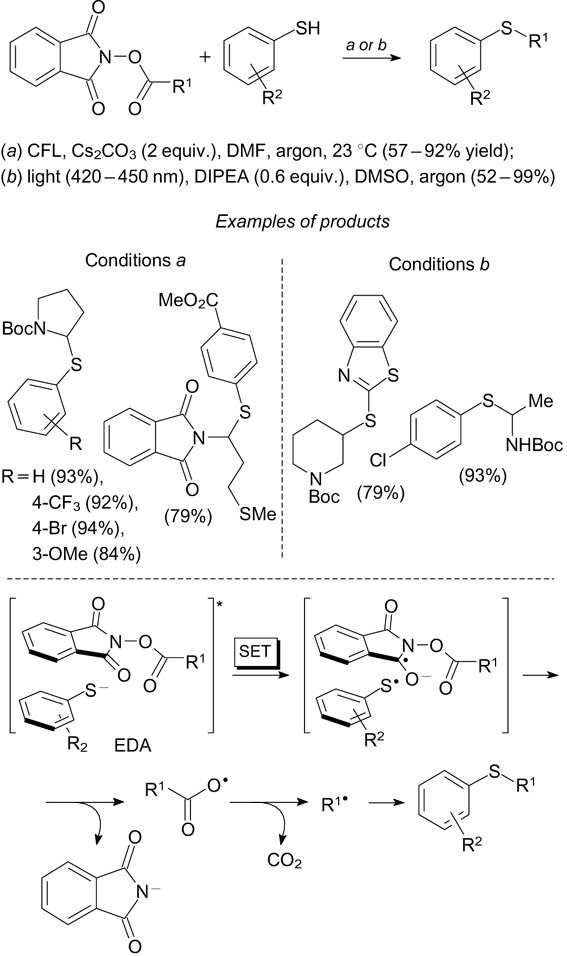
A number of methods have been developed to insert a C–S bond into the molecule via transformations of functional groups present in the molecule that contain no sulfur atoms, e.g., carboxyl, carbonyl and amino groups. For example, reactions of redox-active N-(acetoxy)phthalimides, 59, 60 which are readily formed from the corresponding carboxylic acids and N-hydroxyphthalimide, with thiophenols in the presence of Cs2CO3 or diisopropylethylamine under visible light irradiation furnish the products of decarboxylative S-alkylation of thiophenols. 115, 116 This reaction proceeds without a photoredox catalyst via the preliminary formation of the electron donor–acceptor complex from the oxyphthalimide derivative and thiophenolate anion. The single-electron transfer from the thiophenolate anion to N-(acetoxy)phthalimide is accompanied by elimination of the phthalimide ion and generation of a carboxyl radical, which is decarboxylated to be converted to an alkyl radical. Coupling of the alkyl radical with an arylthiyl radical produces S-alkylthiophenols. An important synthetic application of this method is related to modification of natural amino acids to the corresponding sulfides via the intermediate formation of N-(acetoxy)phthalimides (Scheme 37). This approach is compatible with a number of protecting groups used in amino acid chemistry.
Scheme 37
Download figure:
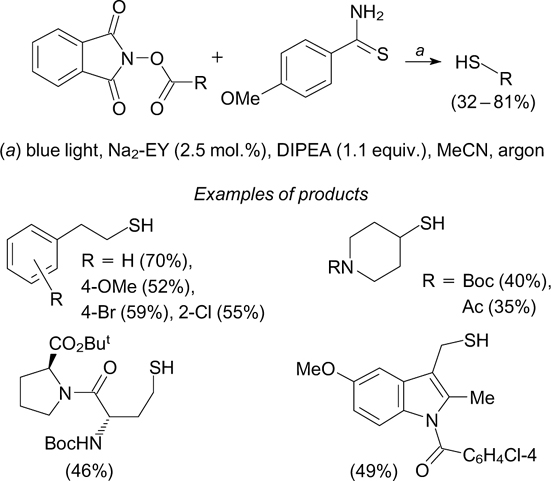
A similar reaction of the redox-active N-(acetoxy)-phthalimides with 4-methoxythiobenzamide under blue light irradiation in the presence of the disodium salt of eosin Y and DIPEA furnishes alkanethiols (Scheme 38). 117
Scheme 38
Download figure:
Yet another way for the introduction of sulfur functional groups into amino acid molecules is based on the visible light-induced deaminative thioesterification of thiobenzoic acid via the preliminary formation of 1-alkyl-2,4,6-tetraphenylpyridinium tetrafluoroborate (Katritzky salts 118 ) (Scheme 39). 119 The reaction also gives initially an electron donor – acceptor complex of the pyridinium cation and thiobenzoic acid anion. Upon the absorption of visible light, it is brought to the excited state and undergoes a single-electron transfer; elimination of 2,4,6-triphenylpyridine gives rise to two radicals: thienyl and alkyl radicals. Coupling of these radicals finally gives S-alkyl derivatives of thiobenzoic acid.
Scheme 39
Download figure:
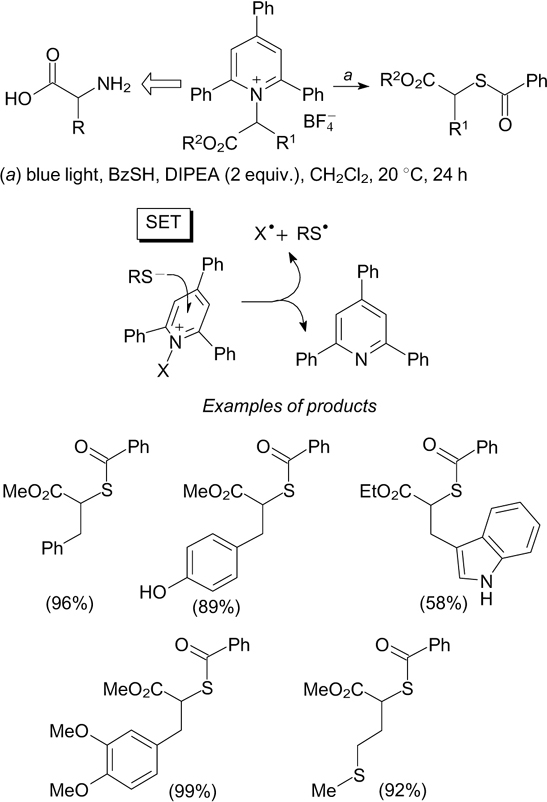
Photochemical transformations involving Hantzsch esters (4-alkyl-1,4-dihydropyridines), which are readily obtained from alkyl aldehydes, can also serve for the generation of C–S bond. 120 The reaction of 4-alkyl-1,4-dihydropyridines with S-aryl(alkyl)thiosulfonates induced by blue light irradiation in the presence of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as a photoredox catalyst under inert atmosphere affords sulfides. When this reaction is conducted in air, with other conditions being the same, sulfoxides can be prepared (Scheme 40). 121
Scheme 40
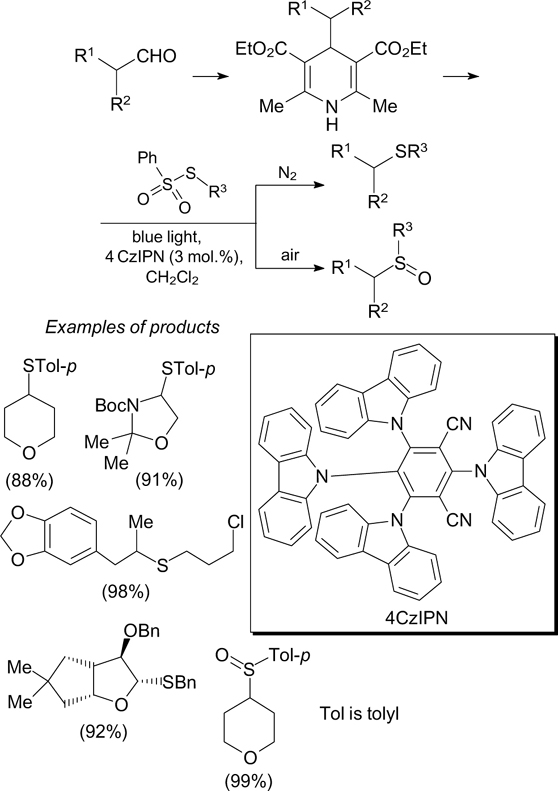
Download figure:
Several methods for the construction of C–S bond without transition metal catalysis were developed using aryl halides as the starting compounds. These methods are a good alternative to the approaches using transition metal-based catalysts, and, in most cases, they are favourably distinguished by environmental safety and cost efficiency. Diaryl sulfides were prepared by the visible light-induced cross-coupling of aryl halides with thiols without a photoredox catalyst. 122, 123 This reaction proceeds most efficiently for aryl bromides and iodides (Scheme 41). Conducting the reaction in the presence of aliphatic compounds capable of generating a stable radical upon hydrogen atom elimination furnishes aryl alkyl sulfides. In this case, aryl halides act as auxiliary reagents generating an aryl radical, which then detaches a hydrogen atom from the aliphatic substrate (HAT, see Scheme 41). 124
Scheme 41
Download figure:

This method for C–S bond formation was used for C–H sulfenylation of nitrogen-containing heterocycles. First, iodination of heterocyclic compounds with the I2 – TBHP system was carried out, and then the iodo derivative generated in situ was coupled with thiophenols in the presence of a base under visible light irradiation. This two-step one-pot procedure resulted in the synthesis of 4-arylthioisoquinolines, 3-arylthioquinolines and 3-arylthio-7-azaindoles in high yields. 125
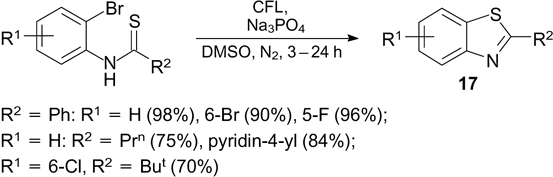
The cyclization of N-(2-bromophenyl)thioamides induced by CFL irradiation in the presence of Na3PO4 as a base has been reported. The method is applicable to the thioamides of various aromatic and aliphatic acids. Nearly quantitative yields of 2-substituted benzothiazoles 17 are attained in the cyclization of aromatic acid thioamides; the cyclization of thioamides of aliphatic acids is also fairly efficient (41 – 82% yields of products) (Scheme 42). 126
Scheme 42
Download figure:
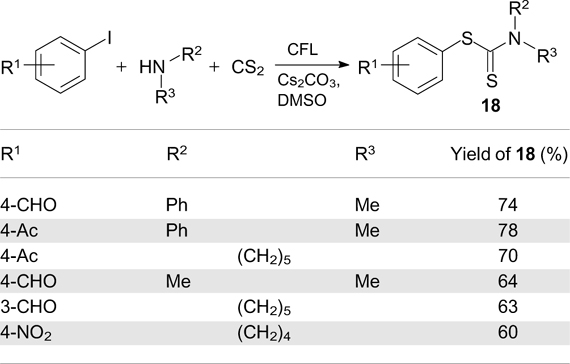
A one-pot synthesis of S-aryldithiocarbamates 18 is based on the reaction of secondary amines with carbon disulfide and aryl iodides carried out under irradiation with CFL in the presence of caesium carbonate (Scheme 43). 127 The reaction consists of two steps: first, the secondary amine and CS2 react to give an adduct, which reacts with aryl iodide via the preliminary formation of an electron donor – acceptor complex according to the mechanism considered above (see Scheme 41).
Scheme 43
Download figure:
The visible light-induced C–S coupling of aryl iodides with potassium ethyl xanthate was used to prepare thiochromanes by intramolecular cyclization. 128 The formation of an electron donor – acceptor complex and the subsequent photoinduced single-electron transfer serve as the key steps of coupling of aryl halides with sodium arylsulfinates on exposure to light at 365 nm, resulting in the formation of sulfones (Scheme 44). The highest yields of the coupling products are attained when aryl bromides and iodides were used. 129 A similar process takes place when a mixture of aryl halides and arylsulfinates is irradiated with light at 435 – 455 nm in the presence of diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (Hantzsch ester) and a base. The Hantzsch ester acts as an auxiliary reagent, which forms an electron donor – acceptor complex with aryl halide. On excitation with visible light, this complex undergoes a single-electron transfer and thus generates an aryl radical. 130
Scheme 44
Download figure:

The cross-coupling of aryl halides with S-nucleophiles of various types in the absence of photoredox catalysts usually gives high product yields in the case of aryl iodides and bromides with electron-withdrawing substituents and electron-deficient heterocyclic compounds. The coupling of halogenated electron-rich heterocycles with thiols requires the presence of a photoredox catalyst. Although iridium complexes are most efficient catalysts for this reaction, an organic photoredox catalyst based on isophthalonitrile, 4CzIPN, ensures the product formation in a relatively high yield and with high selectivity via selective coupling of dichloro-substituted substrate (Scheme 45). 131 The key step of formation of the C–S bond is redox-neutral homolytic aromatic substitution involving the thiyl radical generated from the corresponding thiols upon visible light-induced photochemical reaction involving 4CzIPN.
Scheme 45
Download figure:

2.3. Formation of C–P bond
The important applications of organophosphorus compounds for various fields of human life activity stimulates the continuous search for effective methods for their synthesis. In recent years, new methodological approaches to C–P bond formation by visible light-induced reactions have been developed. 132, 133 These reactions can be divided into two main types. In reactions of the first type, a phosphorus-containing nucleophilic reagent reacts with a highly reactive intermediate (C-radical or C-cation) generated via a photochemical transformation. In the second type of reactions, the P-radical generated via a photochemical transformation reacts with compounds containing carbon– carbon multiple bonds or aromatic systems. Many reactions resulting in the formation of C–P bonds are similar in their mechanisms to the reactions of C–N bond formation considered above.
Quint et al. 134 reported α-phosphorylation of tertiary N-arylamines with secondary phosphine oxides in the presence of N-alkoxypyridinium salt. The reaction is induced by visible light irradiation at 460 nm and conducted in the presence of a base. In the key step, the amine molecule and the pyridinium cation form an electron donor – acceptor complex, which undergoes single-electron transfer on exposure to light thus giving the tertiary amine radical cation. The elimination of a hydrogen atom from the radical cation gives key intermediate 19, an iminium cation, which subsequently reacts with secondary phosphine (Scheme 46).
Scheme 46
Download figure:

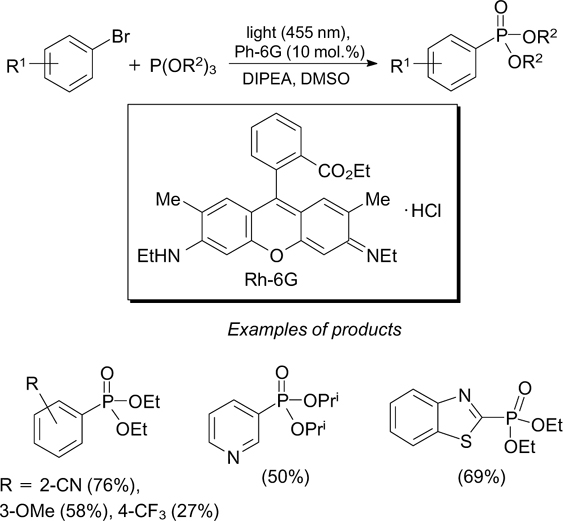
The reaction of aryl halides with trialkyl phosphites in the presence of DIPEA and rhodamine 6G (Rh-6G) as the photoredox catalyst induced by blue light irradiation at 455 nm gives rise to arylphosphonates. Electronically excited rhodamine 6G oxidizes DIPEA and is thus converted to the radical anion, which is able to reduce aryl halides to generate the corresponding aryl radicals (Scheme 47). 135 The reaction is efficient in the case of aryl halides and triflates of (hetero)arenes.
Scheme 47
Download figure:
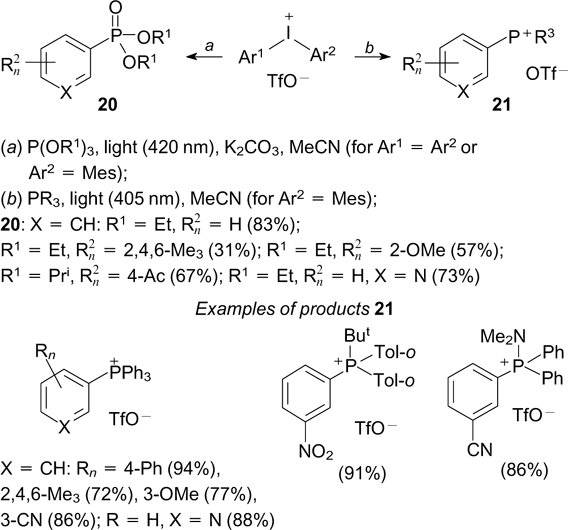
The P-arylation of trialkyl phosphites with diaryliodonium salts to give arylphosphonates is implemented upon blue light irradiation at 420 nm in the absence of photosensitizers. Presumably, the reaction proceeds through the preliminary formation of an electron donor – acceptor complex from the reactant molecules. The light-induced single-electron transfer from the phosphorus atom to the iodine atom in this complex finally gives the aryl radical and trialkyl phosphite radical cation. The subsequent recombination and elimination of the alkyl substituent gives arylphosphonates 20 (Scheme 48). 136 The successful arylation of the phosphorus atom can also be accomplished using unsymmetrical diarylphosphonium salts; in this case, the selectivity of the transfer of aryl substituent is controlled by steric factors. The arylation of tertiary phosphines involving aryl(mesityl)iodonium triflates affords structurally diverse quaternary arylphosphonium salts 21. This reaction was also implemented without an external photosensitizer and apparently proceeds via the formation of a photosensitive electron donor – acceptor complex. Unlike other known methods for the arylation of tertiary phosphines, the course of arylation involving ArMesIOTf is not affected by bulky substituents at the phosphorus atom (see Scheme 48). 137
Scheme 48
Download figure:
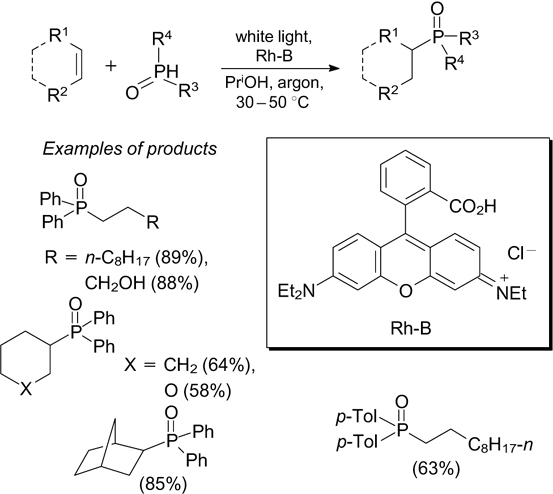
The synthesis of diaryl(heteroaryl)phosphine oxides is based on the reaction of aryl halides with diarylphosphine oxides in the presence of potassium tert-butoxide under blue light irradiation. 138 This procedure gives phosphine oxides in high yields when the substrates are bromides of electron-deficient heterocyclic compounds such as pyridines, quinolines, pyrimidines and pyrazines. The key step of the reaction is the formation of an electron donor – acceptor complex from heteroaryl halide and potassium tert-but-oxide molecules. Upon visible light excitation, single-electron transfer takes place in the complex, resulting in aryl and tert-butyl radicals. The latter participates in the generation of P-radical, which in turn recombines with the aryl radical to give the reaction product. 138 Analogous reactions can be carried out for alkylphosphinates when 9-mesityl-1-methylacridinium perchlorate is used as the photoredox catalyst and diphenyliodonium triflate serves as the oxidant. 139 The addition of secondary phosphines to non-activated alkenes takes place in the presence of rhodamine B (Rh-B) as the photoredox catalyst and under irradiation with white light in an argon atmosphere. 140 In the key step of this reaction, P-radical is generated via single-electron oxidation of the secondary phosphine with excited rhodamine B (Scheme 49). Secondary phosphine oxides containing strong electron-donating substituents do not react in this way.
Scheme 49
Download figure:
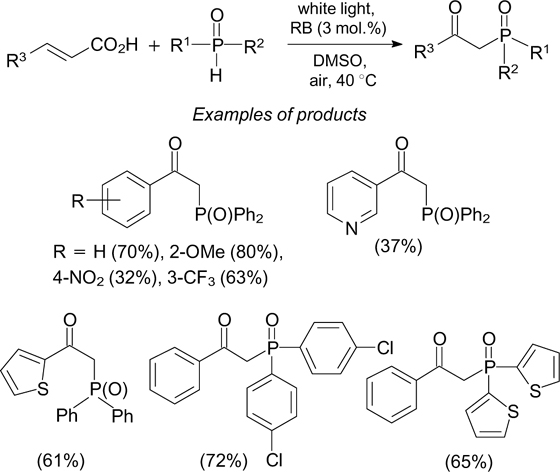
The reaction of cinnamic acids with diarylphosphine oxides under white light irradiation in air in the presence of rose bengal results in the formation of β-ketophosphine oxides via the decarboxylative oxidation (Scheme 50). 141 It is noteworthy that simple unsaturated carboxylic acids do not react in this way, nor secondary phosphines with potent electron-donating substituents. β-Ketophosphine oxides were also obtained upon the visible light-induced oxidative phosphinylation of terminal arylalkynes in an oxygen atmosphere using rhodamine B as the photoredox catalyst. 142
Scheme 50
Download figure:
The reaction of 1,1-disubstituted allyl alcohols with secondary phosphine oxides induced by irradiation at 450 nm in the presence of eosin Y gives β-aryl-γ-ketophosphine oxides. This product is formed as a result of 1,2-migration of aryl group in the radical intermediate arising during the reaction (Scheme 51). 143
Scheme 51
Download figure:

Direct C–H phosphorylation of thiazole derivatives with diarylphosphine oxides can be readily performed using eosin B (EB) 144 or eosin Y disodium salt 145 as the photoredox catalyst (Scheme 52).
Scheme 52
Download figure:
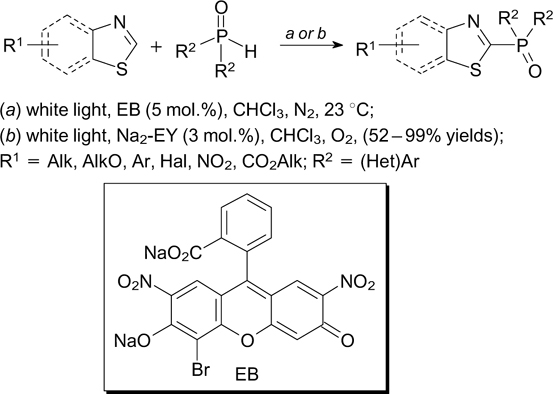
Similar C–H phosphorylation reactions were described for quinoxalin-2(1H)-ones, 146 coumarins, 147 2H-indazoles 148 and imidazopyridines 149 in the presence of oxidants and eosin B, eosin Y, rose bengal or rhodamine B, respectively. The C–H phosphorylation of quinolinones and coumarins in the presence of 1-ethoxy-2-methylpyridinium tetrafluoroborate was accomplished in the absence of external photoredox catalysts or oxidants. In this case, the role of photosensitizer was played by the substrates and reaction products. 150
The addition of P-radicals to multiple bonds forms the basis for the synthesis of (Z)-vinylphosphine oxides by the reaction of secondary phosphine oxides with terminal and internal alkynes in the presence of eosin Y in water under green light irradiation. 151 A similar reaction involving internal alkynes and diarylphosphine oxides induced by green light in the presence of 1-ethoxy-2-methylpyridinium tetrafluoroborate as the oxidant furnishes benzo[b]phosphole oxides. According to the reaction mechanism postulated by the authors, the donor–acceptor complex formed initially from eosin Y and pyridinium cation 22+ undergoes an intramolecular single-electron transfer upon irradiation, which leads to its fragmentation to give an ethoxy radical. The latter detaches a hydrogen atom from diarylphosphine oxide, thus generating a P-centred radical. The P-radical adds to a triple bond to give an alkenyl C-radical, which cyclizes to form a C–C bond and cyclohexadienyl radical, which is then oxidized with the pyridinium salt to the corresponding cation and is deprotonated (Scheme 53). 152
Scheme 53
Download figure:
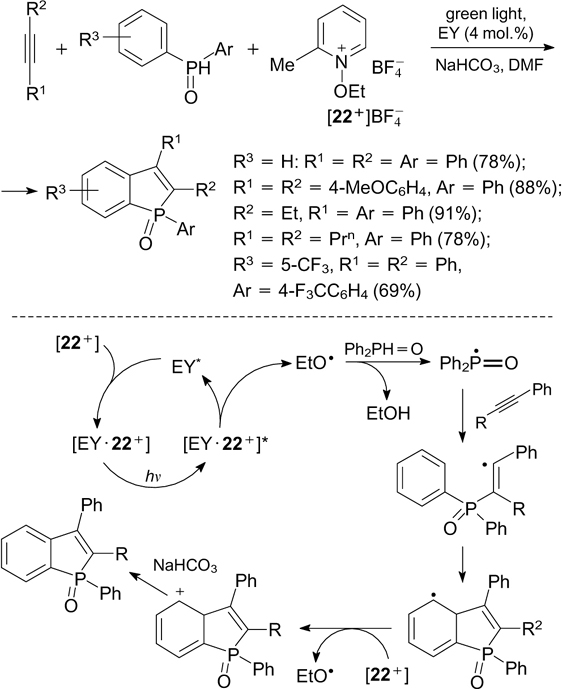
An analogous cyclization reaction initiated by the addition of P-radical to alkynes and finally giving 3-phosphorylated coumarins has been reported. 153
The reaction of 1,1-diarylethanols with secondary phosphine oxides catalyzed simultaneously by a protic acid and a photoredox catalyst (rhodamine B) furnishes (2,2-diarylvinyl)diarylphosphine oxides (Scheme 54). 154
Scheme 54
Download figure:

2.4. Formation of C–O bond
9-Mesityl-10-methylpyridinium perchlorate was used as the photoredox catalyst for the visible light-induced hydration of alkenes, which follows the anti-Markovnikov pathway in the presence of diphenyl disulfide. 155 The mechanism of this reaction (considered in relation to styrene) starts with the oxidation of alkenes to radical cation 23, which directly participates in the C–O bond formation via the intermediate formation of hydroxyalkyl radicals 24a,b (Scheme 55).
Scheme 55
Download figure:
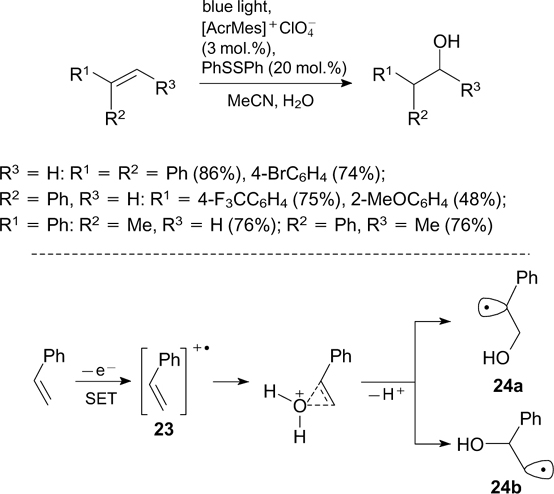
The intermolecular anti-Markovnikov addition of carboxylic acids to alkenes 156 and intermolecular and intramolecular cyclizations giving heterocyclic compounds 157, 158 take place under similar conditions.
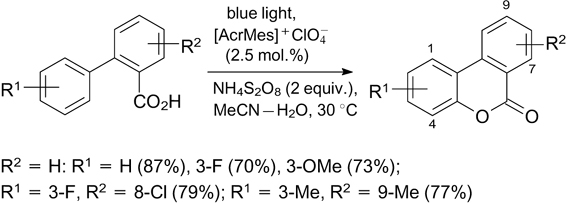
The  photocatalyst was also used for CH-lactonization of o-arylbenzoic acids giving benzo-3,4-coumarins.
159
According to the mechanism proposed by the authors, the intramolecular formation of C–O bond takes place as a result of 6-endo-trig-cyclization of the carboxyl radical generated upon elimination of a hydrogen atom from the carboxyl group (Scheme 56; positions of substituents in the product are indicated). It is noteworthy that in the case of R1=3-OMe, R2 = H, the reaction gave 2- and 4-methoxy isomers (92 : 8). A similar CH-lactonization preceded by E–Z-isomerization was described for (E)-cinnamic acid upon irradiation with light at 402 nm in the presence of (–)-riboflavin.
160
photocatalyst was also used for CH-lactonization of o-arylbenzoic acids giving benzo-3,4-coumarins.
159
According to the mechanism proposed by the authors, the intramolecular formation of C–O bond takes place as a result of 6-endo-trig-cyclization of the carboxyl radical generated upon elimination of a hydrogen atom from the carboxyl group (Scheme 56; positions of substituents in the product are indicated). It is noteworthy that in the case of R1=3-OMe, R2 = H, the reaction gave 2- and 4-methoxy isomers (92 : 8). A similar CH-lactonization preceded by E–Z-isomerization was described for (E)-cinnamic acid upon irradiation with light at 402 nm in the presence of (–)-riboflavin.
160
Scheme 56
Download figure:
The hydroxylation of imidazopyridines with primary and secondary alcohols is carried out by irradiation of a reactant mixture with white (visible) light in the presence of rose bengal. 161 This reaction is suitable for introducing both primary and secondary alkoxyl substituents, in particular containing halogen atoms, carbon–carbon multiple bonds and benzyl groups, into position 3 of product 25. The C–O bond is formed upon the addition of an alcohol molecule to the radical cation, which is initially generated from imidazopyridine via single-electron oxidation with rose bengal in the electronically excited state (Scheme 57). This method is also effective for hydroxylation of benzimidazothiazoles.
Scheme 57
Download figure:

One of approaches to the synthesis of cyclic ethers and lactones by cyclization of alcohols is based on visible light-induced generation of benzyl radicals. The authors proposed a mechanism according to which the initial reaction of electronically excited 2,4,6-triphenylpyrylium trifluoroborate with the substrate gives the C-radical cation via single-electron oxidation. The presence of a positive charge in the aromatic ring considerably increases the CH acidity of the benzylic position. The deprotonation of the radical cation results in generation of the benzyl radical (Scheme 58). 162
Scheme 58
Download figure:
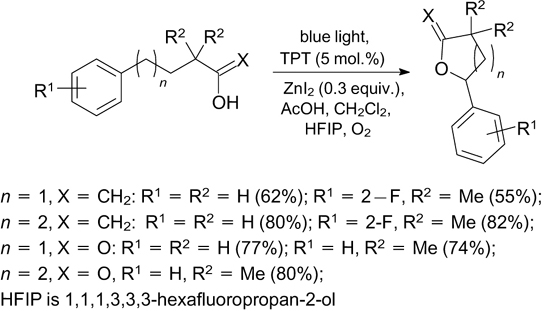
The photooxidation of the benzylic position of diarylmethanes with oxygen proceeds via a similar step of generation of the benzyl radical; the expected ketones are formed when 9-mesityl-10-methylpyridinium perchlorate and blue light irradiation are used. 163
The synthesis of furans 26 is based on oxidative cyclization of 1,3-dicarbonyl compounds and alkynes. 164 The reaction occurs when the reaction mixture is irradiated with blue light in the presence of methylene blue as a photoredox catalyst and ammonium persulfate as an oxidant. The mechanism of furan ring formation implies the initial radical generation from 1,3-dicarbonyl compound, the subsequent radical addition to alkyne to give vinyl radical and cyclization of the latter (Scheme 59).
Scheme 59
Download figure:

3. Electrochemical reactions
The oxidative cross-coupling is a strategy extensively used to form C–heteroatom bonds from accessible precursors. In the last two decades, huge research efforts have been concentrated in this field and great advances have been made. However, the formation of chemical bonds usually requires stoichiometric amounts of oxidants. Currently, considerable interest of the chemical community is attracted by oxidative cross-coupling reactions in the absence of external oxidants. Electrochemical synthesis is a potent and environmentally safe approach that not only provides the oxidative formation of chemical bonds without external oxidants, 165 – 177 but, in some cases, gives an additional valuable product, that is, hydrogen gas. 178
3.1. Formation of C–N bond
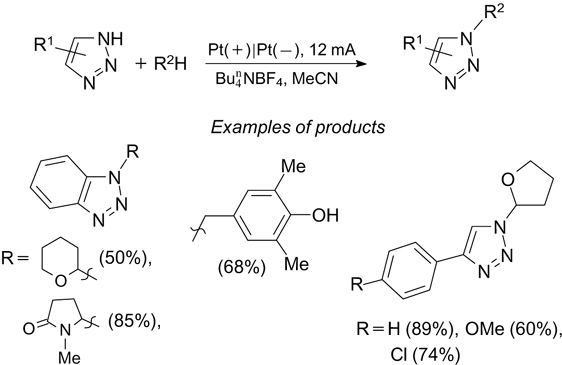
In recent years, electrochemical oxidation 179, 180 has been used as a versatile and environmentally safe alternative synthetic tool for the formation of chemical bonds by direct C–H-activation. 181 – 188 Among direct oxidative electro-chemical amination reactions of C–H bonds, 189 – 198 the C(sp3)–H/N–H cross-coupling involving the C(sp3)–H bond is used most rarely. 199 An example of this type is anodic oxidative coupling of azoles with tetrahydrofuran and some related aliphatic compounds (Scheme 60 † ). 199
Scheme 60
Download figure:
One more example of the C(sp3)–H/N–H cross-coupling is the electrochemical amination of the benzylic carbon atom of alkylbenzene with H2 evolution (Scheme 61). 200 This strategy implies anodic cleavage of the benzylic C–H bond to give a carbocationic intermediate, which is trapped by nitrogen-containing nucleophile, giving rise to the C–N bond.
Scheme 61
Download figure:

A new procedure for the preparation of 9-azoloxanthenes 27 by electrochemical C(sp3)–H/N–H cross-coupling of xanthenes with azoles was reported (Scheme 62). 201
Scheme 62
Download figure:

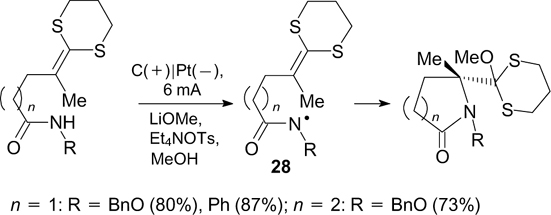
The amidyl radical, which is generated from amides under mild electrooxidative conditions, can also serve as the source of nitrogen in such processes. For example, the cyclization of radicals 28 involving an electron-rich double bond gives five- and six-membered rings (for example, lactams) (Scheme 63). 202
Scheme 63
Download figure:
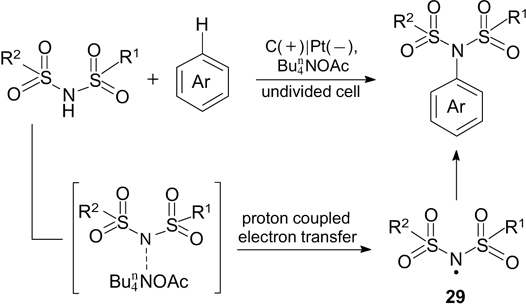
A similar reaction was used for regioselective electrochemical oxidative intermolecular cross-coupling of aromatic compounds with sulfonimides, which proceeds via the addition of the imidyl N-radical. 203 This reaction can be carried out for diverse arenes and heteroarenes (Scheme 64).
Scheme 64
Download figure:

Study of the mechanism of this reaction by cyclic voltammetry indicates that the imidyl radical 29 is generated as a result of proton coupled electron transfer (PCET) upon simultaneous action of tetra-n-butylammonium acetate and anodic oxidation (Scheme 65).
Scheme 65
Download figure:
A method for the synthesis of formamides by decarboxylative N-formylation of primary and secondary aromatic and aliphatic amines in the presence of glyoxalic acids has been developed. The reaction proceeds in an undivided cell with platinum electrodes. A variety of functional substituents (F, Cl, Br and I atoms; OCF3, CN, CF3, NO2, CO2Et and Ac groups) are tolerant to the reaction conditions, and the yields of products vary from 38 to 93%. 204
In recent years, the electrochemical oxidative amination methodology has also been used to prepare nitrogen-containing heterocyclic compounds via the formation of the C–N bond. 205 In particular, the preparation method for functionalized tetracyclic benzimidazoles 30 is based on the formation of N-radicals as a result of anodic cleavage of the N–H bond and the subsequent intramolecular cyclization of the N-radicals (Scheme 66). 206 This atom-economic C–H/N–H-cross-coupling reaction is suitable for substrates with diverse functional groups and is characterized by high regioselectivity.
Scheme 66
Download figure:

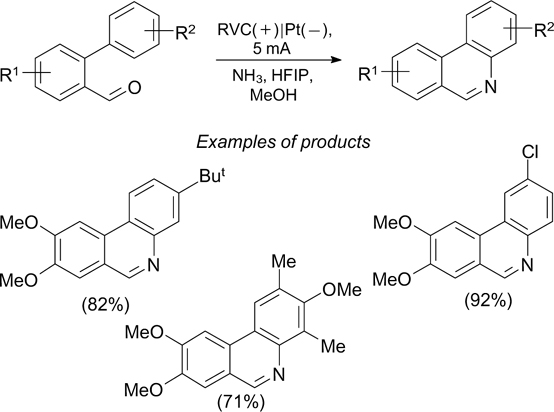
Yet another example of electrochemical amination is the synthesis of phenanthridines and related heterocyclic compounds from biaryl aldehydes and ammonia by C–H/N–H-cross-coupling (Scheme 67). 207
Scheme 67
Download figure:
The electrochemical intermolecular amination was also used to prepare derivatives of nitrogen-containing fused systems — 10H-benzo[4,5]imidazo[1,2-a]indoles 31 (Scheme 68). 208
Scheme 68
Download figure:

The electrochemical dehydrogenative amination of the C–H bond served for the synthesis of N-benzenesulfonylcarbazoles (Scheme 69). 209
Scheme 69
Download figure:
The oxidative cyclization of hydrazones accompanied by C–N bond formation under the action of electrical current makes it possible to prepare 1,2,4-triazolo[4,3-a]pyridines and related heterocyclic systems from α-hydrazinoazines and aliphatic or (hetero)aromatic aldehydes (Scheme 70). 210 The process can be carried out in the one-pot version without isolation of the presynthesized hydrazones. This method was applied for the synthesis of a commercial anxiolytic drug Xanax (45% yield).
Scheme 70
Download figure:

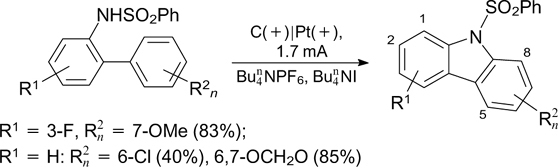
Apart from being used for the synthesis of heterocyclic structures, electrochemical amination is equally widely employed to prepare anilines and heteroarylamines upon intermolecular cross-coupling reactions. A number of 3-amino derivatives of imidazo[1,2-a]pyridines were synthesized by electrochemical oxidative C–H/C–N-cross-coupling, with (benzo)imidazoles being used as the aminating agents (Scheme 71). 208 The corresponding amination products were also obtained in the case of morpholine, while attempts to carry out this reaction for n-butylamine were unsuccessful. According to the mechanism proposed by the authors, the product of amination is formed as a result of recombination of the N-radical, generated by anodic oxidation of deprotonated imidazole, with the C-radical, which is formed upon anodic oxidation of imidazopyridine and subsequent deprotonation.
Scheme 71
Download figure:

The dehydrogenative C–H/C–N-cross-coupling of imidazopyridines with diarylamines of this type results in the regioselective formation of triarylamines (Scheme 72). 211
Scheme 72
Download figure:
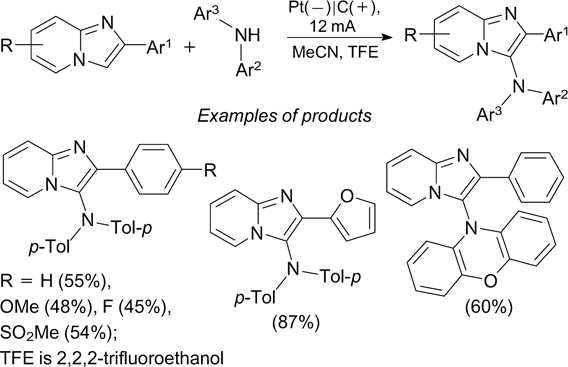
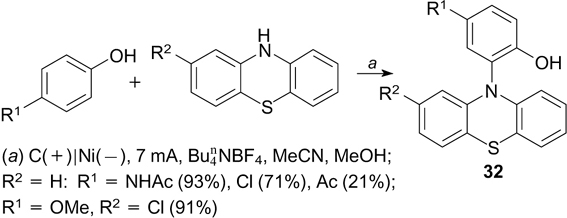
The anodic oxidation of phenothiazines giving the corresponding radical cations is the key step of the electrochemical oxidative amination of phenols with phenothiazines, which gives products 32 (Scheme 73). 212 The amination proceeds most efficiently for phenols containing electron-donating groups, but it has low sensitivity to the electronic nature of substituents in phenothiazines. The amination of phenols with phenoxazine gives products in comparable yields.
Scheme 73
Download figure:
Phenothiazines were also used as aminating agents for N,N-disubstituted anilines. The reaction occurs selectively involving the para-position of anilines and results in 10-aryl-10H-phenothiazines (Scheme 74). 213 Presumably, the process includes the electrochemical oxidation of anilines to give the corresponding radical cations, anodic oxidation of phenothiazines generating N-radicals and the subsequent radical recombination and deprotonation reactions.
Scheme 74
Download figure:
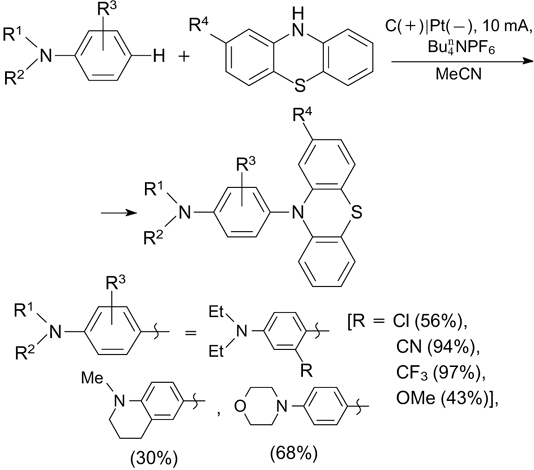
Morofuji et al. 189 developed a C–H-amination method for aromatic compounds based on their electrochemical oxidation in the presence of pyridine (divided H-cell, 10 mA). First, N-arylpyridinium salts are formed and then they are reacted with piperidine (pPy), which gives arylamines (Scheme 75). The key step of the reaction is anodic oxidation of aromatic systems to yield radical cations and cations.
Scheme 75
Download figure:
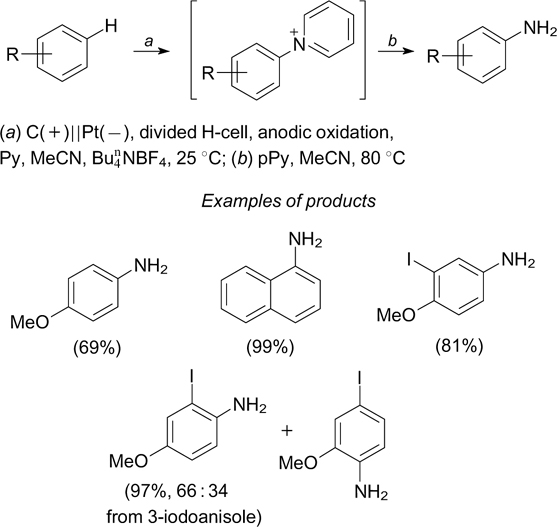
The electrochemical method for the synthesis of anilines by amination of arenes with aliphatic azides accompanied by C–C bond cleavage also includes anodic oxidation of the aromatic ring to give the corresponding radical cations. The reaction is carried out in an undivided electrochemical cell with graphite electrodes. 214
The reaction reported by Malviya et al. 215 does not fit into the classification of electrochemical amination methods accepted in this review. This is electrochemical oxidative cross-coupling of amines and aliphatic isonitriles, which affords carbodiimides in good yields (Scheme 76).
Scheme 76
Download figure:
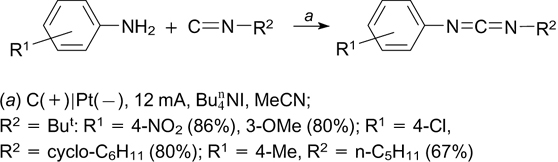
3.2. Formation of C–S bond
Electrochemical methods for the formation of C–S bond occupy an important place in the chemistry of organic sulfur compounds; however, virtually all examples of this type refer to cross-coupling of sulfur-containing reagents with unsaturated compounds. For example, Yang et al. 216 described a simple and efficient electrochemical thiocyanation of enaminones in an undivided electrochemical cell. This gives polyfunctionalized alkenes and chromenes in moderate to high yields with KSCN being used as the thiocyanating agent (Scheme 77).
Scheme 77
Download figure:
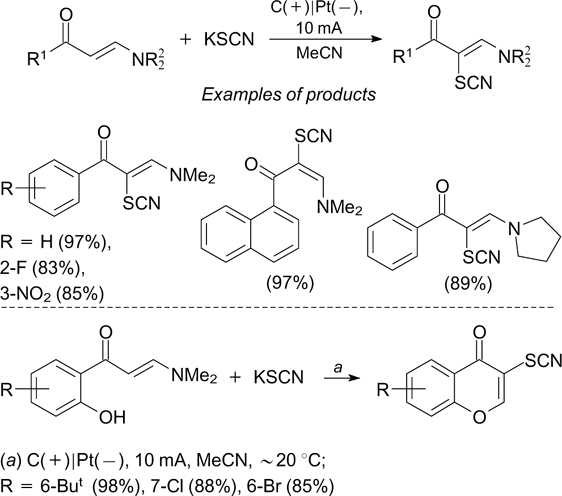
The electrochemical anodic thiocyanation involving inorganic thiocyanates was used for regioselective synthesis of thiocyanated derivatives of pyrrole, indole, pyrazole, isoxazole 217 and pyrazolo[1,5-a]pyrimidine. 218, 219
A stereoselective method for the synthesis of (E)-vinyl sulfones is based on the electrochemical oxidative cleavage of the N–S bond in aromatic sulfonylhydrazines followed by cross-coupling with cinnamic acids, which results in the formation of the C–S bond (Scheme 78). 220
Scheme 78
Download figure:
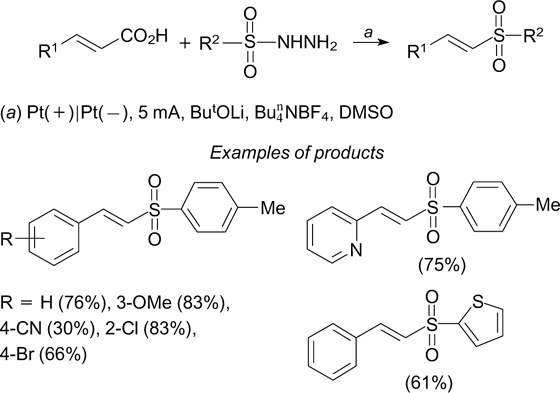
An electrochemical synthesis of vinyl thiocyanates from cinnamic acids and NH4SCN has been developed. The reaction proceeds in an undivided cell with platinum electrodes. 221
Electrochemical C(sp2)–H-sulfenylation of enaminones with thiophenols to give substituted vinylthiols in up to 87% yields is also known (Scheme 79). 222 It is notable that thiophenols are selectively coupled with enaminones, even if the latter contain other alkenyl groups.
Scheme 79
Download figure:
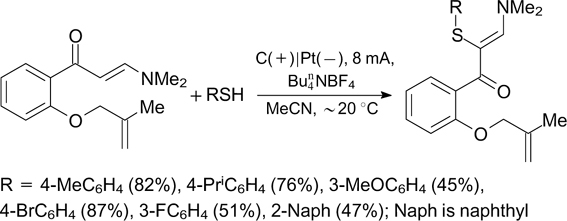
In addition, these products can be easily converted to α-sulfur-containing isoxazoles (Scheme 80), which exhibit various types of biological activity. 222
Scheme 80
Download figure:
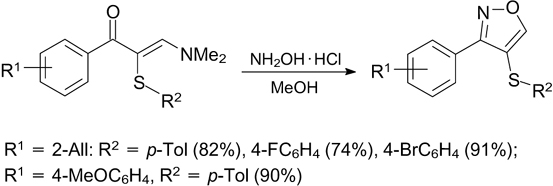
This is not the only example of application of compounds containing an enamine moiety in cross-coupling reactions to form C–S bonds. A method for electrochemical oxidative coupling of enamines with thophenols and hetarylthiols has been developed (Scheme 81). 223
Scheme 81
Download figure:
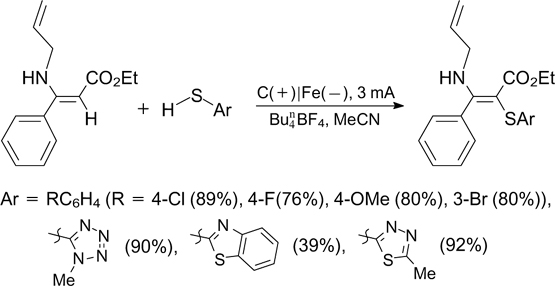
Mo et al. 224 reported electrochemical C(sp)–H/S cross-coupling of terminal alkynes and sulfonyl hydrazides to form alkynyl sulfones on a reticulated vitreous carbon electrode (Scheme 82). Most of products synthesized in this way possess antitumour activities on HeLa cells.
Scheme 82
Download figure:
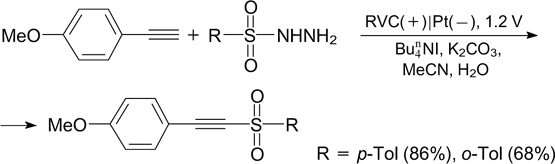
In the electrochemical method for the synthesis of β-ketosulfones (51 – 90% yields) from substituted terminal alkynes or aryl methyl ketones, sodium arylsulfinates served as sources of S-radicals. The electrolysis of the reaction mixture was carried out at a constant current of 40 mA using a steel cathode and a graphite anode. 225
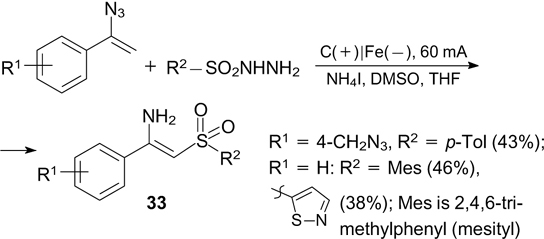
A method for the synthesis of sulfonyl-substituted enamines with a free amino group has been developed. 226 The sulfonylation of vinyl azides involves the intermediate formation of electrophilic iodine-containing intermediates, which oxidize sulfonyl hydrazides to the corresponding S-radicals. The yields of products 33 reach 87% (Scheme 83).
Scheme 83
Download figure:
Terent'ev et al. 227 performed the sulfonylation of alkenes in which sulfonyl hydrazides served as the source of sulfonyl radicals. The reaction was carried out in an undivided cell with a C anode and an Fe cathode; the yields of vinyl sulfones were 25 to 95%. This reaction also proceeds in the presence of a number of functional groups in the substrate (F, Cl, Br, I, NO2, OMe).
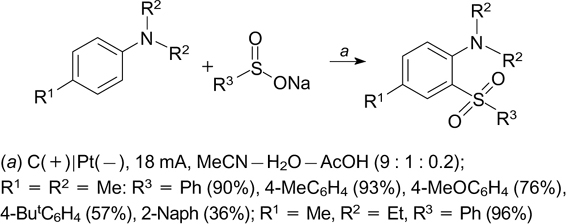
In most of the reported examples of oxidative electrochemical C(sp2)–H/S cross-coupling reactions, (hetero)-aromatic compounds were used both for introduction of sulfur-containing functional groups in the molecules and for the intramolecular cyclizations to give fused sulfur-containing heterocycles. Various sodium sulfinates were utilized in the electrochemical oxidative C–H sulfonylation of anilines, which gave the target o-aminoaryl sulfones in up to 96% yields (Scheme 84). 228
Scheme 84
Download figure:
Benzothiophenes with an arenesulfonyl group in position 3 were prepared using sodium sulfinates and 2-alkynylthioanisoles. 229 The reaction was carried out in an undivided electrochemical cell with C(+) and Pt(–) electrodes at a constant current of 8 mA. A variety of functional groups (F, Cl, Br, I, CO2Me) were tolerant to these reaction conditions; the yields of products varied from 45 to 85%.
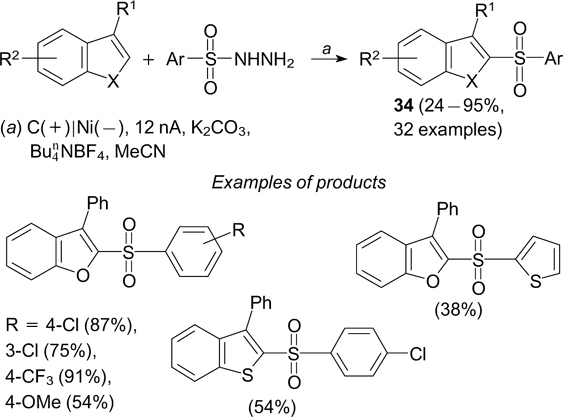
Heteroaryl sulfones 34 can also be obtained via radical electrochemical sulfonylation of C–H bonds in heteroarenes with arenesulfonyl hydrazides (Scheme 85). 230
Scheme 85
Download figure:
The selective oxidative sulfenylation of imidazopyridines is efficiently attained by using both arene- and alkanethiols (Scheme 86). 231
Scheme 86
Download figure:
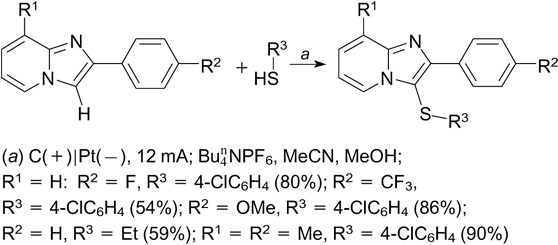
Preliminary quantum chemical calculations for a similar reaction of indole derivatives demonstrated that the electrochemically generated aryl radical cation is the key intermediate (Scheme 87). 232
Scheme 87
Download figure:
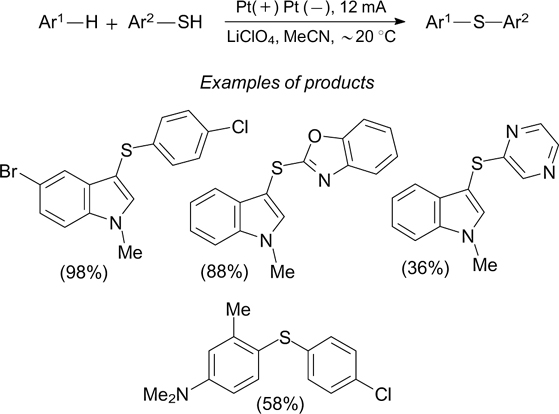
A method for direct thiolation of arenes containing, in particular, alkoxy groups on treatment with aromatic thiols has been developed. 233 It should be emphasized that the product yields do not depend on the steric factors of either arenes or thiols: for example, the yield of the product in the reaction of MesH with p-TolSH is 95%, while for MeOPh and 2,6-Me2C6H3SH, the yield is 99%. The highest yield of reaction products is attained with platinum electrodes in the constant-current electrolysis at 5 mA.
The sulfonylation of 2H-imdazoles into position 3 was carried out using sulfonyl hydrazides as the source of sulfonyl radicals. 234 This reaction proceeds in the presence of air oxygen at room temperature; it can be scaled up to gram amounts and is characterized by high tolerance to the presence of several functional groups (F, Cl, NO2, OMe) in the substrate. The electrolysis is conducted in an undivided cell at a constant current of 10 mA with a graphite anode and a nickel foam cathode. Similar products can be obtained by sulfonylation of 2H-indazoles with aryl- and alkylsulfinic acids in an undivided cell with C(+) and Pt(–) electrodes at 7 mA. 235
Three-component cross-coupling using thiocyanates as the source of sulfur atoms and methanol as the methylating agent formed the basis of the strategy of electrochemically induced regioselective thiomethylation of anilines, anisole, indole and indolizine (Scheme 88). 236
Scheme 88
Download figure:

Under these conditions, many functional groups in the substrate are retained. This method was used for regioselective C(3)-thiomethylation of imidazopyridines (Scheme 89). 237
Scheme 89
Download figure:
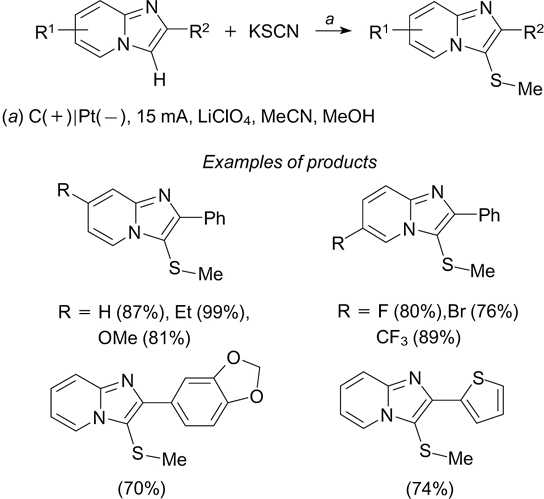
A related sulfonylation – heteroarylation process involving sulfinic acids and alkenes is implemented via the step of ipso-migration of the heteroaryl moiety. 238
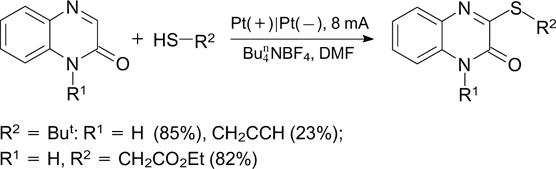
Zhou et al. 239 described an electrochemical method for C(sp2)–H-functionalization of quinoxalin-2(1H)-ones with primary, secondary and tertiary thiols giving 3-alkylthioquinoxalin-2(1 H)-ones (Scheme 90).
Scheme 90
Download figure:
A series of substituted 2-aminobenzothiazoles 35 were prepared by electric current-induced intramolecular dehydrogenative C–H/C–S coupling involving N-aryl-N', N'-dialkylthioureas, which are formed upon the reaction of secondary amines with aryl isothiocyanates (Scheme 91). 240
Scheme 91
Download figure:
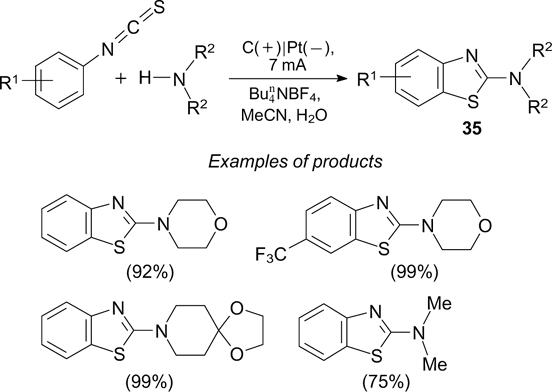
The mechanism proposed for this reaction is depicted in Scheme 92 in relation to the formation of 3-morpholinobenzothiazole (35a).
Scheme 92
Download figure:

The nucleophilic addition of morpholine to isothiocyanate yields a thiourea derivative, which is then deprotonated with the hydroxide anion, generated by the cathodic reduction of water, thus giving ionic intermediate 36. The anodic SET oxidation of intermediate 36 produces radical 37, which rapidly undergoes intramolecular cyclization to be converted to intermediate 38. The subsequent anodic oxidation and deprotonation of this intermediate furnishes the target product 35a.
In the presence of sodium benzoate, the synthesis of benzothiazoles from N-arylthioamides can be carried out under the same conditions (Scheme 93). 240
Scheme 93
Download figure:
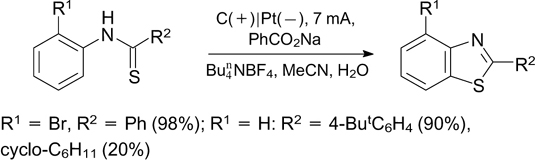
In addition, there is known TEMPO-catalyzed intramolecular cyclization of N-(hetero)arylthioamides, resulting in benzothioazoles and thiazolopyridines. 241 The reaction is carried out in an undivided electrochemical cell at 10 mA with a glassy carbon anode and a platinum cathode.
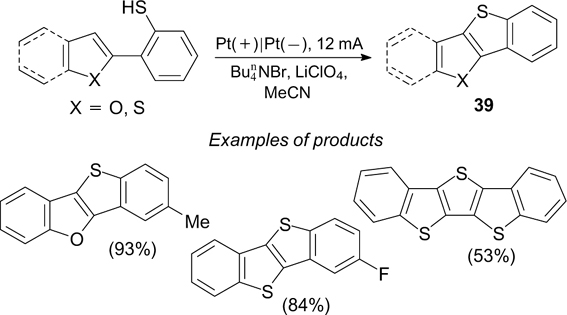
In the electrochemical dehydrogenative synthesis of fused systems 39 from sulfanylarenes, a significant factor is the addition of  , which catalyzes the reaction and serves as the source of bromide ions (Scheme 94).
242
, which catalyzes the reaction and serves as the source of bromide ions (Scheme 94).
242
Scheme 94
Download figure:
Benzo-annulated six-membered S-heterocycles were synthesized by intramolecular dehydrogenative C–S coupling with appropriate thioamides in a modular flow electrolytic cell (Scheme 95). 243 The electrolysis was conducted at a constant current of 35 mA; the flow cell was equipped with a Pt cathode and an anode made of graphite and poly(vinylidene fluoride) (PVDF).
Scheme 95
Download figure:

A similar electrochemical aminoselenation of styrenes is based on participation of secondary amines, imides and azoles as aminating agents in the presence of diselenides. The reaction was carried out in an undivided electrochemical cell equipped with C(+) and Pt(–) electrodes. 244
3.3. Formation of C–P bond
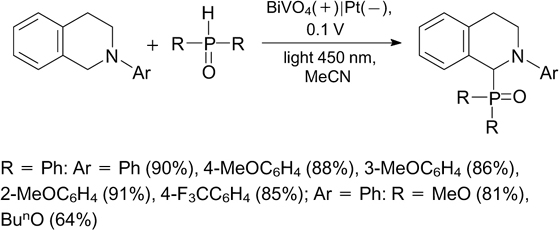
An original method for simultaneous of photo- and electrochemical processes was reported in 2019. 245 The authors designed a new photoelectric cell to carry out C–P cross-coupling with evolution of hydrogen. N-Aryltetrahydroisoquinolines and various phosphine oxides were used as the starting compounds (Scheme 96). ‡
Scheme 96
Download figure:
The intramolecular cyclization of diarylphosphine oxides in an electrochemical process [Pt(+)|Pt(–) undivided cell, constant current of 2.5 mA] affords diarylphosphole oxides in good yields (24 – 72%). 247
The other few examples of C–P bond formation in electrochemical processes are concerned with cross-coupling reactions involving aromatic compounds with C–H and C–halogen bonds. For example, an electrochemical approach was developed for the phosphorylation of the C(sp2)–H bond in quinoxalin-2(1H)-ones via C–H/P–H cross-coupling. This procedure is applicable to a broad range of substrates and gives products 40 in high yields (up to 99%) (Scheme 97). 248
Scheme 97
Download figure:

The electrochemical cross-coupling of aryl halides with trialkyl phosphites gives arylphosphonates in moderate to good yields (Scheme 98). 249 In the case of aryl iodides, the products are formed in higher yields, whereas in the case of bromides, the yields are moderate. 4-Iodonitrobenzene and 4- and 2-iodopyridines do not give the expected coupling products, but are further reduced under conditions of electrolysis.
Scheme 98
Download figure:

Cowper et al. 250 successfully performed the Arbuzov reaction of aryl chlorides under electrophotocatalytic conditions; the reaction included the intermediate formation of aryl radicals.
3.4. Formation of C–O bond
Although recent publications considering the use of electrochemical reactions for the formation of C–O bonds are few, they are nevertheless useful for evaluation of the future scope of applicability of this method. For example, a process has been developed for selective dehydrogenative electrochemical functionalization of substituted toluenes into the benzylic position to form the C–O bond using 1,1,1,3,3,3-hexafluoropropan-2-ol as the source of oxygen and boron-doped diamond (BDD) electrodes (Scheme 99). 251 The benzyl ethers 41 thus formed are versatile precursors for the subsequent functionalization, because in protic acids, they act as benzyl cations. The benzyl cations are generated on treatment of compound 41 with acids. The reactions with aromatic nucleophiles afford practically valuable diarylmethanes.
Scheme 99
Download figure:
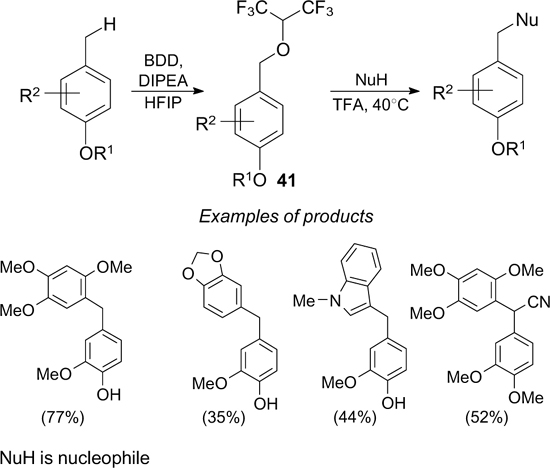
Bityukov et al. 252 developed an electrochemical method for the preparation of α-acyloxy-β-dicarbonyl compounds (36 – 92% yields) as a result of intermolecular C–O coupling of β-dicarbonyl compounds with carboxylic acids (Scheme 100).
Scheme 100
Download figure:
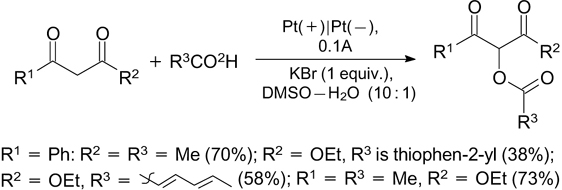
In addition, syn-stereoselective acetoxyhydroxylation of alkenes under electrophotocatalytic conditions has been described. 253
O-(Phthalimid-1-yl)oximes were obtained upon simultaneous formation of N–O and C–O bonds via the reaction of vinyl azides with N-hydroxyphthalimide. 254 During the reaction, O-radicals and N-imidoxyl radicals are generated. 255, 256 This method is characterized by high yields of coupling products (up to 84%) and tolerates substrates with some labile and functional groups (F, NO2, CF3, N3, OMe). Esters of aryl- and alkylcarboxylic acids, including sterically hindered ones, were synthesized from N-alkoxyamides by electrolysis in an undivided Pt(+)|Cu(–) electrochemical cell. 257
The other examples of C–O bond formation are electrochemical intramolecular cross-coupling reactions of compounds containing aromatic C–H bonds; these reactions afford oxygen-containing heterocycles. An electrochemical synthesis of benzoxazoles from anilides giving products in good yields has been described (Scheme 101). 258 Presumably, the process is initiated by anodic oxidation of the deprotonated amide nitrogen atom to give an amidyl radical resonance-stabilized by the aromatic ring. The interaction of the radical centre with the carbonyl group leads to generation of a heterocyclic radical, which is converted to benzoxazole via the subsequent oxidation and deprotonation. 258
Scheme 101
Download figure:

Another example of activation of the aromatic C–H bond is the synthesis of lactones 42 from 2-diarylbenzoic acids (Scheme 102). 259 This reaction can also be carried out for 2-hetaryl-substituted benzoic acid and also can be scaled-up and used to prepare analogues of natural products.
Scheme 102
Download figure:

A putative mechanism of this reaction is depicted in Scheme 103 in relation to the formation of product 42a. The initial anodic oxidation of the substrate via single-electron transfer gives rise to an aromatic radical cation 43. Then this radical cation attacks the carboxyl group to form radical intermediate 44, which rapidly aromatizes via SET and is deprotonated via the anodic oxidation step, thus giving target compound 42a.
Scheme 103
Download figure:
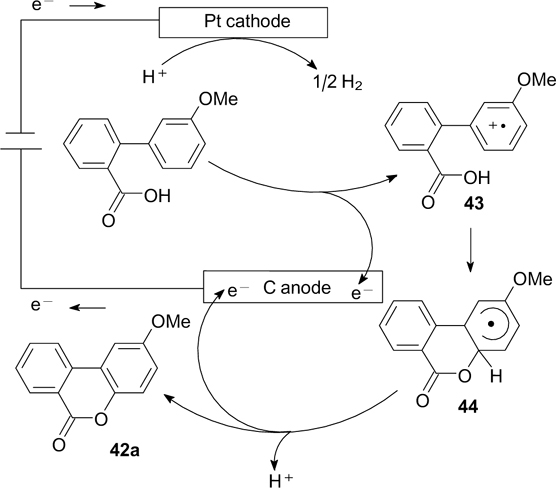
An analogous reaction was performed by electrolysis of solutions of 2-arylbenzoates in an undivided electrochemical cell using platinum electrodes and using a constant current of 23 mA. 260
A highly regioselective synthesis of 4H-1,3-benzoxazines is based on the intramolecular cyclization of N-benzylacylamines, resulting in the formation of regioisomeric products (45 – 79%, 22 examples) (Scheme 104). 261
Scheme 104
Download figure:
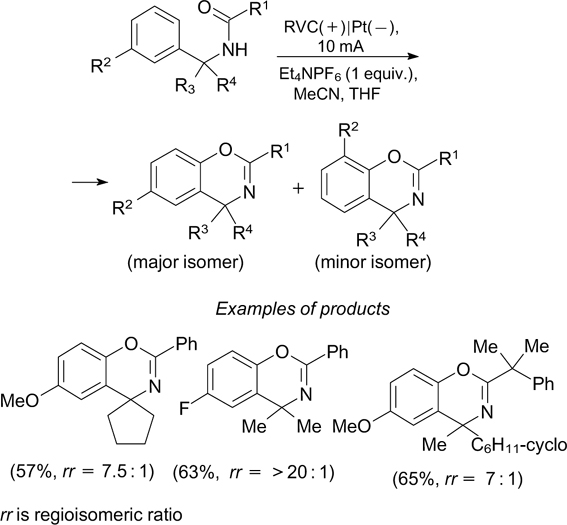
The introduction of oxygen-containing functional groups into various molecules can be accomplished by cross-coupling involving not only the C–H bond, but also the carbon–heteroatom bond. The desulfurization and C–O bond formation take place, for example, in the electrochemical oxidation of 3,4-dihydropyrimidine-2(1H)-thiones (including thiourea) in the presence of alcohols, which serve simultaneously as solvents and sources of alkoxy groups. 262 The reaction gives 2-alkoxypyrimidine derivatives (Scheme 105).
Scheme 105
Download figure:
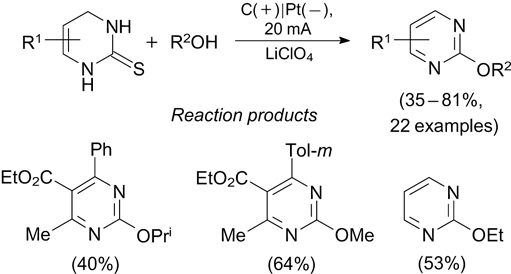
Nokami et al. 263 reported a one-pot synthesis of oligosaccharides based on anodic oxidation of the thioglycoside residue. This residue is converted to the trifluoromethanesulfonic acid derivative, which reacts with another thioglycoside molecule containing a hydroxyl group to give diglycoside. Methods for hydroxylation of toluene derivatives were reported. 264 The synthesis of diaryl ketones, benzaldehydes and diketones 265 by electrochemical oxidation of diarylmethanes and alkenes, proceeding via the formation of alkoxysulfonium ions, 266 was developed.
The above successful examples of application of electrochemical methods for C–O bond formation attest to the necessity of further development of this area with the goal to synthesize practically useful products.
4. Reactions of aryl halides initiated by alkali metal bases
A mechanism of carbon–halogen bond activation in the absence of transition metals alternative to the photo- and electrochemical initiation is related to the single-electron reduction of aryl halide to give aryl radical anion and subsequent elimination of halide ion. 267 These reactions are initiated by alkali metal bases and organic additives (amines, diamines, diols and other organic compounds). Quite of few such reactions have been reported, which refer to cross-coupling to form the carbon – carbon bonds. 21, 22, 268 – 271 The initial reaction of the inorganic base with the additive furnishes a complex — a strong organic electron donor, which induces the reduction of aryl halide via the single-electron transfer. The role of the additive can also be performed by the organic solvent (DMSO, DMF) or the substrate. 272 Conversely, reactions of aryl halides that involve single-electron reduction of the carbon – halogen bond to give a C–N bond are few and are mainly limited to examples of intramolecular amination of aryl halides, resulting in the formation of heterocyclic products. Some transition metal-free reactions of aryl halides giving C–N bond are considered in this part of the review.
4.1. Electron as a catalyst concept
In 2014, Studer and Curran 273 introduced the paradigm according to which the electron is a catalyst to describe the mechanisms of some preparatively significant reactions discovered in the last decade. At the first glance, these reactions have little in common, except for two features: they are free from transition metal compounds and, most likely, they involve the formation of radical intermediates. In particular, various radical cascade reactions can be assigned to this type, e.g., unimolecular radical aromatic substitution, 274, 275 base-induced homolytic nucleophilic substitution, 276 radical Heck reaction 277 and so on.
According to this concept, the general mechanism of the carbon – heteroatom bond formation can be represented in the following way (Scheme 106). The first stage is initiation of the catalytic cycle by the electron transfer from an electron-rich species, which in this case can be considered as a catalyst precursor, i.e., an electron (step 1). Then the substrate ArX molecule is reduced to give the corresponding (ArX)−• radical anion (step 2), the fragmentation of which leads to the Ar. radical and the X− halide ion (step 3). The reaction of the Ar. radical with a nucleophilic reagent yields a new radical anion, (ArNu)−• (step 4), which in turn lends an electron to the catalytic cycle and is thus converted to the final product (step 5). Steps 3 and 4 may occur concertedly; in this case, free radical is not formed as an intermediate. In general, this reaction pathway is similar to the traditionally accepted mechanism of unimolecular aromatic nucleophilic substitution SRN1, which can be considered as an example of electron-catalyzed reactions. Unlike classical metal- or radiation-induced reactions that follow the SRN1 mechanism, the reactions discussed below usually do not require such conditions and readily proceed upon thermal activation. The participation of radical intermediates was shown experimentally for many of these reactions, despite the fact that they have little in common regarding the mechanism. The use of the Studer and Curran concept allows these reactions to be considered as examples of electron-catalyzed reactions.
Scheme 106
Download figure:
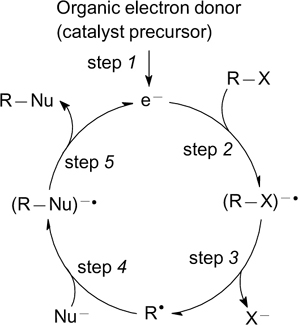
4.2. N-Arylation of benzamides
One of the few examples of intermolecular formation of C–N bond upon transition metal-free amination of aryl halides is related to the reaction of substituted benzamides and their heterocyclic analogues with aryl iodides in the presence of K2CO3 and N, N'-dimethylethylene-1,2-diamine (DMEDA). The arylation does not take place in the absence of DMEDA. As the arylating agents, ortho- and para-halo-substituted aryl iodides are used. No cine-substitution products are detected, which rules out the aryne mechanism of the reaction (Scheme 107). 278
Scheme 107
Download figure:
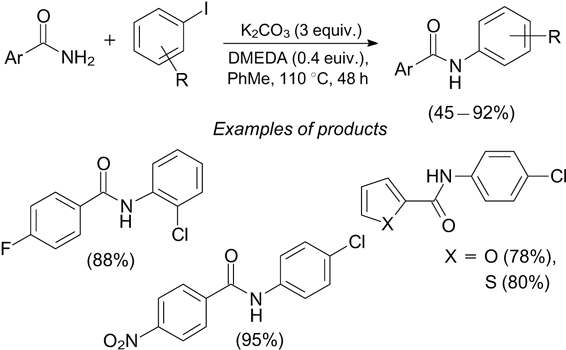
4.3. Synthesis of five-membered heterocyclic compounds
A feasible approach to the synthesis of substituted 9H-carbazoles is based on the heterocyclization of 2-amino-2'-bromobiphenyls. High yields of 9H-carbazoles are attained when potassium tert-butoxide is used as a base (Scheme 108; the position of substituents in the substrate is indicated). 279
Scheme 108
Download figure:
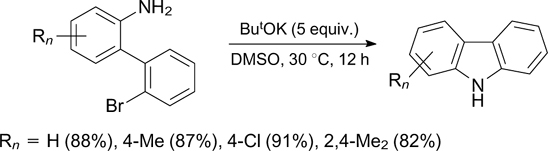
The cyclizaion of 3-amino-2-(2-bromophenyl)acrylates as a result of copper or iron salt-catalyzed intramolecular amination of aryl halides forms the basis of the known method for the synthesis of esters of N-substituted indole-3-carboxylic acids, valuable building blocks for the preparation of various indole derivatives, in particular, pharmacologically active compounds. 280, 281 The cyclization of the same substrates can be induced without transition metal catalysis (Scheme 109). 282 For example, indoles were prepared by initial conversion of enols to enamines and subsequent cyclization induced by the ButOK–DMF system. The efficiency of this method of synthesis of N-aryl indole derivatives is comparable with or is higher than the efficiency of known catalytic reactions. 280, 281 In addition, this reaction can be used to prepare indole in gram amounts and to synthesize a chiral product from (S)-(–)-α-methylbenzylamine without the loss of enantiomeric purity.
Scheme 109
Download figure:
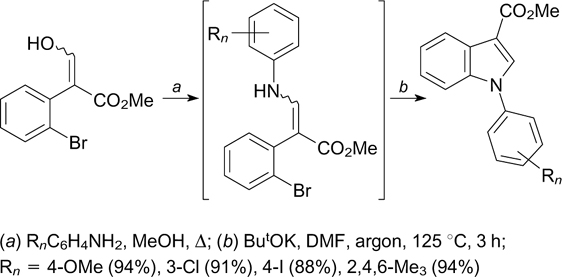
Relying on the concept of electron-catalyzed coupling reactions, the authors proposed the following catalytic cycle for the formation of indole (Scheme 110). The transformation starts from the release of electron, which acts as the catalyst. The possible sources of electron are the ButOK–DMF system or the aza-allyl anion resulting from deprotonation of the substrate. The subsequent reduction of aryl bromide via single-electron transfer results in the corresponding radical anion 45, which is converted to aryl radical 46 and bromide ion upon fragmentation. The intramolecular capture of radical 46 by N-nucleophilic moiety results in generation of radical anion 47. In the final stage of the cycle, radical anion 47 is converted to the product and transmits an electron to a next substrate molecule, thus closing the catalytic cycle. This sequence is analogous to the SRN1 mechanism. Alternatively, radical anion 47 can be obtained from radical anion 45 via a concerted process without intermediate formation of aryl radical 46, which corresponds to the SRN2 mechanism. Although the true mechanism of the reaction is not clear as yet, high selectivity observed with the use of multihalo-substituted substrates suggests that conversion of radical anion 45 to radical anion 47 follows, most likely, a concerted mechanism.
Scheme 110
Download figure:
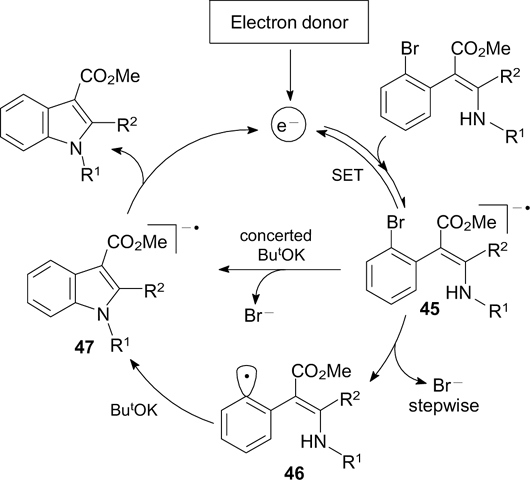
The intramolecular cyclization of ortho-halo-substituted N-arylamidines induced by KOH in DMSO furnishes substituted benzimidazoles 48 (Scheme 111). 283 This method is suitable for substrates containing labile substituents and functional groups and efficient for the preparation of halo-substituted benzimidazoles. It is noteworthy that the heterocyclization may involve various halo derivatives. Fluoro- and iodo-substituted substrates have comparably high reactivities, whereas bromo and chloro derivatives are considerably less reactive. This indicates that the mechanism of heterocyclization depends on the nature of the halogen in the starting ortho-halo-substituted N-arylamidine. Apparently, in the case of fluorine-containing substrates, the reaction follows the SN Ar mechanism, while iodo derivatives react through the formation of radical intermediates.
Scheme 111
Download figure:

For comparison, given below are the yields of substituted benzimidazoles 48a – c obtained upon the palladium-catalyzed intramolecular amination of aryl bromides under the following conditions: Pd2(dba)3 (0.5–1 mol.%), Ph3P (4 – 8 mol.%), NaOH (2 equiv.), H2O, DME, MW, 160 °C; dba is dibenzylideneacetone, DME is dimethoxyethane, MW is microwave radiation. 284
Although the product yields in the two types of processes are comparable, the catalyzed reaction requires the use of microwave radiation and tolerates a smaller range of substituents (in particular, it is inapplicable for the preparation of bromo-substituted benzimidazoles). Furthermore, although the Ph3P ligand is used in the palladium-catalyzed reaction in a minor amount, the authors noted that traces of this compound were present in the reaction products. A special procedure using the Amberlist 15 ion exchange resin was developed for the removal of these impurities. 284
Structures 48
Download figure:
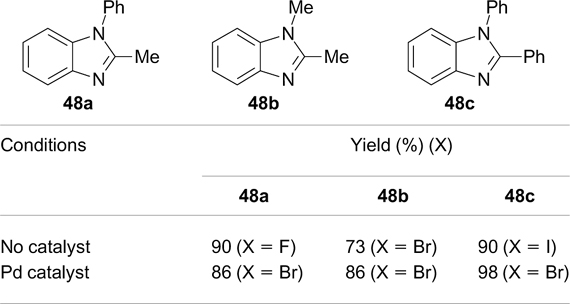
A similar intramolecular cyclization of N-(2-iodophenyl)arylamidines carried out in the presence of K2CO3 (2 equiv.) in water at 100 °C for 30 h (Scheme 112) affords 2-(hetero)arylbenzimidazole derivatives in moderate to high yields. 285 This reaction smoothly proceeds for iodides and bromides, whereas chlorides and fluorides remain inert under the same conditions. In addition, the benzimidazole ring is not formed when strong electron-withdrawing groups are present in the benzene ring of the aromatic substituent R2. The choice of base also considerably affects the efficiency of cyclization: for example, sodium derivatives and organic bases do not facilitate the formation of the desired product.
Scheme 112
Download figure:
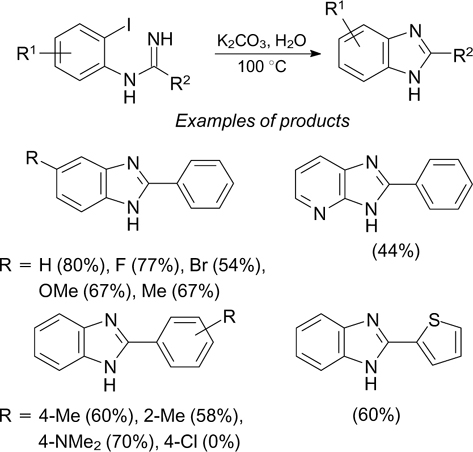
A similar heterocyclization was used to prepare pyrido[1,2-a]benzimidazoles from N-(2-halophenyl)-2-aminopyridines (Scheme 113). 286
Scheme 113
Download figure:
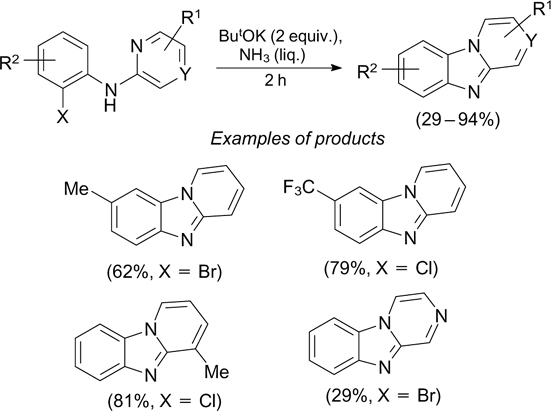
The transition metal-free intramolecular cyclization of ortho-iodo(bromo)-substituted N-arylureas serves as a convenient preparative route towards benzimidazol-2-ones (Scheme 114). 287 This method is applicable for the synthesis of structurally diverse benzimidazol-2-ones, including brominated derivatives and compounds with bulky substituents at the nitrogen atom. The use of ureas containing two unsubstituted NH groups as the starting compounds does not give the cyclization product. The highest yields of products are obtained for iodo- and bromo-substituted substrates. The cyclization of fluoro-substituted ureas proceeds smoothly, while chloro derivatives are least reactive. These facts suggest that the heterocyclization mechanism depends on the nature of the halogen atom acting as the leaving group. In the case of fluorinated substrates, the SNAr mechanism predominates, while the substitution of iodo and chloro derivatives involves radical intermediates.
Scheme 114
Download figure:
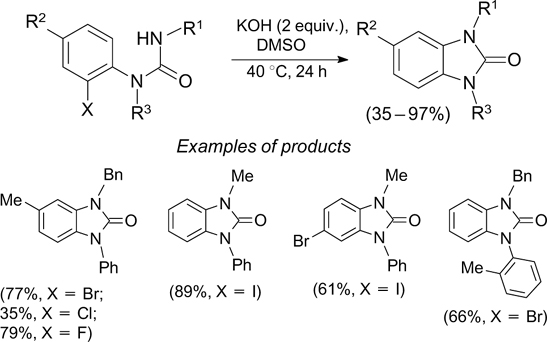
One of early studies 288 related to the synthesis of heterocyclic compounds by amination of aryl halides free from transition metal-based catalysts describes the heterocyclization of (Z)-o-bromoacetophenone N-tosylhydrazones (Scheme 115). The reaction takes place under the action of K2CO3 in the presence of 10 mol.% trans-N,N'-dimethyl-1,2-diaminocyclohexane (49) and yields N-substituted indazoles. Other bases (KOH, ButOK, Na2CO3) and other amines were inefficient or less efficient. While analyzing the mechanisms of heterocyclization, the authors assumed the radical mechanism to be most likely, which was confirmed by studying the reaction mixture by EPR spectroscopy.
Scheme 115
Download figure:
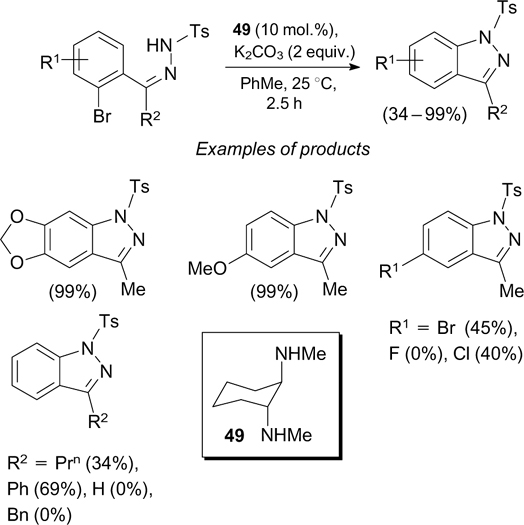
A similar heterocyclization reaction of hydrazides of ortho-halogenated benzoic acids gave 1H-indazolones (Scheme 116). 289 The heterocyclization proceeds most efficiently for hydrazones of o-iodobenzoic acids, whereas in the case of chloro- and fluorobenzoic acids, no cyclization products are formed. The optimal conditions include the use of potassium tert-butoxide as a base and DMSO as the solvent in the presence of 40 mol.% proline. The reaction is sensitive to the electronic nature of substituents: electron-withdrawing substituents present in either benzoic acid moiety or hydrazine moiety lead to decreasing yields of 1H-indazolones. The cyclization presumably follows a radical mechanism, as indicated by considerable decrease in the yield of the cyclization product upon the addition of radical scavengers into the reaction medium. 289
Scheme 116
Download figure:
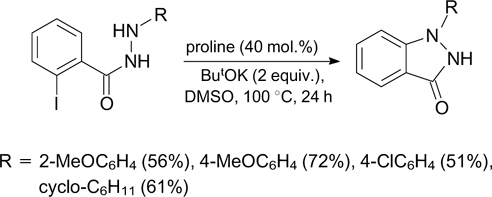
Heating of 2-(2-bromophenyl)acetyl-N-tosylpyrroles 50 in DMF in the presence of 2 equiv. of Cs2CO3 affords dihydropyrrolizino[3,2-b]indol-10-ones 51 (Scheme 117). 290 In this reaction, DMF is not only a solvent, but also a reactant. The mechanism proposed for this reaction includes the formation of tricyclic intermediate 52 (see Scheme 117), which undergoes base-induced intramolecular cyclization. The subsequent demethylation of quaternary ammonium salt furnishes product 51.
Scheme 117
Download figure:

The reaction of bromoacetylenes with benzamidines in the presence of K2CO3, 2,2'-bipyridine (bipy) and water underlies the synthesis of substituted imidazoles (Scheme 118). 291 According to the mechanism proposed by the authors, the reaction starts with the formation of amidine and bromoacetylene adducts, compounds 53, which then cyclize to substituted imidazoles. The reaction is applicable to substituted aromatic amidines and aryl- or alkyl-substituted bromoacetylenes; using N-monosubstituted amidines, the corresponding N-substituted imidazoles can be obtained only in low yields.
Scheme 118
Download figure:
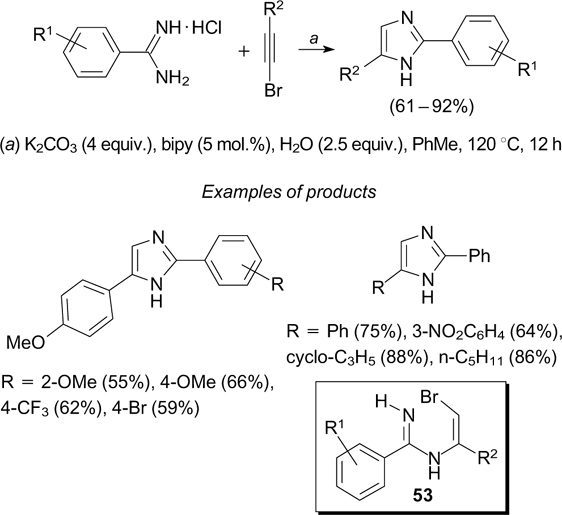
The strategy towards the formation of nitrogen-containing five-membered heterocycles based on base-catalyzed intramolecular cross-coupling reactions can be successfully used to prepare benzofuran and its fused derivatives. For example, the intermolecular addition of the alkyl radical to the carbonyl group followed by the intramolecular addition of the alkoxy radical to o-phenoxymethylhaloarenes affords substituted benzofurans in high yields under catalysis with a strong base (Scheme 119). 292
Scheme 119
Download figure:

The radical mechanism of this reaction depicted in Scheme 120 was confirmed by free radical trapping experiments and analysis of EPR spectra.
Scheme 120
Download figure:

One more example of this type of C–O bond formation takes place in the ButOK-initiated intramolecular cyclizations of 2-(2-bromoaryl)benzyl alcohols and -benzoic acids to give the corresponding 6H-benzo[c]chromene and 6H-benzo[c]chromen-6-one derivatives, respectively (Scheme 121). 279
Scheme 121
Download figure:
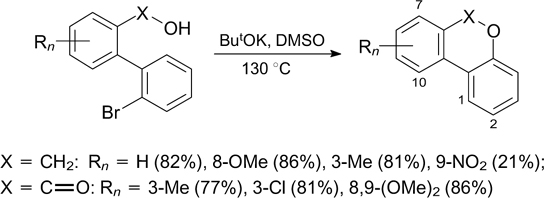
There are also known examples of the preparation of sulfur-containing five-membered heterocycles using the above-discussed reactions. The domino synthesis of 2-substituted benzothiazoles 54 includes base-induced one-pot addition of derivatives with an active methylene group to o-iodoaryl isothiocyanates and subsequent intramolecular formation of the C–S bond in the intermediate thioamidate anion 55 (Scheme 122). 293 A radical (SRN 1) mechanism was proposed for this cyclization. The reaction at room temperature proceeds within 1 – 3 h and gives products 54 in good yields.
Scheme 122
Download figure:
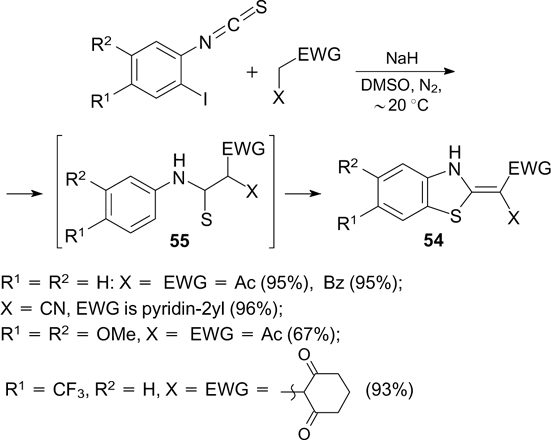
The efficient synthetic pathway to 2,3-disubstituted benzothiophenes and their heterocyclic analogues developed by the same authors 294 (Scheme 123) includes a tandem reaction consisting of base-induced condensation of o-iodoarylacetonitriles, -acetates or -ketones with (hetero)aryl dithioesters and intramolecular C–S bond formation.
Scheme 123
Download figure:
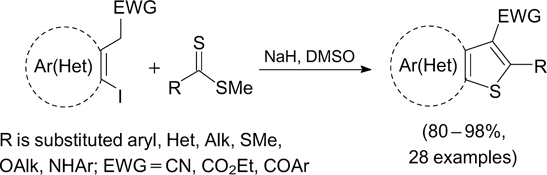
4.4. Synthesis of six-membered heterocyclic compounds
Non-catalyzed heterocyclization of N-acylated aryloxyanilines to give C–N bonds underlies an efficient approach to the synthesis of phenoxazines. The highest yields of cyclization products are attained with the K2CO3–DMEDA system (Scheme 124). 295 The cyclization products are obtained only from iodo derivatives. This method is inapplicable to substrates containing an alkyl or tosyl group instead of the acetyl group at the nitrogen atom. The nature of the substituent in the aniline moiety of the starting compound barely influences the cyclization; conversely, the presence of electron-donating substituent in the iodoaryl moiety increases the yield of phenoxazines, although to a minor extent. This method is also applicable to the preparation of some aza analogues of phenoxazine.
Scheme 124
Download figure:
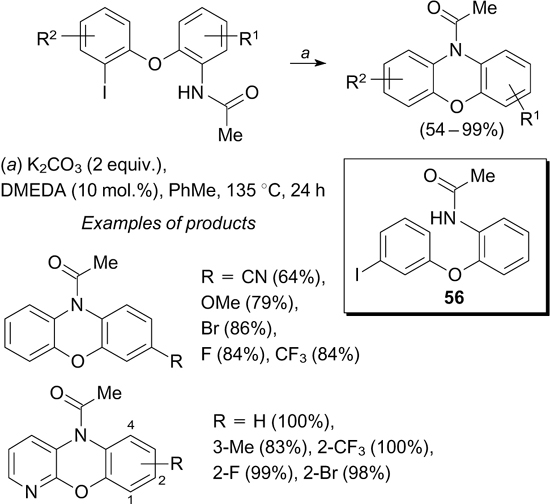
In order to elucidate the mechanism of heterocyclization, the authors carried out a number of experiments, which did not unambiguously clarify the reaction mechanism, but provided the following conclusions. First, phenoxazines are formed exclusively in the presence of DMEDA or trans-N,N'-dimethyl-1,2-diaminocyclohexane (49). Second, the use of potassium carbonate of high purity (⩾99.999%, according to atomic absorption spectroscopy) virtually precluded the possibility of participation of trace amounts of transition metals in the reaction. Third, the aryne mechanism was also ruled out, because substrate 56 containing m-iodophenoxyl moiety (see Scheme 124) remained unchanged under the reaction conditions; particularly, it did not undergo cyclization and/or protodehalogenation. Fourth, the reaction proved to be insensitive to the presence of an equimolar mixture of radical scavengers in the reaction mixture; this indicates that the formation of radical intermediates is unlikely.
The tandem reaction of N-(2-iodophenyl)acetamides with 2-bromothiophenols in the presence of potassium carbonate in DMF gives rise to phenothiazines. 296 The reaction occurs without additional initiators and includes two successive steps of carbon – heteroatom bond formation. Initially, intermolecular formation of the carbon – sulfur bond takes place, yielding acyclic intermediate 57 (Scheme 125), which is the major product formed in the case of short-term heating. The second step is heterocyclization upon the intramolecular formation of the carbon–nitrogen bond, and the final step is the removal of the acetyl group. Experiments using radical scavengers demonstrated that the reaction apparently occurs without the formation of free radicals.
Scheme 125
Download figure:
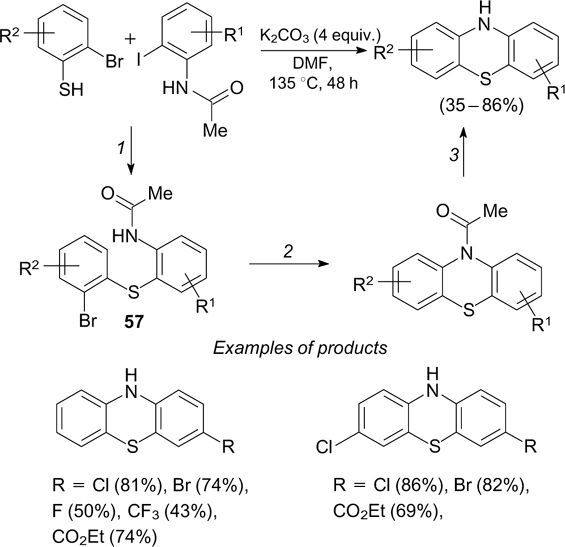
Phenothiazines are also formed as a result of a similar heterocyclization of N,S-diacetyl-2-aminobenzothiols with 1,2-dihalo-substituted benzenes induced by caesium carbonate in DMF (Scheme 126). 297 When radical scavengers were added to the reaction mixture, the yield of cyclization products significantly decreased. On the basis of experiments, the authors suggested that the reaction starts with base-promoted S-deacetylation of the substrate, which is followed by the intermolecular formation of the C–S bond. The heterocycle is formed upon the subsequent intramolecular formation of the C–N bond.
Scheme 126
Download figure:

One-pot synthesis of pyrazolo[1,5-a]quinoxalines from 1-(2-chlorophenyl)-5-(ethoxycarbonyl)pyrazoles and aliphatic primary amines includes the initial formation of pyrazolecarboxylic acid amides 58 (Scheme 127) and the subsequent intramolecular formation of carbon – nitrogen bond. 298 Experiments involving radical scavengers suggest that no radical intermediates are formed during this reaction. Base-promoted heterocyclizations of this type can also be used to prepare imidazo[1,5-a]quinoxaline derivatives upon cyclization of halo derivatives of imidazole-2-carboxylic acid N-arylamides. 299
Scheme 127
Download figure:

The domino reaction of 2-sulfanylbenzimidazoles with 2-halo-substituted benzyl bromides affords imidazobenzothiazines 59 via the successive formation of C–S and C–N bonds (Scheme 128). 300 The tetracyclic product is formed most efficiently in the presence of the Cs2CO3–DMF system. In the case of unsymmetrically substituted 2-sulfanylbenzimidazoles, the reaction gives a mixture of regioisomers.
Scheme 128
Download figure:
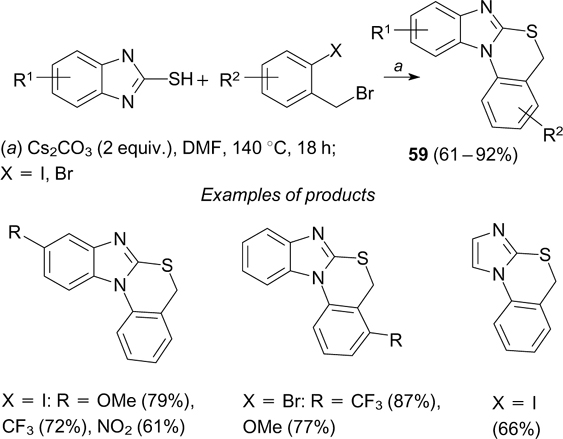
The reaction of substituted 1-(2-aminophenyl)-8-iodonaphthalenes with K2CO3 and excess MeI in DMSO furnishes π-fused systems 60 under mild conditions via intramolecular formation of the C–N bond. It was shown in special experiments that the N,N-dimethylation product is formed initially, and the subsequent heterocyclization of this compound involves tertiary amine and is accompanied by demethylation. Radical scavengers slightly decrease the yield of products 60, but the experiments carried out by the authors rule out the aryne mechanism for the formation of cyclization products as well (Scheme 129). 301
Scheme 129
Download figure:
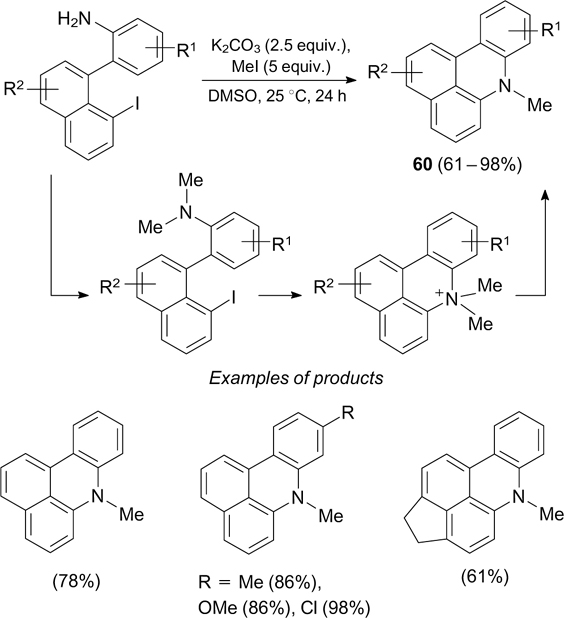
The papers considered in this part of the review were mainly published between 2010 and 2020, and the presented reactions are characterized by high preparative value owing to their efficiency, cost effectiveness and easy implementation. The key benefit of these reactions is that they do not require expensive and toxic catalysts based on transition metals, which can be successfully replaced by simple and readily available inorganic bases, organic ligands and solvents. Although special experiments aimed at making assumptions about the mechanism of amination of aryl halides were carried out almost in each study, reactions of this type cannot be assigned to any of known types of nucleophilic aromatic substitution. Nevertheless, the following general patterns can be distinguished for these reactions.
- The reactions proceed efficiently in the presence of inorganic bases such as alkali metal carbonates, hydroxides and tert-butoxides. In most cases, the highest efficiency is attained by using potassium derivatives, although there are examples in which sodium and caesium bases perform better than potassium compounds.
- The reactions proceed more easily in the case of iodo- and bromo-substituted substrates, while chloro derivatives have low reactivity. Fluoro-substituted substrates often show high reactivity, which may attest to a change in the reaction mechanism on going from iodo and bromo derivatives to fluorine compounds. In the latter case, the reactions may follow the SNAr mechanism or, more likely, the mechanism of concerted aromatic nucleophilic substitution. 302
- Apart from inorganic bases, for these reactions to proceed smoothly, special additives (aliphatic amines, heterocyclic compounds, etc.) should be present in the reaction mixture, or the reactions should be carried out in solvents containing donor heteroatoms (DMF, DMSO). The additives and solvents, in combination with the inorganic base used, play a key role in these reactions. In some cases, this role can probably be played by substrates; in these cases, the reactions successfully proceed in inert hydrocarbon solvents without the use of foreign additives.
5. Conclusion
The strategy of organic synthesis based on transition metal-free cross-coupling reactions has been intensively developed in recent years. A number of synthetic problems that could not be solved until recently without the use of transition metal-based catalysts are easily solved today using alternative methods for bond activation in organic molecules, in particular, SET processes. The use of visible light and electric current to induce single-electron oxidation and reduction of organic compounds allows the carbon – heteroatom bond-forming reactions to be conducted at room temperature for substrates containing functional groups incompatible with traditional cross-coupling reactions. In some cases, this activation increases the reactivity of organic molecules. The practical value of these methods is based on not only cost effectiveness associated with the absence of expensive catalysts. The reactions induced by visible light and electric current can often proceed without strong oxidants. If these reactions are based on air oxygen as the oxidant, they can be assigned to green chemistry reactions. The attraction of these approaches to initiation of organic reactions is also related to the use of simple experimental equipment (glass reactors; diode and luminescent lamps, including household devices, as sources of visible light; and simple sources of direct current).
Special mention should be made of carbon – heteroatom bond formation reactions initiated by alkali metal bases. Despite the fact that these reactions often require drastic conditions, their main benefit is that they involve the same substrates and give the same products as classical cross-coupling reactions giving carbon – heteroatom bonds.
The further development of transition metal-free cross-coupling strategy is certainly of considerable fundamental and practical interest. In particular, a relevant task is the search for new reactions of this type that would provide efficient synthesis of compounds inaccessible by classical catalytic methods. In addition, it is worth concentrating on the development of new organic photoredox catalysts, in particular, to replace the expensive transition metal complexes that are widely used now. An equally important line of research is the development of new reagents that would allow reactions to be conducted using their own photochemical properties or without photocatalysts using electric current activation.
This review was prepared with the financial support of the Russian Foundation for Basic Research (Project No. 20-13-50056).
6. List of abbreviations and symbols
The following abbreviations and symbols are used in the review:
| 2-But-AQN | — 2-tert-butylanthraquinone; |
| 3DPAFIPN | — 3,5-dicyano-2,4,6-tris(diphenylamino)-fluorobenzene; |
| 4CzIPN | — 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; |
| [AcrMes]+ | — 9-mesityl-10-methylacridinium cation; |
| All | — allyl; |
| AQN | — anthraquinone; |
| BDD | — boron-doped diamond electrode; |
| Boc | — tert-butyloxycarbonyl; |
| BPA | — 1,1'-binaphthyl-2,2'-phosphoric acid; |
| bipy | — 2,2'-bipyridine; |
| Cbz | — benzyloxycarbonyl; |
| CFL | — compact fluorescent lamp; |
| col | — 2,4,6-collidine (2,4,6-trimethylpyridine); |
| Cp | — cyclopentadienyl; |
| DABCO | — 1,4-diazabicyclo[2.2.2]octane; |
| DABSO | — DABCO complex with SO2; |
| dba | — dibenzylideneacetone; |
| DBU | — 1,8-diazabicyclo[5.4.0]undec-1-ene; |
| DCA | — 9,10-dicyanoanthracene; |
| DCE | — dichloroethane; |
| DIPEA | — N,N-diisopropylethylamine; |
| DMA | — N,N-dimethylacetamide; |
| DME | — dimethoxyethane; |
| DMEDA | — N,N'-dimethylethylene-1,2-diamine; |
| DNs | — dinitrophenylsulfonyl; |
| dr | — diastereomeric ratio; |
| EB | — eosin B; |
| EDA | — electron donor – acceptor complex; |
| EWG | — electron-withdrawing group; |
| ee | — enantiometic excess; |
| EY | — eosin Y; |
| Fmoc | — 9-fluorenylmethyloxycarbonyl; |
| Fu | — furyl; |
| HAT | — hydrogen atom transfer; |
| HFIP | — 1,1,1,3,3,3-hexafluoropropan-2-ol; |
| m-CBA | — 3-chlorobenzoate; |
| Mes | — 2,4,6-trimethylphenyl (mesityl); |
| Ms | — methanesulfonyl (mesyl); |
| MS | — molecular sieves; |
| MW | — microwave radiation; |
| Na2-EY | — disodium salt of eosin Y; |
| Naph | — naphthyl; |
| NBS | — N-bromosuccinimide; |
| Ns | — 4-nitrobenzenesulfonyl; |
| NuH | — nucleophile; |
| PC | — photoredox catalyst; |
| PC* | — photoredox catalyst in the excited state; |
| PCET | — proton coupled electron transfer; |
| PHT | — N-phenylbenzo[b]phenothiazine; |
| PHTN-NH | — NH-phthalimide; |
| pPy | — piperidine; |
| PVDF | — polyvinylidene fluoride; |
| Py | — pyridine; |
| Tol | — tolyl (methylphenyl); |
| Pyf | — 2,3,5,6-tetrafluoropyridin-4-yl; |
| RB | — rose bengal; |
| Rh-6G | — rhodamine 6G; |
| Rh-B | — rhodamine B; |
| rr | — regioisomeric ratio; |
| RVC | — is reticulated vitreous carbon electrode; |
| S | — substrate; |
| SET | — single-electron transfer; |
| TBHP | — tert-butyl hydroperoxide; |
| TBP | — tri-n-butyl phosphate; |
| TEMPO | — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, |
| Tf | — trifluoromethanesulfonyl (triflyl); |
| TFA | — trifluoroacetic acid; |
| TFE | — 2,2,2-trifluoroethanol; |
| TMS | — trimethylsilyl; |
| TPT | — 2,4,6-triphenylpyrylium tetrafluoroborate; |
| Ts | — p-toluenesulfonyl (tosyl). |
Footnotes
- †
The notation Pt(+)|Pt(–) means undivided electrochemical cell with the indicated anode and cathode.
- ‡
Note that the most recent achievements in the photoelectrochemistry are summarized in a review by Barham and König. 246