


Abstract
The review summarizes literature data on the methods for the introduction of fluorine atoms and fluoralkyl groups into different ligands to construct metathesis-active ruthenium carbene complexes. It also analyzes the influence of fluorinated ligands on the catalytic activity of the complexes. The choice, structure and positions of fluorinated substituents in NHC ligands are generally dictated by the desire to increase the electrophilicity of the ruthenium atom due to the electron-withdrawing effect of fluorine atoms and fluoroalkyl groups, resulting, as a rule, in an increase in the activity of the ruthenium complex. In catalysts with unsymmetrical fluorine-containing NHC ligands, there is a possibility of additional Ru–F coordination, making the complexes much more stable and, consequently, more active. The presence of fluorine in chelating alkylidene ligands provides an increase in the catalyst initiation rate due to a weakening of the ruthenium – heteroatom bond. Besides, the introduction of polyfluoroalkyl groups into ligands solves the problem of catalyst recovery using fluorous biphasic systems for reuse.
The bibliography includes 172 references.
Export citation and abstract BibTeX RIS
| S.M.Masoud. PhD in Chemistry, Researcher at the Laboratory for Ecological Chemistry, INEOS RAS. |
| E-mail: skh.masoud@gmail.com |
| Current research interests: organic and organometallic chemistry, homo geneous catalysis. |
| D.V.Vorobyeva. PhD in Chemistry, Senior Researcher at the same Laboratory. |
| E-mail: d-20@mail.ru |
| Current research interests: organic, organofluorine and organometallic chemistry, homogeneous catalysis. |
| D.A.Petropavlovskikh. PhD student at the same Laboratory. |
| E-mail: dmitrypetrpavl@gmail.com |
| Current research interests: organic, organofluorine and organometallic chemistry, homogeneous catalysis. |
| Christian Bruneau. PhD, Professor, Director of the Green Chemistry and Catalysis Department at the Institute of Chemical Sciences at the University of Rennes. |
| E-mail: christian.bruneau@univ-rennes1.fr |
| Current research interests: organometallic chemistry, green catalysis. |
| S.N.Osipov. Doctor of Chemical Sciences, Head of the Laboratory for Ecological Chemistry, INEOS RAS. |
| E-mail: osipov@ineos.ac.ru |
| Current research interests: organic, organofluorine and organometallic chemistry, homogeneous catalysis. |
1. Introduction
In recent years, the alkene metathesis has become one of the most important tools for the carbon – carbon double bond formation in the synthesis of diverse molecules, from simple alkenes 1 to biologically active compounds 2–4 and macromolecules. 5 The importance of the development of the metathesis method in organic synthesis has been recognized by the award of the 2005 Nobel Prize in Chemistry jointly to Y.Chauvin (France), R.Grubbs (USA) and R.Schrock (USA). Ruthenium, 6 molybdenum and tungsten carbene complexes 7 are the most efficient alkene metathesis catalysts.
Numerous types of metathesis catalysts are currently known. However, ruthenium carbene complexes, commonly known as Grubbs catalysts, (Fig. 1) are the most popular due to high air and moisture stability combined with tolerance to a wide range of functional groups (e.g., hydroxy, carboxy, carbonyl, etc.) in substrates. 8 The first ruthenium-based metathesis catalyst with the known structure was synthesized in 1992. 9 Since that time a large number of different modifications were described. 10 This area of research has attracted great attention of chemists and, despite significant advances, the design of novel, more efficient and selective metathesis catalysts is still of great interest.

Figure 1. Commercially available ruthenium metathesis catalysts: the first-generation Grubbs catalyst (G-I), the second-generation Grubbs catalyst (G-II) and the second-generation Hoveyda – Grubbs catalyst (HG-II).
Download figure:
Standard imageHérisson and Chauvin 11 were the first to suggest the mechanism of metal-catalyzed olefin metathesis, according to which the [2+2]-cycloaddition of the starting olefin to the carbene centre of the metal complex giving a metallacyclobutane intermediate and the subsequent retro-[2+2]-cycloaddition (Scheme 1) are key steps in the metathesis. It is worth noting that each step of this catalytic cycle is reversible.
Scheme 1
Download figure:

In the general case, the reaction affords an equilibrium mixture of all possible metathesis products. Therefore, the metathesis can be successfully applied as a preparative method only if the equilibrium is shifted in a particular direction, for instance, via the opening of strained rings or, on the contrary, the closure of stable five- or six-membered carbon rings or heterocycles. Depending on the structure of the starting substrates, several main types of metathesis were described, such as the cross-metathesis (CM), ring-closing metathesis (RCM) of diolefins, ring-closing enyne metathesis (RCEYM), ring-opening metathesis polymerization (ROMP), acyclic diene metathesis polymerization (ADMET), etc. Among these types of catalytic reactions, RCM, ROMP and CM have the most practical value. The outcome of the cross-metathesis is the most difficult to predict because of the absence of strong thermodynamic restrictions associated with the irreversibility of the reaction and E/Z-product selectivity. Despite the fact that the E isomer of the alkene product is thermodynamically favourable, the cross-metathesis generally does not afford this isomer in pure form, and the impurity of the Z isomer is often hard to separate. On the other hand, many biologically active and natural compounds contain carbon – carbon double bonds in the Z configuration. Therefore, the synthesis of new selective catalysts capable of providing the formation of pure E or Z isomers of alkenes is one of the challenging problems in the methodological development of metathesis reactions.
The chemistry of fluorine-containing compounds is a rapidly growing field of research. 12 Due to the unique properties of the fluorine atom, such as low polarizability, the small atomic radius, high electronegativity, the ability to form hydrogen bonds and coordinate to transition metals 13 and a strong carbon – fluorine bond, organofluorine compounds often exhibit properties significantly different from those of the non-fluorinated parent compounds. High fluorine abundance in the Earth's crust and the easy detection of fluorine by 19F NMR spectroscopy (due to the high gyromagnetic ratio and monoisotopic nature 14 ) makes the chemistry of organofluorine compounds highly attractive for synthetic chemists. Hence, organofluorine compounds are widely used in different fields, such as medicinal chemistry, materials science and agriculture. Finally, of special attention are organoperfluorine derivatives capable of forming a non-polar fluorous phase that is immiscible with most organic solvents and thereby facilitating the recovery of catalysts or isolation of reaction products from the reaction mixture. 15
In addition to the above-mentioned fluorophilicity, which is caused by the presence of perfluoroalkyl substituents, the introduction of fluorine atoms into ligands of ruthenium catalysts can result in the formation of an intramolecular Ru–F bond, enhance the electron-withdrawing properties or increase the steric bulkiness of the ligands. The changes caused by the introduction of fluorine into the ligand can significantly alter the catalyst activity and selectivity.
In the general case, Grubbs catalysts contain several different ligands (Fig. 2). In these catalysts, the reactive carbene ligand (most often arylidene) plays a key role, being directly involved in the [2+2]-cycloaddition step in the metathesis. Second- and third-generation catalysts contain an N-heterocyclic carbene (NHC) ligand. Due to the unique steric and electronic properties of the latter, catalysts bearing this ligand have high stability and activity. The phosphine (first- and second-generation Grubbs catalysts) or pyridine (third-generation Grubbs catalysts) ligands are most often employed as L-type ligands; a chloride ion is used as an X-type ligand in the majority of complexes.
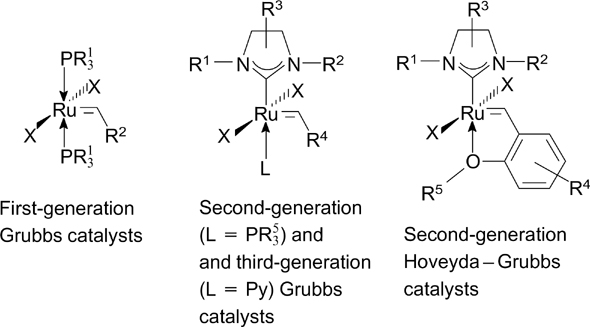
Figure 2. Grubbs and Hoveyda – Grubbs catalysts.
Download figure:
Standard imageAll the above-mentioned ligands (alkylidene, NHC, L, X) can be modified in different ways depending on the task at hand. In recent years, various modifications of Grubbs catalysts were described, 10, 14 the main goals of which were to increase the catalyst selectivity and stability, improve catalyst recyclability and recoverability and simplify product isolation. However, some of these problems are still challenging. Certain problems can be addressed using fluorine-containing substituents. The present review summarizes available methods for the synthesis of fluorine-containing ruthenium alkene metathesis catalysts, their structural features and catalytic activity. The review is divided into sections according to the type of fluorine-containing ligands. The data in each section are presented in chronological order.
2. Fluorine in NHC ligands
Complex 1, synthesized by A.Fürstner group 16 in 2001 (Scheme 2), was the first example of second-generation Grubbs catalysts bearing a fluorinated group in the NHC ligand. The fluorine-containing precursor of this ligand was synthesized by the alkylation of easily accessible N-mesitylimidazole. 17 Complex 1 was synthesized by a standard procedure involving the deprotonation with potassium tert-butoxide. Due to the presence of a perfluoroalkyl substituent, catalyst 1 has good solubility and shows high catalytic activity in the ring-closing metathesis of N,N-dimethallyl-N-tosylamide in supercritical carbon dioxide.
Scheme 2
Download figure:

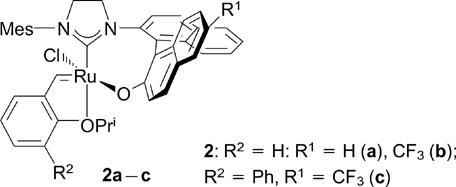
In 2002, A.Hoveyda and co-workers 18 reported a method for the synthesis of catalyst 2a bearing a chiral bidentate binaphthyl NHC ligand. Catalyst 2a proved to be air-stable and exhibited high activity in asymmetric metathesis reactions (> 98% ee). More recently (in 2003), this research group described the synthesis and catalytic activity of six further modifications of catalyst 2, two of which (2b,c) contained a CF3 group. 19
Structures 2a – c
Download figure:
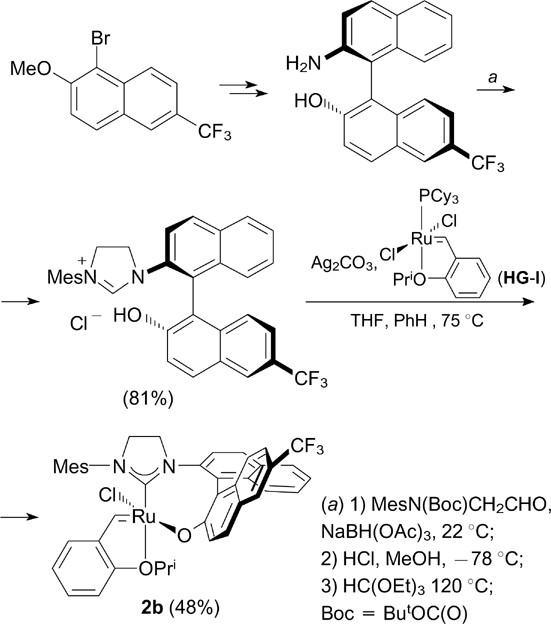
A fluorinated binaphthyl NHC ligand for complexes 2b,c was prepared by a multistep procedure starting from a substrate that already contained fluorine (Scheme 3). Target complex 2b was synthesized by transmetalation of the appropriate silver NHC complex, which was generated in situ from the trifluoromethyl-containing binaphthyl-bridged imidazolinium salt and Ag2CO3.
Scheme 3
Download figure:
The presence of the trifluoromethyl group causes a significant increase in the efficiency of the catalysts. For example, the rate of asymmetric AROCM of norbornenedicarboxylic anhydride and styrene increases three- and 160-fold in the presence of catalysts 2b and 2c, respectively, compared to non-fluorinated catalyst 2a (Table 1).
Table 1. AROCM of norbornenedicarboxylic anhydride and styrene. 16

| |||
|---|---|---|---|
| Catalyst | Time | Conversion (%) | Yield (%) |
| 2a | 66 h | > 99 | 35 |
| 2b | 22 h | > 99 | 55 |
| 2c | 25 min | > 99 | 85 |
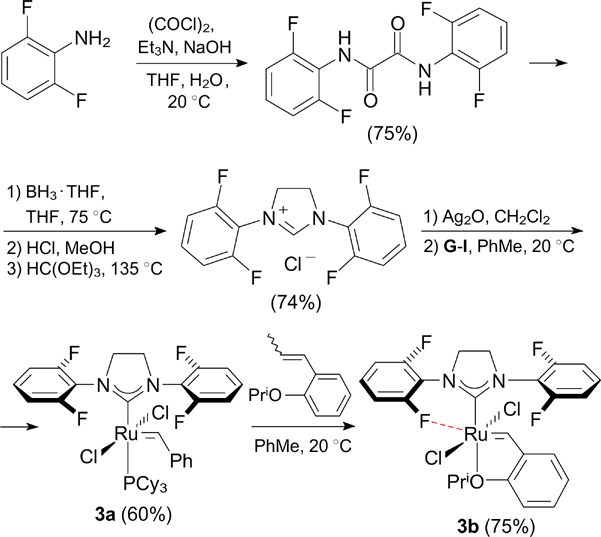
In 2006, Grubbs and co-workers 20 characterized complexes 3a,b containing fluorine in the ortho positions of aryl substituents of the NHC ligand (Scheme 4). Catalyst 3a exhibited higher activity in RCM of diethyl diallylmalonate (DEDAM) compared to the related catalyst G-II. Meanwhile, catalyst 3b proved to be less active than HG-II. This significant difference in the activity was attributed to the Ru⋯F coordination established by X-ray diffraction. This coordination leads to an increase in the stability of the 14-electron complex in the step of initiation of catalyst 3a; however, in the case of catalyst 3b, this coordination bond hinders the access of an alkene molecule to the metal centre. Despite relatively low activity, catalyst 3b proved to be efficient in RCM reactions giving tetrasubstituted alkenes. 21 The corresponding fluorine-containing ligand was synthesized from commercially available 2,6-difluoroaniline.
Scheme 4
Download figure:
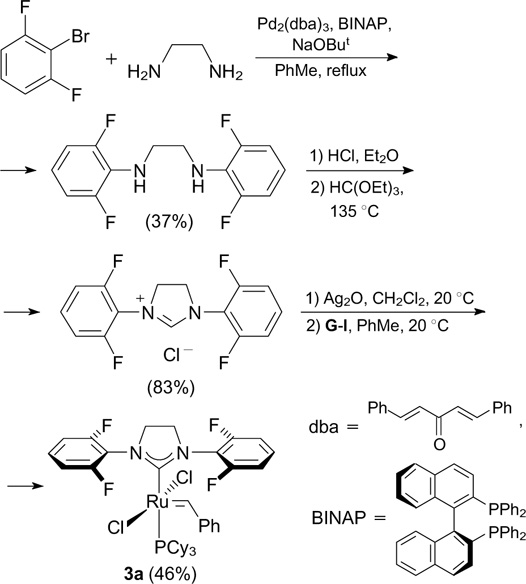
An alternative approach to the synthesis of complex 3a using 1-bromo-2,6-difluorobenzene as the starting compound was proposed by Costabile and co-workers 22 (Scheme 5). The resulting complex was tested in different RCM, ROMP and CM reactions.
Scheme 5
Download figure:
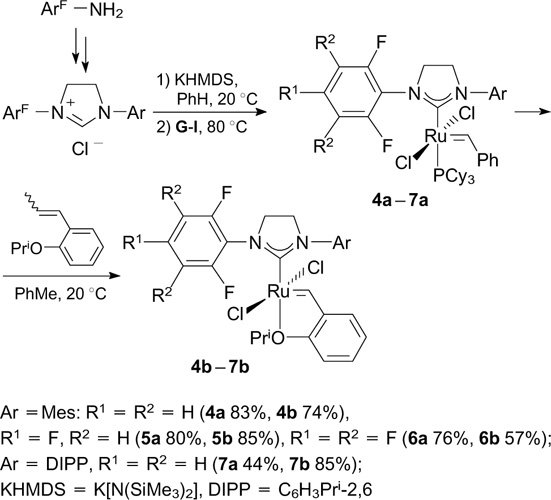
Similar trends in the above-mentioned reactions were found for related unsymmetrical complexes 4 – 7; in all cases, phosphine-containing catalysts 4a – 7a exhibited higher activity than phosphine-free catalysts 4b – 7b (Scheme 6). 23, 24 In the cross-metathesis of allylbenzene with cis-1,4-diacetoxy-2-butene, catalysts 5 – 7 showed higher E/Z selectivity with a conversion of > 60%. Precursors of NHC ligands were synthesized starting from appropriate anilines by a procedure involving the following steps: 1) the treatment of fluorine-containing aniline with oxalyl chloride or its monoesters; 2) the addition of a second aniline molecule giving oxalyl amides; 3) the reduction of carbonyl groups with BH3 ⋅ THF and 4) the cyclization to the unsymmetrical imidazolinium salt upon the treatment with triethyl orthoformate. The deprotonation of imidazolinium salts with potassium hexamethyldisilazide afforded complexes 4a – 7a, the ligand-exchange reaction of which under standard conditions gave phosphine-free complexes 4b – 7b (see Scheme 6).
Scheme 6
Download figure:
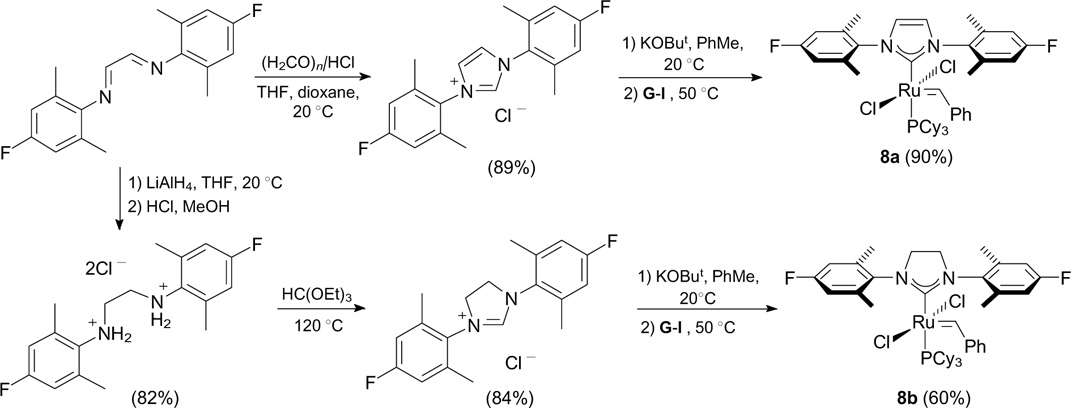
Plenio and co-workers 25 synthesized second-generation Grubbs complexes with NHC ligands containing a fluorine atom in the para position of N-aryl moieties (8a and 8b, Scheme 7). However, the catalytic activity of the new complexes was not evaluated and only some physicochemical properties were studied. Fluorine atoms were introduced into the aromatic ring by the Schiemann reaction.
Scheme 7
Download figure:
Grela and co-workers 26 synthesized catalysts 9 and 10 bearing chiral 1,2,4-triazol-5-ylidene ligands with pentafluorophenyl groups and evaluated their activity in ARCM and AROCM reactions (Scheme 8). Complexes 9 and 10 showed high enantioselectivity in AROCM of norbornenedicarboxylic anhydride with styrene, but the product yields and stability of these complexes were lower compared to the non-fluorinated analogues. Besides, because of low stability, these complexes were generated in situ for all catalytic tests.
Scheme 8
Download figure:

This research group also reported the synthesis of an indenylidene catalyst bearing an unsymmetrical NHC ligand with a pentafluorophenyl group (11, Scheme 9). 27 In RCM of DEDAM performed in air, 28 catalyst 11 proved to be more active but less stable than commercially available M2 (trade name, by Umicore). The ligand was synthesized starting from pentafluorobenzaldehyde. Complex 11 was prepared using two different approaches: 1) the deprotonation of the corresponding imidazolium tetrafluoroborate and 2) the decomposition of the pentafluorophenyl adduct. The former approach was found to be more efficient.
Scheme 9
Download figure:
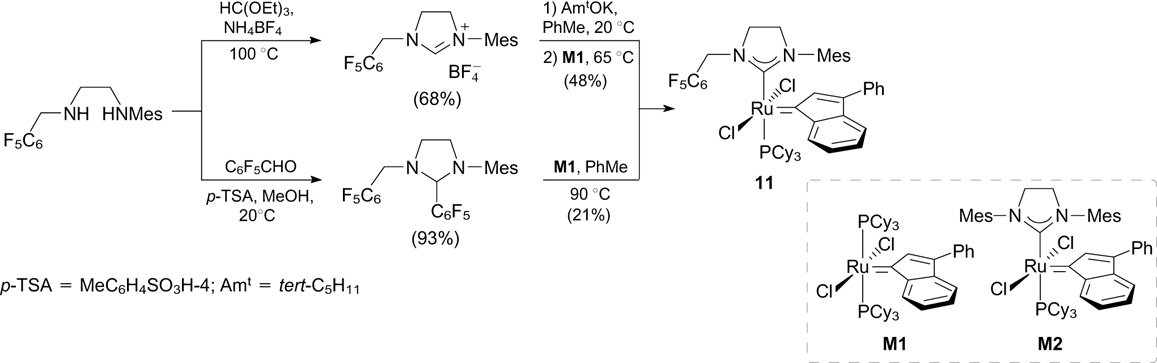
Kvíčala et al. 29 reported the modification of a second-generation Hoveyda – Grubbs catalyst with perfluoroalkyl moieties to increase its fluorophilicity. The mesityl groups of NHC ligands and the benzylidene ligands were replaced with perfluoroalkyl groups (see the next Section). A perfluoroalkyl-containing moiety was introduced by the Heck reaction of iodine-containing diimine with perfluorooctylethylene followed by the closure of the imidazolium ring and the catalytic double-bond hydrogenation (Scheme 10).
Scheme 10
Download figure:
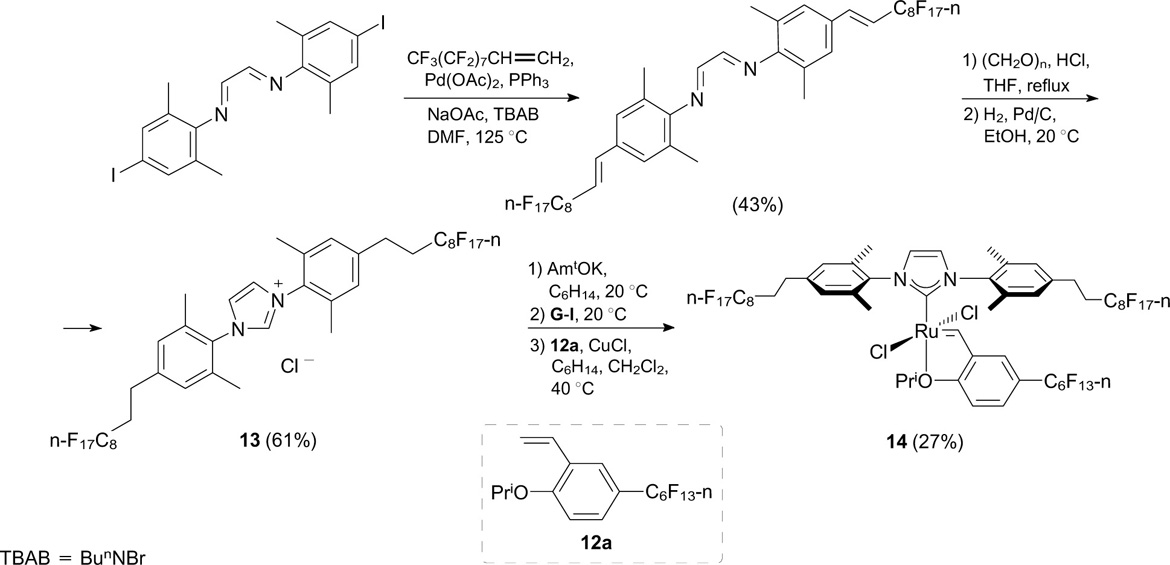
Despite the presence of three polyfluoroalkyl substituents in complex 14, the partition coefficient Pi (FBS) [the ratio of the concentrations of the substance in each phase of the fluorous biphasic system (FBS)] between perfluoro(methylcyclohexane) (PFMC) and toluene was as low as 0.16, which is too small for the catalyst regeneration. The catalytic activity of complex 14 in RCM of DEAMM was not higher than the activity of available HG-II. It is worth noting that attempts to coordinate a ruthenium atom to N,N'-dipolyfluoroalkyl NHC ligands lacking the mesityl moiety failed due apparently to low stability of sterically unhindered NHC complexes (Scheme 11). 30
Scheme 11
Download figure:
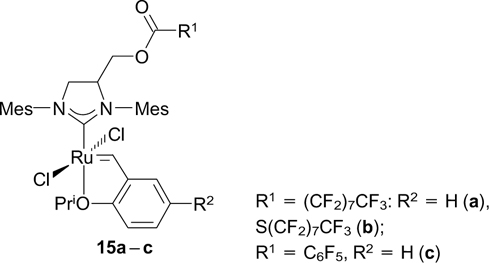
Grela and co-workers 31, 32 made efforts to synthesize fluorous metathesis catalysts. However, perfluoroalkyl-containing complexes 15a – c 31 also showed low fluorophilicity. Thus, complex 15a was insoluble in PFMC, and complex 15b formed only a stable emulsion. 31 Complexes 15 were not tested as catalysts because of low fluorophilicity.
Structures 15a–c
Download figure:
Another approach to increase the fluorophilicity was described by Kvíčala and co-workers 33 in 2015. They were the first to introduce a fluorine-containing substituent directly into the imidazolium ring using the reaction of 2-(perfluorohexyl)ethyllithium with diimines 16 (Scheme 12). The reaction resulted in the selective formation of the threo isomer (an enantiomeric mixture) in high yields.
Scheme 12
Download figure:

Diamines 17a,b were used to prepare a series of second-generation Hoveyda – Grubbs catalysts containing fluorine substituents in NHC ligands (19 and 20, Scheme 13). Catalyst 19 was synthesized via the replacement of tetrafluoroborate with a chloride ion on an Amberlite 400 (A-400) ion-exchange column.
Scheme 13
Download figure:

The partition coefficient for catalyst 19 was low [Pi (FBS) = 0.055], while it was higher than unity for 20 [Pi (FBS) = 1.1]. Therefore, a heavy fluorous Grubbs catalyst bearing fluorinated substituents only in the NHC ligand was synthesized for the first time. Both complexes showed activity in RCM of DEAMM and DEDMM comparable with that of commercially available HG-II. It should be noted that complex 19 can be reused for five cycles with a slight loss of activity (from 86 to 81% within 1 h in RCM of DEAMM) in a mixture of dichloromethane and highly fluorinated ethers HFE-7100 (a mixture of methyl perfluorobutyl and methyl perfluoroisobutyl ethers).
Recently, Kvíčala and co-workers 34 reported the synthesis of three new fluorous complexes 22 – 24 (Schemes 14, 15). Complex 22 was prepared by a standard procedure from the previously known ligand 18b and appropriate HG-I-type catalyst 21a (see Section 3, Scheme 53).
Scheme 14
Download figure:
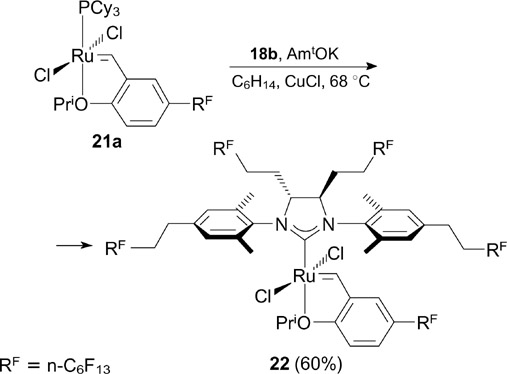
Scheme 15
Download figure:

Complexes 23 and 24 (Scheme 15) were synthesized by a special procedure assuming that the –CF2CH2– unit is a potential source of instability. The authors replaced the linear substituent CF3(CF2)5(CH2)2 with the more stable tertiary moiety CF3(CF2)4CMe2, which was synthesized by the reaction of trimethylaluminium with a perfluorohexyl group.
Complex 22 exhibited high fluorophilicity [Pi (FBS) = 14], good activity in RCM of DEDAM and reusability (the yield of the metathesis product was 89% over five cycles). The fluorophilicity of complex 23 [Pi (FBS) = 0.5] was much lower; however, this complex showed high activity in RCM of sterically hindered DEDMM. Besides, Kvíčala and co-workers 34 described fluorous modifications of Grubbs catalysts bearing perfluoroalkyl substituents in arylidene ligands and X-type ligands. These complexes are considered in Sections 3 and 4, respectively.
In 2016, Togni and co-workers 35 reported the synthesis of a series of new catalysts 25a – f containing an N-CF3 group in the benzimidazole-based NHC ligand (Scheme 16). The synthesis of the corresponding benzimidazolium salts is based on the electrophilic arylation or alkylation of N-CF3-benzimidazole. Target complexes 25a – f were prepared by the deprotonation of the salts with NaHMDS in situ in toluene in the presence of a stoichiometric amount of G-I [RuCl2(=CHPh)(PCy3)2].
Scheme 16
Download figure:
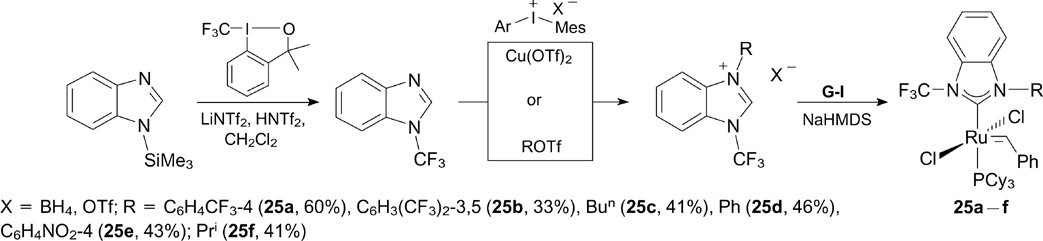
All complexes were tested in RCM of DEDAM and in CM of allylbenzene with 1,4-diacetoxybut-2-ene. However, these complexes were found to be somewhat less active than the commercial catalyst G-II. On the other hand, these complexes showed excellent chemoselectivity (up to 97% alternating dyads) in alternating norbornene – cyclooctene copolymerization and unusually high selectivity (80 – 90%) to terminal olefins in the ethenolysis of ethyl oleate.
The authors suggested that the activity of the complexes can be associated with the electronic and steric effects of the N-CF3 group in the NHC ligand, the more so that the X-ray diffraction data for complexes 25a,b and 25d provide evidence for the existence of a Ru–F interaction, which is similar to that found by Grubbs for 3b (see Scheme 4) and is responsible for its high initiation rate and activity in RCM.
In the subsequent study, 36 these authors tested the activity of complexes 25a – f and other ruthenium benzylidene complexes with different NHC ligands in the ethenolysis of cyclic alkenes, resulting in the selective formation of α,ω-dienes. It is worth noting that this metathesis transformation involving ruthenium initiators is usually accompanied by the competing ROMP reaction, giving mainly polymeric products and low yields of the target terminal dienes. Among all the N-CF3 systems under study, complex 25c proved to be the most efficient in the ethenolysis of cyclooctene, providing a high conversion (96%) and good selectivity for the formation of ethenolysis product 26 (53%) (Scheme 17).
Scheme 17
Download figure:
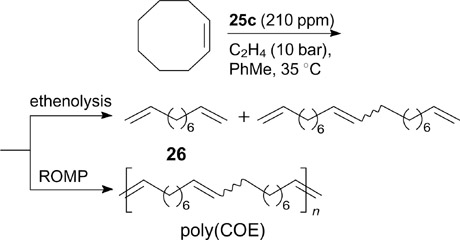
Moreover, 25c did not show the formation of even trace amounts of polycyclooctene [poly(COE)] in ROMP in the absence of ethylene. On the contrary, the use of the commercial catalyst G-II resulted in the formation of poly(COE) as the major product, while the target product was obtained in 12% yield.
These authors also evaluated the activity of 25c in the ethenolysis of challenging substrates, such as norbornene derivatives, which are the most popular monomers in ROMP due to high strain of their bicyclic structures. An efficient method was developed for the synthesis of valuable functionalized α,ω-dienes in yields higher than 70%. 36 Quantum chemical calculations were performed in order to account for the observed selectivity in the ethenolysis. These calculations confirmed the authors' hypothesis that the control of the selectivity is determined mainly by the ρ-acceptor properties of the unsymmetrical N-CF3-containing NHC ligand, which increase the relative rate of degenerate metathesis and increase the selectivity of the ethenolysis of cyclic alkenes.
In general, the presence of an unsymmetrical NHC ligand bearing the strongly electron-withdrawing CF3 group in complexes 25 and the Ru–F interaction are key factors providing high selectivity in the above-described transformations, which can be used to modulate the desired catalytic properties.
As mentioned above, the key step in the metathesis affords a metallacyclobutane intermediate, and the orientation of the substituents in the latter has a decisive effect on the stereochemical outcome of the reactions, particularly, of cross-metathesis reactions. The control of the geometry of this intermediate is an important condition for both stereoselective and stereoretentive olefin metathesis. 37,38 Recently, Endo and Grubbs 39 described the synthesis of fluorine-containing derivatives of Z-selective catalyst 28 (Scheme 18) with a chelating (through CH activation) adamantyl-based NHC ligand and catalyst 31 40, 41 with a dithiolate ligand (Scheme 19). All complexes were synthesized starting from appropriate commercially available fluorine-containing anilines. Adamantyl complexes 28a – d were tested in cross-metathesis and homodimerization reactions, in which they showed high activity and Z-selectivity; however, these parameters are not better than those of non-fluorinated analogue 27. 42
Scheme 18
Download figure:
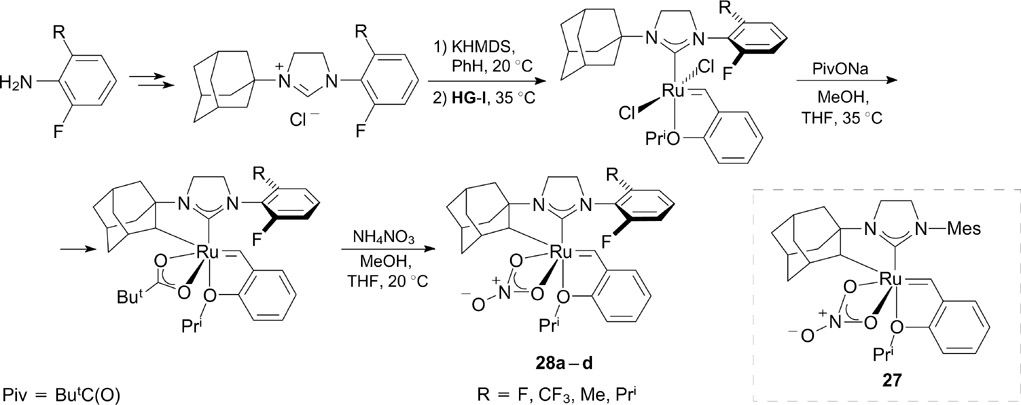
The replacement of methyl groups in the ortho positions of N-aryl substituents in thiolate complex 29 with fluorine atoms (catalysts 30 and 31, see Scheme 19) led to an increase in the yields in the metathesis of trans-alkene substrates 43 and high Z- and E-selectivity in the synthesis of trisubstituted allyl alcohols and ethers in the cross-metathesis. 44 Dithiolate ligands in complexes 30 and 31 were introduced by the transmetalation of zinc dithiolate.
Scheme 19
Download figure:
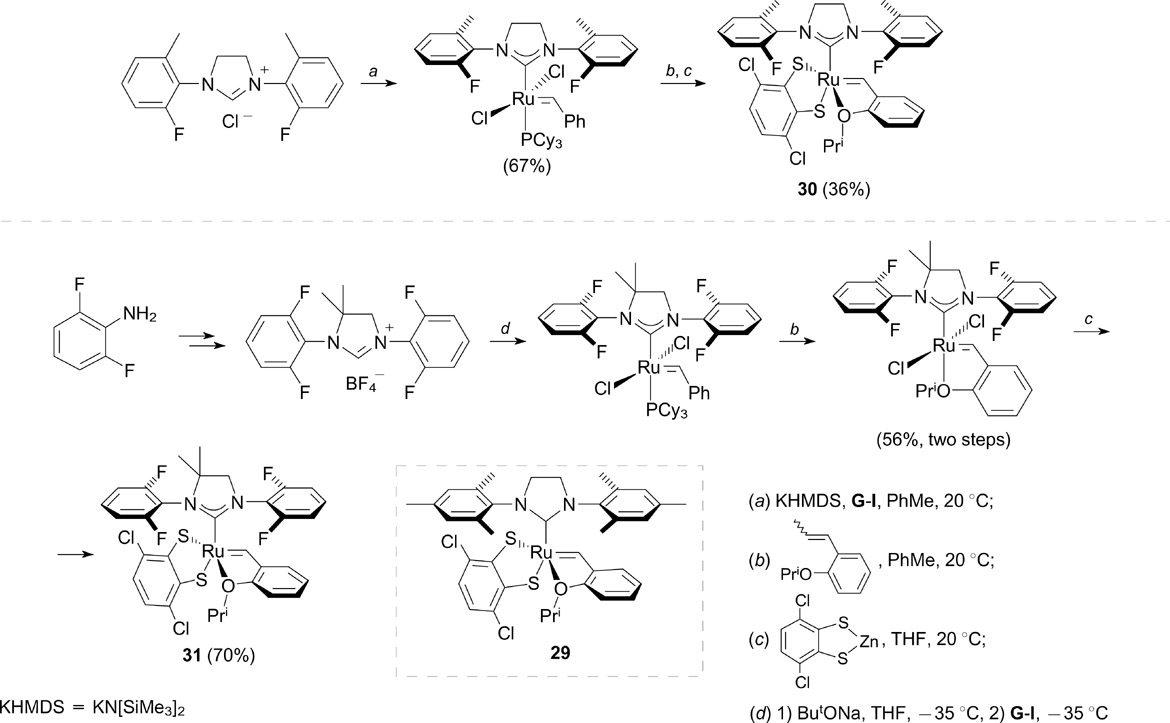
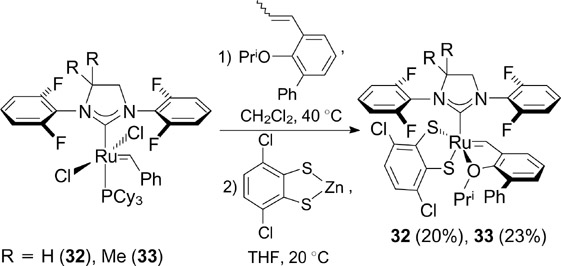
The introduction of an additional phenyl group into the benzylidene ligand of catalysts 30 and 31 (catalysts 32 and 33, Scheme 20) resulted in an increase in the rates of formation of Z isomers. 45
In 2019, Hoveyda and co-workers 46 synthesized complex 34 (Scheme 21), an unsaturated analogue of 32, and evaluated its catalytic activity in the Z-selective cross-metathesis in order to prepare (Z)-α,β-unsaturated esters, carboxylic acids and their amides highly valuable in medicinal chemistry.
Scheme 20
Download figure:
Scheme 21
Download figure:
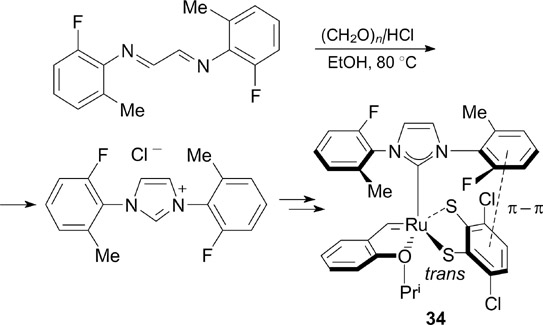
Complex 34 proved to have much higher catalytic activity and Z-selectivity than complex 32, providing the Z/E isomer ratios >98 : 2. Based on the calculated and experimental data, the authors concluded that the observed activity of 34 is associated with a π – π interaction between the N-fluoroaryl substituent and the catecholthiolate ligand due to a shortening of the Ru–CNHC and Ru–S(trans) bonds. Apparently, this provides the optimal stereochemical configuration of the substituents in the key ruthenacyclobutane intermediate.
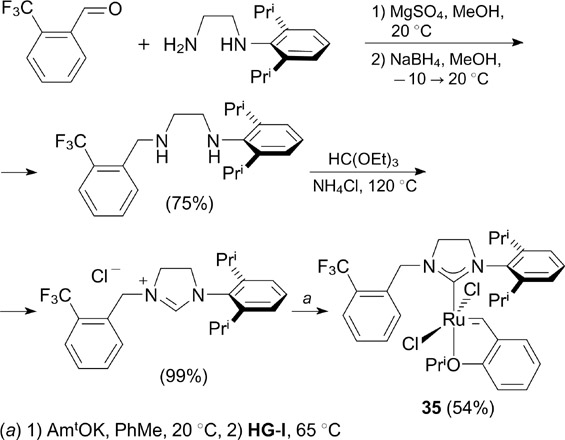
Grela and co-workers 47 reported the synthesis of complex 35 containing a trifluoromethyl group in the NHC ligand (Scheme 22). Complex 35 was prepared in three steps from commercially available N-(2,6-diisopropylphenyl)ethane-1,2-diamine and o-(trifluoromethyl)benzaldehyde. Despite high stability in solution, complex 35 was found to be somewhat less active in benchmark RCM reactions than structurally similar non-fluorinated catalysts.
Scheme 22
Download figure:
Scheme 23
Download figure:
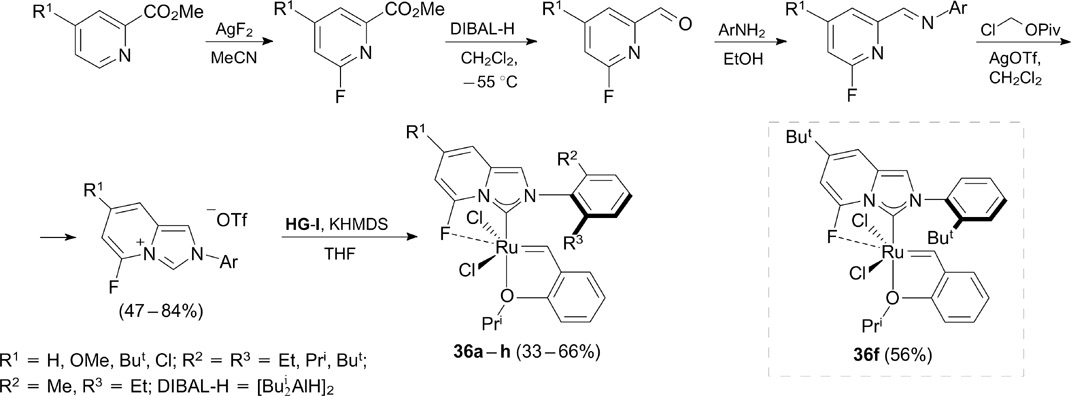
A series of ruthenium-based complexes 36 bearing unsymmetrical fluorinated imidazo[1,5-a]pyridin-3-ylidene ligands were recently synthesized and evaluated in the ethenolysis of methyl oleate. 48 The synthesis of the imidazopyridinium precursor of the NHC ligand includes: 1) the selective Hartwig's fluorination of pyridine-2-carboxylate; 49 2) the reduction of carboxylate to aldehyde followed by the reaction with anilines giving the corresponding imines and 3) the cyclization of the latter with chloromethyl pivalate and silver triflate to form the desired NHC precursors (Scheme 23). The target ruthenium carbene complexes were synthesized under standard ligand-exchange conditions using the first-generation Grubbs – Hoveyda catalyst HG-I.
These authors studied the effect of the NHC ligand structure on the catalytic activity, selectivity and thermal stability of the catalysts. According to the X-ray diffraction, the complexes are stabilized in the solid state by a Ru–F interaction. Complex 36f containing the ortho-(tert-butyl)phenyl substituent at the nitrogen atom and the σ-donor But group in the pyridine moiety was found to be the most efficient catalyst. It showed a high turnover number (TON ⩽ 6700) and high selectivity (up to 86%) to the formation of terminal olefins in the ethenolysis of methyl oleate. This catalyst also proved to be very efficient in the most challenging transformations, such as the ethenolysis of cis-cyclooctene and the natural carotenoid squalene.
Osipov and co-workers 50, 51 developed methods for the introduction of the hexafluoroisopropylalkoxy group in the ortho position of one of the N-aryl substituents in the NHC ligand in order to increase the stability and selectivity of unsymmetrical ruthenium complexes. At first it was expected that the presence of a bulky fluorine-containing group bearing an anionic centre in the vicinity of the ruthenium atom can enhance the stability of the complex through an additional Ru⋯O coordination, and the presence of two trifluoromethyl groups can improve solubility in industrially important monomers or alkanes.
First, diamines 37 were prepared by conventional methods of organic synthesis, and their subsequent heterocyclization to desired imidazolinium salts 38 (see Ref. 50) was successful only after the selective formylation of the sterically more accessible N-arylamino group followed by the intramolecular Vilsmeier – Haack cyclization (Scheme 24, step h).
Scheme 24
Download figure:
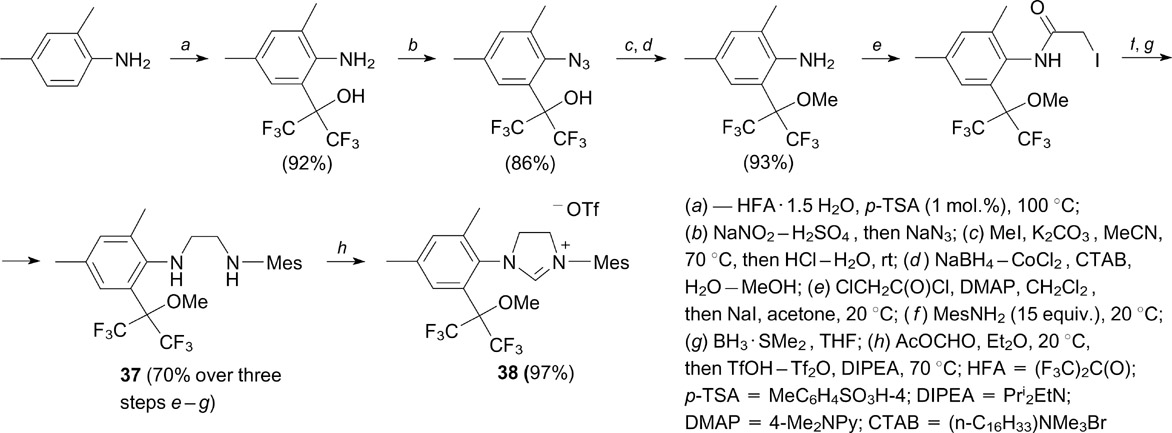
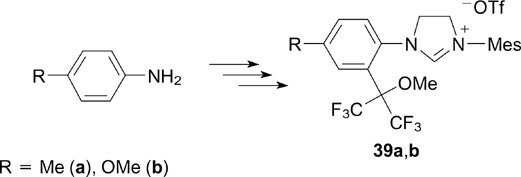
This method was applicable to the synthesis of salts 39a,b, 50 lacking the methyl group at the ortho position, starting from para-toluidine and para-anisidine (Scheme 25).
Scheme 25
Download figure:
Salts 38 and 39a,b were used to synthesize ruthenium complexes 40 and 41 (Scheme 26). Appropriate NHC ligands were generated in situ using KHMDS or AmtOK in toluene in the presence of first-generation Grubbs (G-I) and Hoveyda – Grubbs (HG-I) complexes to accomplish the ligand exchange.
Scheme 26
Download figure:
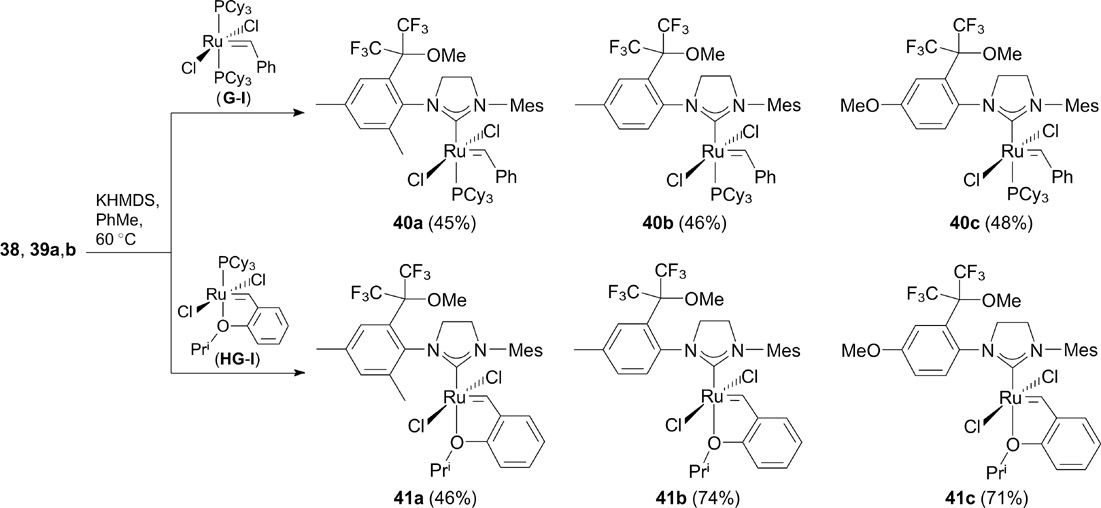
Compounds 40 and 41 were evaluated for catalytic activity in RCM and CM reactions. It was found that the activity of these compounds is comparable with and sometimes even higher than that of the catalysts G-II and HG-II. 50, 51 For instance, fluoroalkyl-containing Hoveyda – Grubbs-type complexes 41b,c bearing a mono-ortho-aryl-substituted NHC ligand exhibit unusually high initiation rate in the cross-metathesis of 1,3-diacetoxybut-2-ene with allylbenzene, resulting in the conversion more than 10% higher than that achieved with HG-II. 51
This research group also developed an efficient approach to the synthesis of similar ruthenium complexes 45 and 46 bearing unsaturated N,N'-diarylimidazolyl-2-ylidene NHC ligands (see Scheme 28). 52 The method for the preparation of NHC precursors is based on an original strategy for the synthesis of sterically hindered imidazolium salts 53 involving the heterocyclic interconversion of oxazolinium salts on treatment with substituted anilines. This strategy is also applicable to fluorine-substituted arylamines containing two nucleophilic centres. Thus, the reaction of Fürstner's oxazolinium salt with ortho-hexafluoroisopropoxyaniline resulted in the selective formation of desired hydroxy-containing imidazolium salt 42 (Scheme 27); this reaction also afforded an insignificant amount of tricyclic compound 43, which is apparently due to the heterocyclization at the hydroxy group. In pure form, salt 42 can be isolated by a single crystallization from a chloroform – diethyl ether mixture in good yields even at high loads. 54
Scheme 27
Download figure:
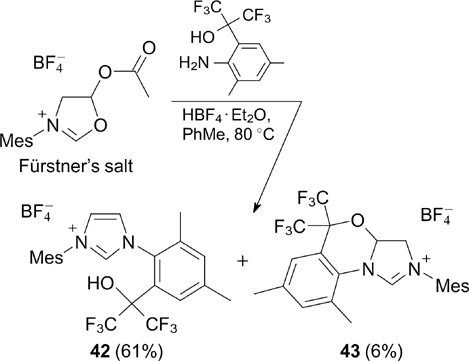
The free hydroxy group in compound 42 can be directly functionalized via O-alkylation in the presence of potassium carbonate giving salts 44a – c in high yields. The latter proved to be suitable NHC precursors in the synthesis of complexes 45 and 46 (Scheme 28).
Scheme 28
Download figure:
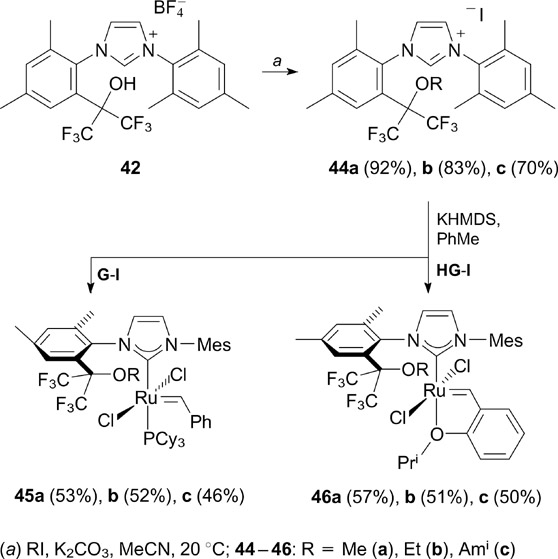
Grubbs-type complexes 45a – c were found to be much more active in RCM reactions of diallyl- and allyl methallyl malonates 52 compared to non-fluorinated unsaturated symmetrical analogue of the complex G-II.
The reaction of Fürstner's oxazolinium salt with fluorinated binucleophilic aniline affords OH-substituted imidazolium salt 42 as the target product along with unusual by-product 43 (see Scheme 27). Due to the rigid tricyclic structure, the latter compound is of interest for the preparation and evaluation of catalytic activity of the corresponding ruthenium complexes. For this purpose, a route to the selective synthesis of these compounds as triflate salts was specially developed. The synthetic sequences included the reaction of fluorine-containing anilines with readily accessible N-aryl dialdehydes 53, 55 – 58 giving the corresponding benzoxazines. The subsequent intramolecular heterocyclization via activation of the formyl group under the conditions found for the synthesis of salts 38 and 39 (see Schemes 24 and 25) gave desired tricyclic products 47a – c in high yields (Scheme 29).
Scheme 29
Download figure:

Stable ruthenium complexes 48a – c bearing sterically rigid NHC ligands were synthesized via a standard route from salts 47a – c and evaluated for catalytic activity. It was found that allylbenzene complexes 48a – c caused almost no isomerization in the self-metathesis (SM) and provided high yields of the target self-metathesis product and excellent selectivity. 54 Apparently, the observed selectivity is attributed to the rigid structure of the NHC ligand, resulting in the close arrangement of the ortho substituent of the aromatic moiety and the ruthenium centre, which interferes with the isomerization via the most probable hydride mechanism. 59 – 61
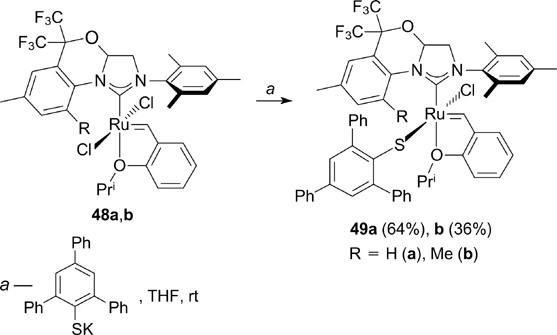
In order to design new metathesis catalysts capable of inducing Z/E selectivity, complexes 48a,b were used in the synthesis of monothiolate derivatives 49a,b (Scheme 30). However, despite moderate activity in RCM and CM reactions, these compounds did not induce significant Z-selectivity in the self-metathesis of allylbenzene. 62
Scheme 30
Download figure:
3. Fluorine in arylidene ligands
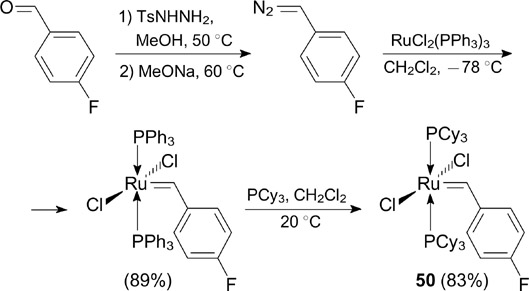
The first G-I-type catalyst bearing a fluorinated arylidene ligand 50 was synthesized in 1996 by Grubbs and co-workers 63 from the diazo derivative of commercially available p-fluorobenzaldehyde (Scheme 31). The presence of a fluorine atom at the para position of the benzylidene ligand was found to have a weak influence on the kinetic profile of the catalyzed reaction.
Scheme 31
Download figure:
Blechert and co-workers 64 synthesized catalysts 51 – 53 bearing a fluorine-substituted arylidene ligand by the replacement of styrene ligands in complexes G-I and G-II with the corresponding fluorinated styrene derivatives (Scheme 32). The phosphine-containing catalysts (51a, 52a and 53a) exhibited higher activity compared to HG-I, whereas the phosphine-free complexes (51b, 52b and 53b) showed activity very similar to that of HG-II.
Scheme 32
Download figure:

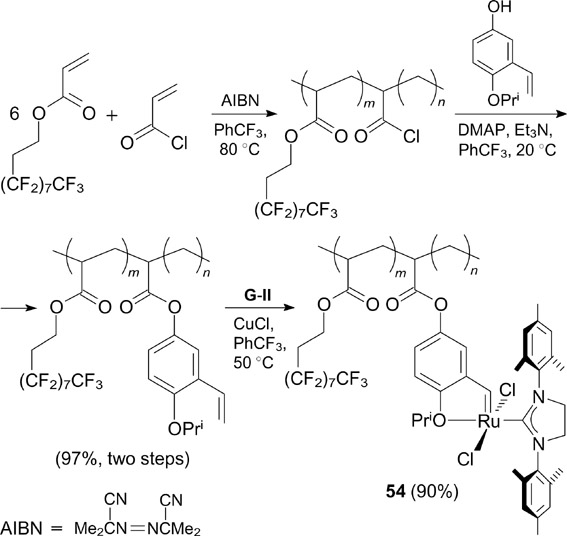
Yao and Zhang 65 described a Hoveyda – Grubbs catalyst immobilized via an arylidene ligand on a perfluoroalkyl-containing polymer. The polymer was synthesized from commercially available heptadecafluorodecyl acrylate (Scheme 33). The synthesized catalyst 54 not only showed excellent solubility in perfluorohexane and trifluoromethylbenzene and high activity in RCM with a number of substrates but also exhibited high recyclability. For example, high activity of this catalyst in RCM of N,N-diallyltosylamide was maintained even after 20 cycles.
Scheme 33
Download figure:
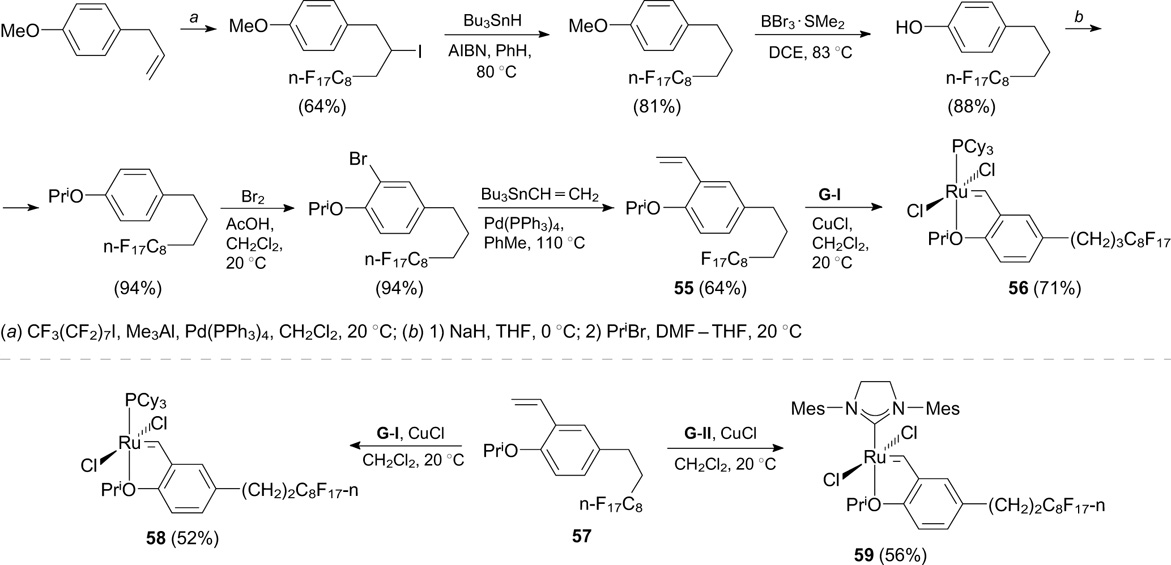
In 2005, Matsugi and Curran 66 described light fluorous versions of first- and second-generation Hoveyda-type catalysts. Styrene 55 was synthesized by the trimethylaluminium-induced addition of perfluoro-n-octyl iodide at the double bond followed by the dehalogenation with tributyltin hydride (Scheme 34). 67 Complexes 58 and 59 were synthesized from commercially available styrene 57. Catalysts 56, 58 and 59 were evaluated for activity in RCM reactions. The complexes exhibited activity similar to that of their non-fluorinated analogues HG-I and HG-II and showed good recyclability.
Scheme 34
Download figure:
Compound 60 homologous to complex 56 was prepared in a similar way, and its activity was evaluated in the ene – diene cross-metathesis (Scheme 35). 68 Complex 60 retained activity similar to that of HG-II after three reuses. The catalyst was recovered from the reaction mixture by fluorous solid-phase extraction (F-SPE).
Scheme 35
Download figure:

Besides, complex 60 was used in the intramolecular metathesis to prepare a series of heterocyclic compounds, including macrocycles, followed by the application of F-SPE for the catalyst regeneration. 69,70
Michalek and Bannwarth 71 developed a method for the synthesis of an HG-II-type complex (61) containing a perfluorooctyl group for non-covalent immobilization on fluorous silica gel (Scheme 36). The catalytic activity of the synthesized catalyst was evaluated in different RCM reactions. The immobilization of catalyst 61 on an amphiphilic polymer conetwork (APCN) made it possible to perform RCM in an aqueous medium. 72 The appropriate perfluoroalkyl-containing styrene was synthesized using trialkylbromosilane. 73
Scheme 36
Download figure:

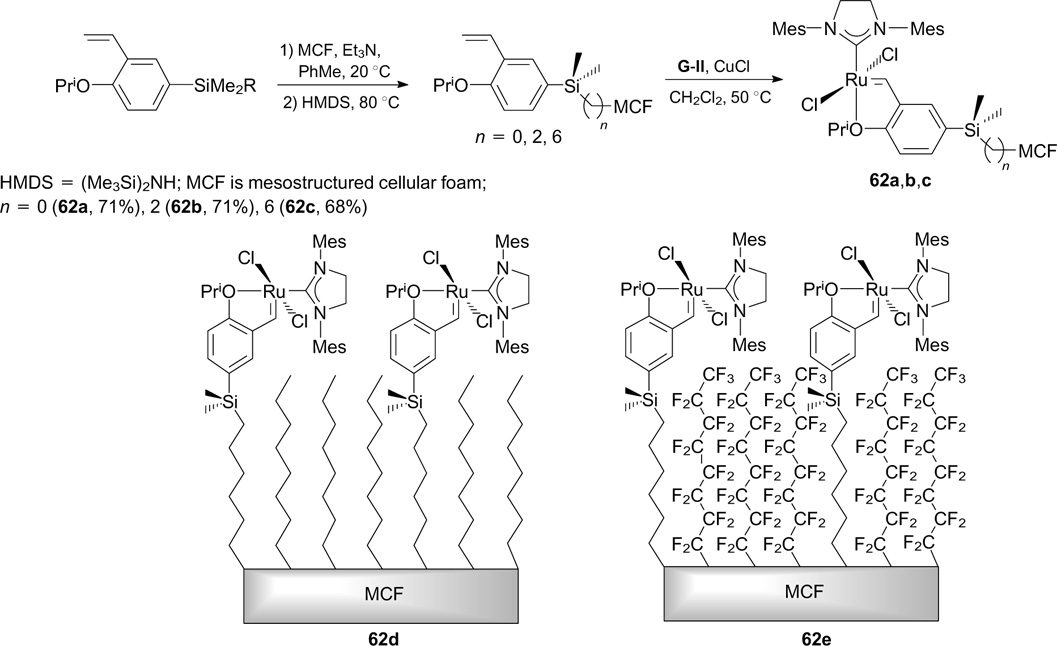
The covalent immobilization of a Hoveyda – Grubbs-type catalyst on mesostructured cellular foam (MCF) containing perfluoroalkyl groups (62e, Scheme 37) as the fluorous solid support was performed through silyl linkages of arylidene ligands. Immobilized catalysts 62a – e showed good recyclability in RCM reactions. 74
Scheme 37
Download figure:
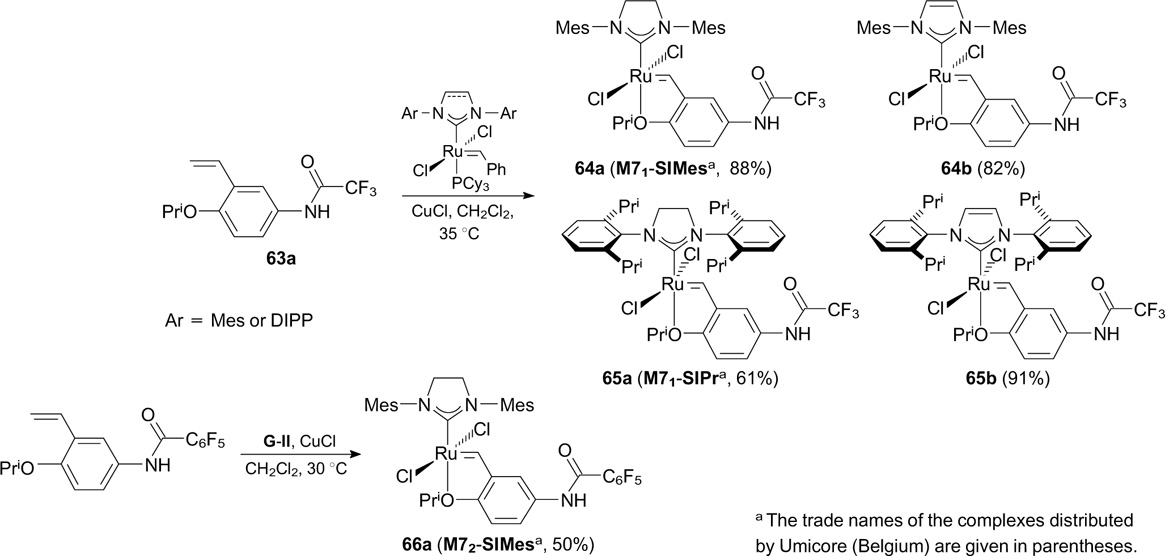
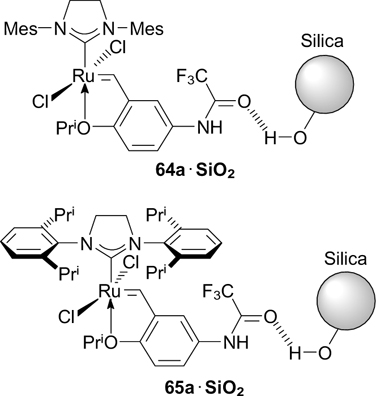
A series of trifluoroacetamide-containing Hoveyda – Grubbs complexes 64 and 65 were synthesized by Nolan's and Mauduit's groups (Scheme 38). 75, 76 Starting styrene 63a was prepared by the acylation of 4-isopropoxy-3-vinylaniline with trifluoroacetic acid. Catalysts 64a,b and 65a,b exhibited high activity in a wide range of metathesis transformations (RCM, RCEYM and CM), including large-scale reactions with alkene substrates. Pentafluorophenyl-containing complex 66a was found to be less active than 64a in RCM of DEDAM. 77 In all cases, the NHC(O)CF3 group had a beneficial effect on the catalytic activity of the complexes. Catalysts 65 with 2,6-diisopropylphenyl-containing ligands proved to be more efficient with sterically non-hindered substrates. Besides, the purification of reaction mixtures by silica gel column chromatography resulted in very low residual ruthenium contamination in the reaction products (less than 2.5 ppm). Such high degree of purification was possible due to the formation of hydrogen bonds between oxygen atoms of trifluoroacetamide groups and silica gel.
Scheme 38
Download figure:
Subsequently, the activity of catalysts 64a and 65a was tested in different RCM, 78,79 CM 80,81 and ethenolysis reactions. 82,32 In all cases, these catalysts were found to be more active than their non-fluorinated analogues. They also showed satisfactory results in the metathesis macrocyclization for the construction of the 16-membered bafilomycin macrolactone core on the model substrate (Table 2). 83
Table 2. Synthesis of bafilomycin-type macrolactone and the results of catalysis.
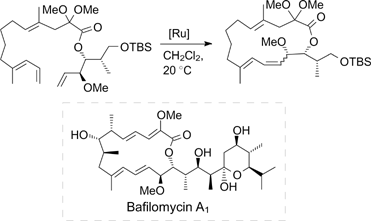
| ||||
|---|---|---|---|---|
| [Ru] | mol.% | Conversion (%) | Z/E | Yield (%) |
| G-I | 20 | ∼10 | 1 : 1 | – |
| G-I | 20+20 | > 90 | 7 : 3 | – |
| 64a | 5 | ∼35 | 7 : 3 | – |
| 65a | 5 | ∼35 | 7 : 3 | – |
| 67 | 5 | ∼35 | 7 : 3 | – |
| 67 | 10+10 | > 90 | 7 : 3 | 48% |
However, the best results were achieved using fluorine-containing first-generation catalyst 67, which was synthesized from the commercially available indenylidene catalyst M1 (Scheme 39). 83
Scheme 39
Download figure:
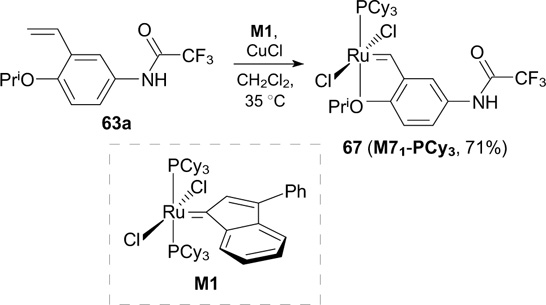
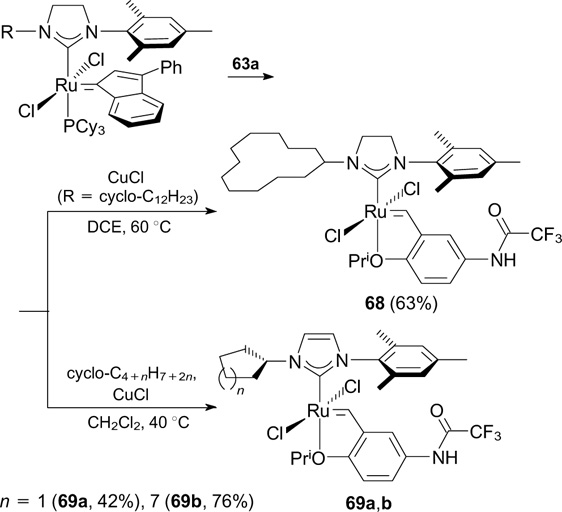
Catalysts 68 and 69a,b bearing a trifluoroacetamide-substituted benzylidene ligand and a cycloalkyl-substituted NHC moiety were synthesized in 2014 (Scheme 40). However, these catalysts showed lower stability and exhibited lower activity in RCM of DEDMM compared to 64a,b 84 (see Scheme 38).
Scheme 40
Download figure:
The synthesis of heterogeneous analogues of catalyst 64a was described in several publications in 2014 – 2016. For instance, complex 70 containing the anthracenyl group was immobilized on modified silica via the formation of a charge-transfer complex, in which the anthracenyl group served as a donor and the electron-deficient 2,4,7-trinitrofluoren-9-one moiety acted as an acceptor (Scheme 41). 85 The immobilized catalyst 70@SiO2 retained high activity in RCM of DEDAM after four reuses, and it was also tested for recyclability in different types of metathesis reactions (RCM, RCEYM, CM), where it showed high reaction yields and selectivity.
Scheme 41
Download figure:

The ability to form hydrogen bonds between trifluoroacetamide-containing complexes and silica gel was employed to prepare heterogeneous systems. Compounds 64a and 65a were immobilized on silica without surface pretreatment. 86,87 Immobilized catalytic systems 64a@SiO2 and 65a@SiO2 showed high activity in RCM, CM and RCEYM reactions in flow conditions. A significant decrease in the catalytic activity was observed after the fifth cycle.
Recently, the immobilization of catalysts bearing the trifluoroacetamide-based styrene ligand onto reduced graphene oxide (RGO) (Scheme 42) was reported. 88 The catalyst 71@RGO proved to be active in RCM of DEDAM, but a significant loss of activity was already observed after the second cycle.
Structures 64@SiO2 and 65@SiO2
Download figure:
Scheme 42
Download figure:
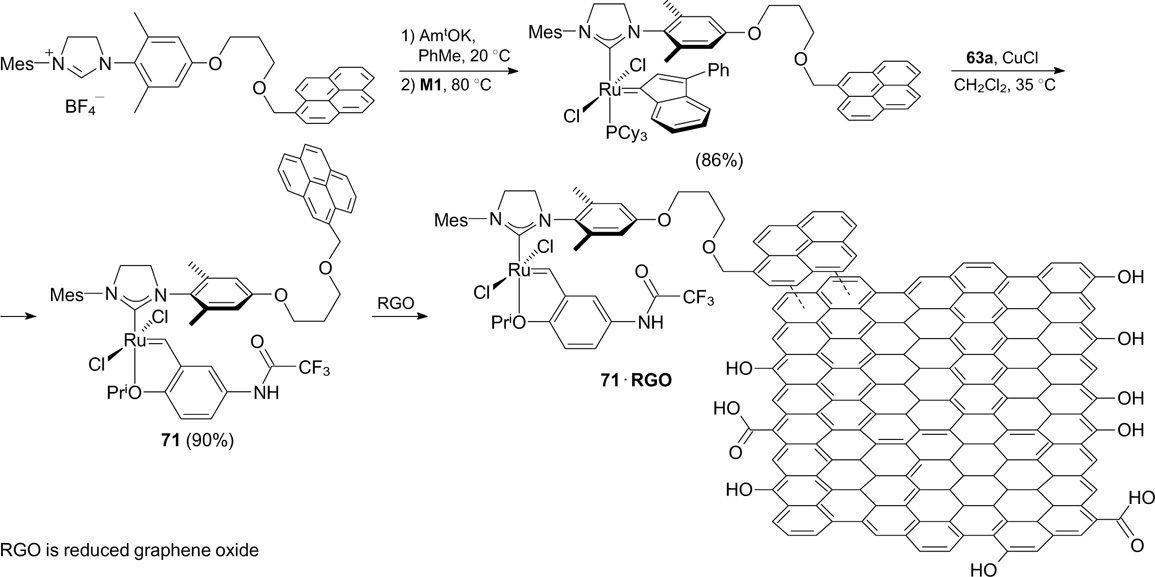
In the past decade, several latent fluorine-containing metathesis catalysts were described. Lemcoff and co-workers 89, 90 synthesized complexes 72a – c bearing the benzylidene ligand with the chelating trifluoromethylthio group in two steps starting from commercially available 2-bromophenyl trifluoromethyl sulfide (Scheme 43).
Scheme 43
Download figure:
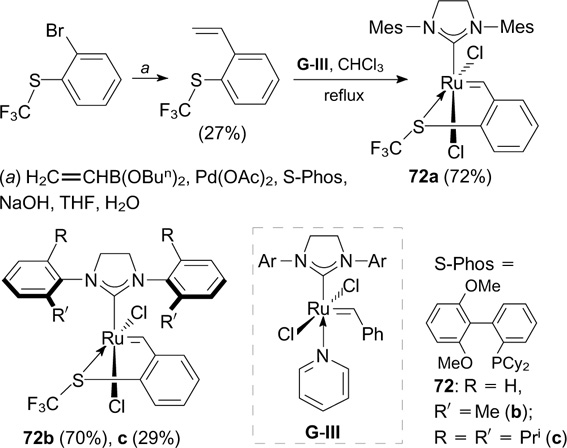
These complexes exist in the cis-dichloro configuration, which is inactive under standard conditions in RCM, CM and ROMP reactions; however, they isomerize to the active trans conformation under UV irradiation or heating (Scheme 44).
Scheme 44
Download figure:

Complexes 72b,c containing the ortho-tolyl and 2,5-diisopropylphenyl substituents, respectively, in the NHC ligand 91 were tested in RCM and CM reactions. Complex 72b showed high activity towards sterically hindered DEDMM or N,N-dimethallyltosylamide under thermal or UV activation conditions. Complex 72c proved to be the most active towards unhindered substrates.
Besides, Lemcoff and co-workers described the application of complexes 72a in the two-step photochemical synthesis of coumarins, 92 butenolides and levulinates 93 (Scheme 45). The UV-activated cross-metathesis was a key step in all cases.
Scheme 45
Download figure:

The introduction of electron-withdrawing substituents in the para position of the styrene ligand leads to a significant decrease in the donor properties of the oxygen atom in catalysts HG-II, resulting in an increase in the initiation rate of the complex due to acceleration of the ligand dissociation. In order to further improve the properties of the leaving group, electron-withdrawing substituents can also be introduced in the ortho-ester function of styrene. Generally, this modification leads to a significant increase in the conversion of RCM, the reaction rate and the product yields with a minimum catalyst load.
Taking into account these facts, Duan et al. 94 designed catalytic systems based on trifluoropyruvate imines, where the chelation occurs through double coordination with imine nitrogen and ester oxygen atoms (Scheme 46). The double coordination was established by X-ray diffraction.
Scheme 46
Download figure:
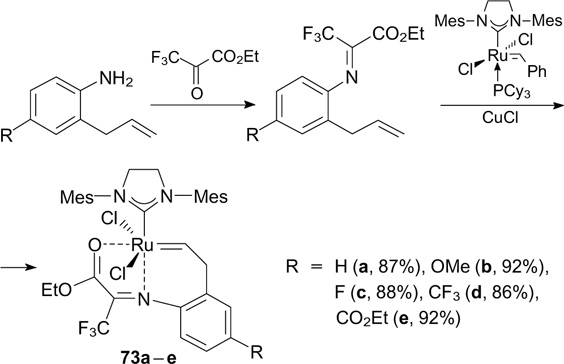
Complexes 73d,e containing additional electron-withdrawing groups (R = CF3, CO2Et) in the para position of the styrene ligand were found to have the highest activity and showed high conversion, initiation rates and yields in RCM reactions with a wide range of functionalized substrates.
Grela and co-workers 95 synthesized catalyst 74 with a covalent chelating trifluoroamide ligand by the replacement of the ligand in the complex M31 (Umicore) in the presence of pyridine (Scheme 47). Catalyst 74 was tested in RCM of DEDAM and DEAMM. Its activity was low in neutral medium, but a significant increase in the activity was observed upon activation with an ethereal solution of HCl. Fluorine-containing catalyst 74 was found to be more active in RCM of DEDAM compared to its non-fluorinated analogue 75.
Scheme 47
Download figure:
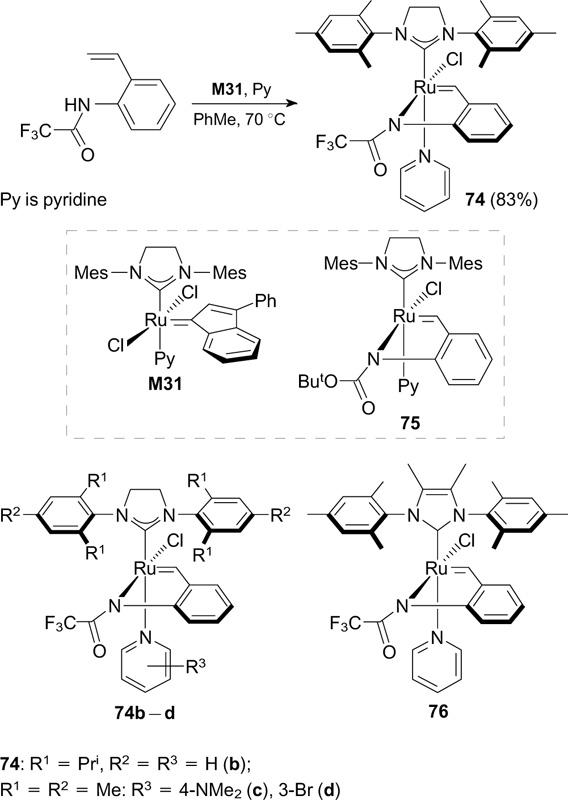
Complexes 74b – d and 76 were prepared in a similar way. Upon activation with HCl or Me3SiCl, these complexes can be used as efficient catalysts in different RCM reactions, enyne cycloisomerization and CM, in particular, with multifunctional substrates. Moreover, due to their latency and possibility of on-demand activation, fluorine-containing amide complexes 74a – d and 76 can be employed in ROMPs of dicyclopentadiene (DCPD) and endo,exo-bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid. However, low solubility of these complexes in non-polar solvents limits their commercial applications. 96
Structurally similar carbamate complexes 77 bearing a para-CF3 group in the styrene ligand (Scheme 48) were synthesized by Pietraszuk and co-workers 97 and were tested in RCM of DEDAM and DEAMM, in ROMP of COD and in CM. In all cases, the complexes showed latent behaviour. Two equivalents of HCl were required for their activation. The presence of the electron-withdrawing trifluoromethyl group promotes an increase in the activity compared to their non-fluorinated analogues.
Scheme 48
Download figure:
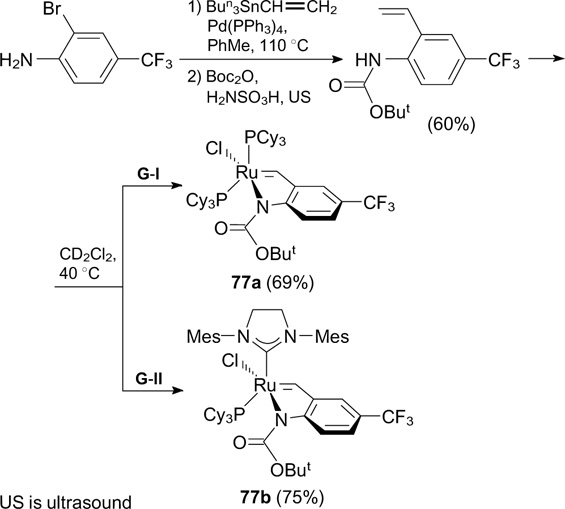
In 2008 – 2010, Matsugi et al. 98, 99 reported the synthesis of catalysts 78 containing a perfluoroalkyl group directly bonded to the aromatic ring of the benzylidene ligand (Scheme 49). The perfluoroalkylation of salicylaldehyde was performed in the presence of the radical initiator V-70L [a fraction of the commercially available initiator V-70, m.p. = 58 °C, 100 (±)-2,2'-azobis(2,4-dimethyl-4-methoxyvaleronitrile) 101 ] and caesium carbonate as a soft base. These conditions made it possible to perform the reaction at room temperature.
Scheme 49
Download figure:
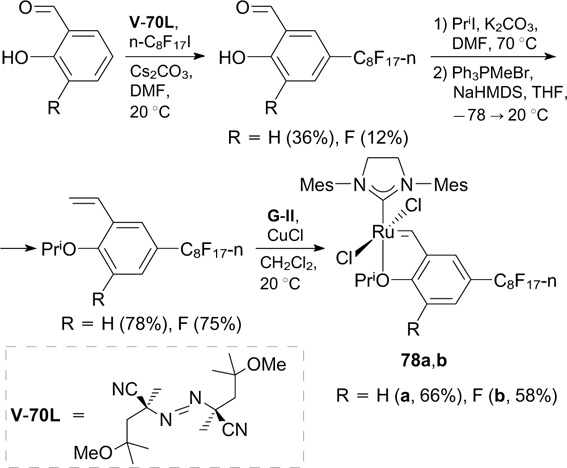
Compared to HG-II and 59 (see Scheme 34), catalysts 78a,b showed higher activity in RCM of DEDAM, DEAMM and N,N'-diallyltosylamide. Catalyst 78a was recovered using F-SPE in each iteration. It was demonstrated that this catalyst can be reused up to five times, with excellent yields of both the recovered catalyst and the reaction products. Meanwhile, complex 78b was not recoverable.
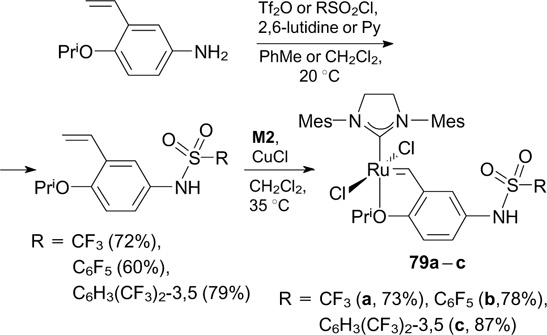
Mauduit and co-workers 102 reported the synthesis of complexes 79a – c bearing fluorinated sulfonamide groups. In this approach, the corresponding styrenes were prepared by the sulfonation of 4-isopropoxy-3-vinylaniline with trifluoromethanesulfonic anhydride or aryl sulfonyl chlorides (Scheme 50). Catalysts 79a – c showed low activity in RCM of DEAMM and in CM with different substrates.
Scheme 50
Download figure:
Caijo and Mauduit 103 patented the synthesis of new catalysts 80a, 81a – c and 82b bearing the 1,4-oxazine moiety in the benzylidene ligand (Scheme 51). This modification provides an approach to the further synthetically facile fine tuning of the complexes. A fluorine-containing moiety was introduced via acylation of the appropriate amine with pentafluorobenzoyl chloride. The commercial indenylidene catalysts M2 and M21 (Umicore) were employed as the starting ruthenium complexes.
Scheme 51
Download figure:
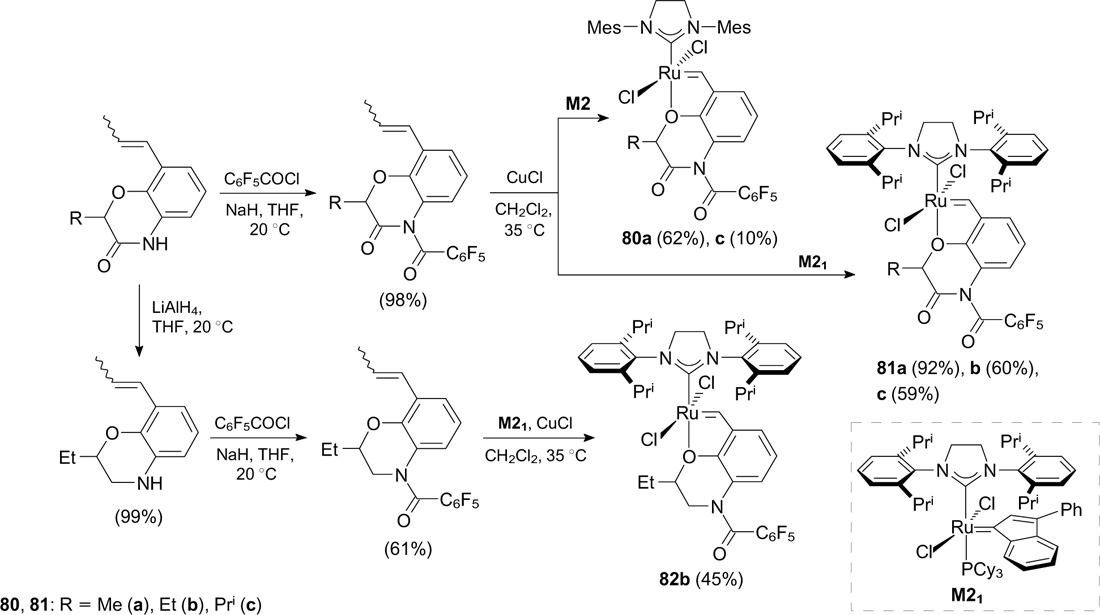
In RCM of DEAMM, catalysts 81b 104 and 82b 105 showed high activity and initiation rates. It was found that the replacement of mesityl groups in complexes 80a,c by the diisopropylphenyl moiety (81a,c) leads to a significant decrease in the initiation rate with a simultaneous increase in the stability of these complexes in solution. It was also demonstrated that the structure of the benzylidene ligand plays a key role in high activity of catalyst 81c. 106
In 2013, Grela and co-workers 107 reported the synthesis and catalytic properties of a series of Hoveyda – Grubbs-type catalysts containing different electron-withdrawing groups. The catalytic properties were fine-tuned using the weak electron-withdrawing perfluoro-n-propylthio group (83a) and the strong electron-withdrawing perfluoro-n-propylsulfonyl group (83b) (Scheme 52). Perfluoroalkyl groups were introduced via the nucleophilic substitution of perfluoropropyl iodide in the presence of potassium carbonate. As expected, the initiation rate of catalyst 83b was high, but the catalyst was found to be unstable even at low temperatures.
Scheme 52
Download figure:

Kvíčala et al. 29 described, apart from the synthesis of imidazolium salt 13 containing two perfluoroalkyl groups (see Scheme 10), the synthesis of perfluoroalkyl-containing styrenes 12a,b and 85 and the corresponding light fluorous catalysts 14, 21a,b, 84 and 86 (Schemes 53 and 54). In all cases, the Ullmann reaction was used to introduce a perfluoroalkyl group capable of increasing the activity of the catalysts and fluorophilicity. Catalysts 14, 21a,b and 84 showed high activity in RCM reactions.
Scheme 53
Download figure:

Scheme 54
Download figure:

Bis-perfluoroalkyl-containing styrene 85 proved to be inactive in the reaction giving complex 86 (Scheme 54) and was not capable of coordinating to the ruthenium atom through an oxygen atom. For Hoveyda – Grubbs-type catalysts, the inability for chelation of benzylidene ligands with low electron density at the oxygen atom was described for the first time by Grela and co-workers 108 in 2004.
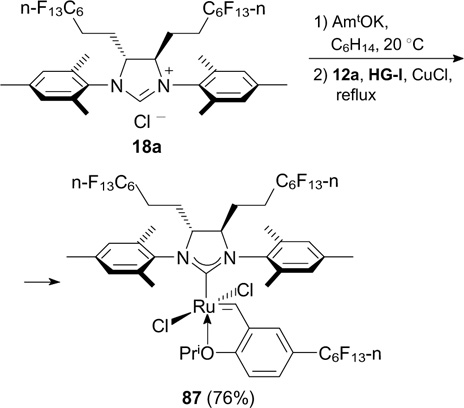
More recently, Kvíčala and co-workers 33 synthesized complex 87 containing perfluoroalkyl substituents both in NHC and benzylidene ligands (Scheme 55). However, the new complex did not act as a heavy fluorous catalyst and was less active in RCM reactions than HG-II.
Scheme 55
Download figure:
The possibility of immobilization of perfluoroalkyl-containing Hoveyda-type catalysts on a polytetrafluoroethylene support was described by Matsugi and co-workers 109 in 2015. Complex 88 containing the perfluorodecyl group was the first example of Hoveyda – Grubbs-type complexes that can transfer between the liquid and solid phases in RCM reactions (Scheme 56), unlike complex 78a synthesized previously. Complex 88 provided excellent yields of both the reaction products and the recovered catalyst in RCM with different substrates.
Scheme 56
Download figure:

In the subsequent study, this research group 110 reported the synthesis of new perfluoroalkyl-containing catalysts 89a – f and 90a,b and their catalytic activity in RCM. Fluorinated substituents were introduced into aromatic substrates by the above-mentioned methods. 1-Naphthyl-containing catalyst 89d showed the highest activity in the metathesis.
Structures 89, 90
Download figure:

4. Fluorine in indenylidene, allenylidene and methylidene ligands
In 2001, Grubbs and co-workers 111 synthesized the first ruthenium difluorocarbene complex 91 (Scheme 57) when studying the metathesis of 1,1-difluoroethylene as one of the challenging substrates in the presence of catalyst G-II. It was found that this reaction was not catalytic because secondary metathesis products, such as ethylene and tetrafluoroethylene, were not detected. However, the double bond in difluoroethylene is initially cleaved in a metathesis fashion and, consequently, it is the first example of difluoroalkene metathesis. Complex 91 was tested in ROMP of cyclooctadiene, in which it showed low catalytic activity.
Scheme 57
Download figure:

Six years later, Johnson and co-workers 112 reported the synthesis of second- and third-generation monofluoromethylidene catalysts (92a and 92b, Scheme 58). β-Fluorostyrene was prepared under mild conditions by the electrophilic fluorination of potassium alkenyltrifluoroborate in the presence of Selectfluor. 113 The catalytic activity of complexes 92a and 92b was higher than that of related difluorocarbene compound 91 but it was still rather low. Catalyst 92b was less stable in solution and had a higher initiation rate than 92a.
Scheme 58
Download figure:

Despite rather high stability in the solid state, complex 92a is prone to form carbide complex 93 in solution (Scheme 59). The reaction occurs slowly in dichloromethane with a long induction period.
Scheme 59
Download figure:

The most recently available ruthenium monofluoromethylidene complex 94 (Scheme 60) was described in 2009. 114 Initially, it was identified as an intermediate in CM reactions of disubstituted alkenes and vinyl fluoride in the presence of HG-II. Subsequently, this catalyst was synthesized as the target product in high yield by the reaction of an excess of vinyl fluoride with HG-II. The dinuclear structure of the complex was established by X-ray diffraction. In CM of 5-decene and vinyl fluoride, complex 94 showed TON = 1.2.
Scheme 60
Download figure:

Dixneuf and co-workers 115 were the first to synthesize cationic allenylidene complex 95 containing fluorine atoms in the para positions of phenyl groups of the allenylidene ligand (Scheme 61).
Scheme 61
Download figure:

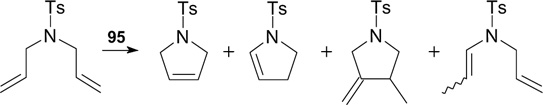
In RCM of N,N-diallyltosylamide, complex 95 showed higher activity than the non-fluorinated analogue but it was less selective. The by-products generated in the reaction are shown in Scheme 62.
Scheme 62
Download figure:
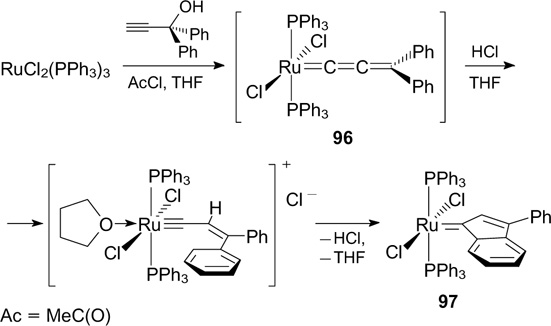
Ruthenium indenylidene complexes are also efficient catalysts for the olefin metathesis, which are rather resistant to severe conditions. An important advantage of such catalysts is that they are readily accessible. Thus, the indenylidene complex was synthesized by the reaction of 1-arylpropargyl alcohol with tris(triphenylphosphine)ruthenium(II) chloride in the presence of Lewis acid (Scheme 63). The reaction proceeds through the formation of allenylidene complex 96, which can also be isolated from the reaction mixture. The method is general for the synthesis of indenylidene complexes of not only ruthenium but also other transition metals. 116 Generally, the subsequent transformations of complex 97 involve the replacement of the triphenylphosphine ligand with the tricyclohexylphosphine one using an excess of tricyclohexylphosphine.
Scheme 63
Download figure:
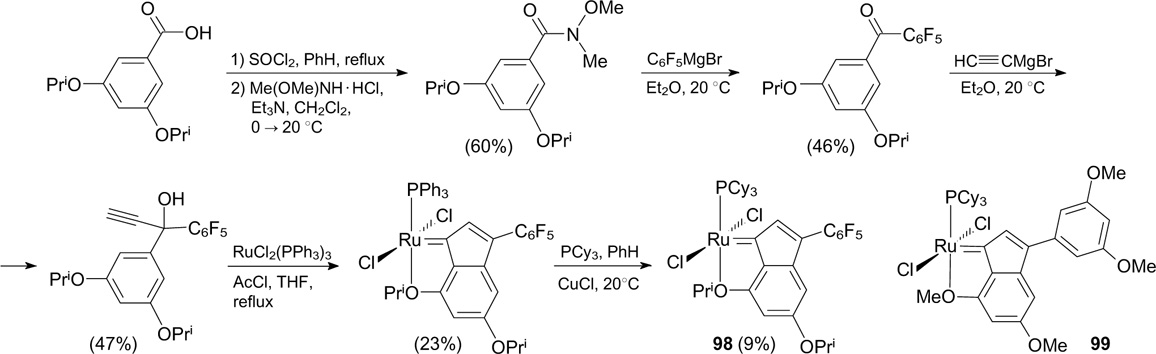
In the patent, Holtcamp and co-workers 117 described the synthesis of indenylidene catalyst 98 bearing the pentafluorophenyl group (Scheme 64). The fluorinated group was introduced via the reaction of 3,5-diisopropoxy-N-methoxy-N-methylbenzamide with pentafluorophenylmagnesium bromide. Catalyst 98 showed a high turnover number in the ethenolysis of methyl oleate (TON = 4300), whereas, e.g., methoxy-containing catalyst 99 had TON = 1800. This large difference can be attributed to the strong electron-withdrawing properties of the pentafluorophenyl group, resulting in a decrease in the strength of the ruthenium – oxygen coordination bond and an increase in the rate of the initiation step of the metathesis. 118
Scheme 64
Download figure:
Bruneau and co-workers 119 described another modification of the chelating indenylidene ligand. A fluorine atom was introduced into the substrate by the reaction of 2,6-difluorobenzaldehyde with 3,5-diisopropoxyphenyllithium (Scheme 65). Carbene complex 100 was prepared in the green solvent dimethyl carbonate. The synthesized complex 101 proved to be efficient in RCM of DEDAM and DEAMM and was found to be thermally stable.
Scheme 65
Download figure:
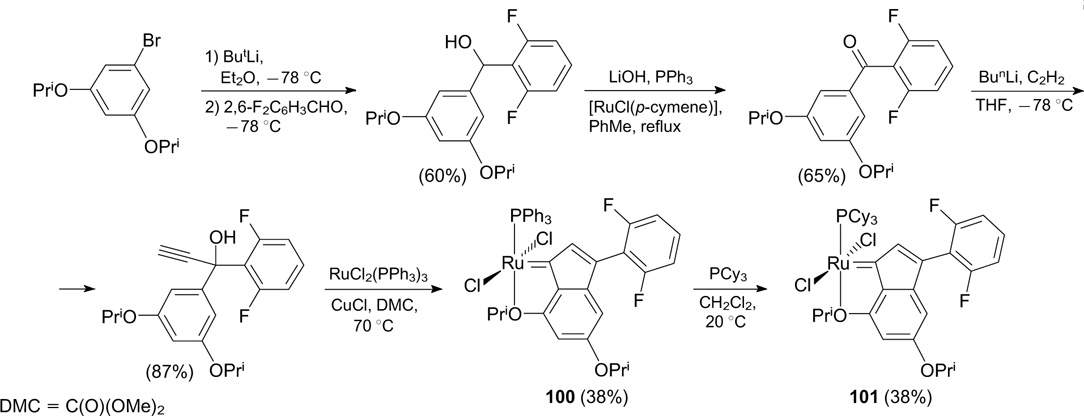
Indenylidene complexes 102a,b containing one fluorine atom in the para position of the benzene ring of the indenylidene ligand were described by Verpoort and co-workers 120, 121 (Scheme 66). The corresponding propargyl alcohol was prepared from commercially available p-fluorobenzophenone according to a literature procedure. 122 Complexes 102a,b were evaluated for catalytic activity in RCM, RCEYM and ROMP reactions. In all cases, catalysts 102a,b showed activity similar to that of the non-fluorinated analogue M2.
Scheme 66
Download figure:
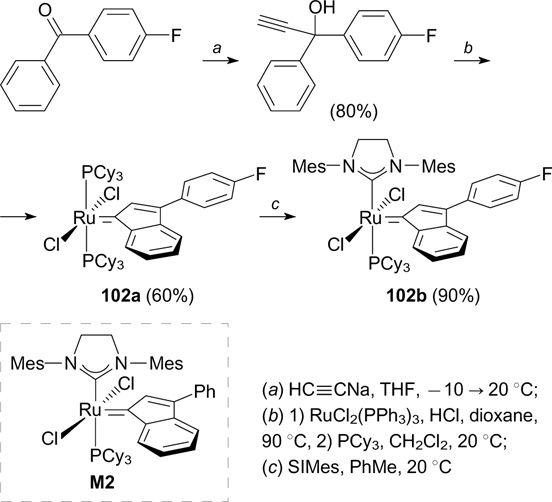
5. Fluorine in L-type ligands
In 1998, Grubbs and co-workers 123 synthesized a series of ruthenium complexes bearing a mixed-type (L and X) salicylaldimine ligand. In particular, they prepared complex 103 containing a fluorinated substituent in the L-type moiety (Scheme 67). The activity of catalyst 103 was similar to that of its analogues, the efficiency of which was low at room temperature but significantly increased upon heating.
Scheme 67
Download figure:

Non-chelating L-type ligands in ruthenium complexes are generally rather labile. Hence, the replacement of an L-type ligand with another one is usually performed by the treatment with an excess of the latter and occurs quite rapidly under mild conditions.
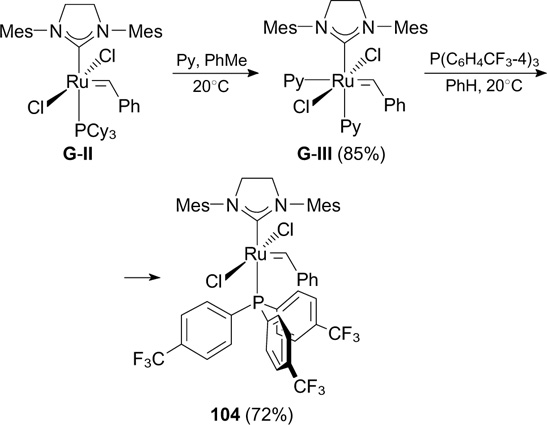
In 2001, Grubbs and co-workers 124 described the first synthesis of a third-generation catalyst (G-III) by the reaction of complex G-II with an excess of pyridine (Scheme 68). Although G-III does not contain fluorine atoms, in this Section it is considered as an important starting compound for the synthesis of other complexes, in particular of those modified with fluorine-containing phosphine ligands. For instance, commercially available electronically pure tris[p-(trifluoromethyl)phenyl]phosphine, with low electron density at the phosphorus atom, was used to synthesize complex 104, which proved to be more active than G-II in the CM reaction of acrylonitrile and allylbenzene and showed a high initiation rate and activity in ROMP of cyclooctadiene (see Scheme 68). 125
Scheme 68
Download figure:
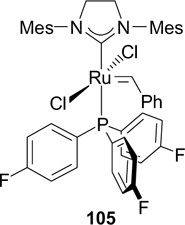
Fluorous phosphine complex 105 was synthesized in a similar way. 126 This complex showed lower activity in RCM of diethyl 2-allyl-2-cinnamylmalonate and in ROMP of cyclooctadiene compared to complex 104. Tris(pentafluorophenyl)phosphine was inactive in the reaction with G-III. 125
Structure 105
Download figure:
In 2006 – 2012, Gladysz and co-workers 127, 128 published a series of studies on Grubbs-type catalysts 107a – d bearing fluorous phosphine ligands. Previously, this research group reported the synthesis of phosphines 106a – d (Scheme 69). 128 The introduction of perfluoroalkyl substituents was performed in two steps. Initially, phosphine was added to the appropriate perfluoroalkylethylene in the presence of azobisisobutyronitrile to form a mixture of the target compound (∼90%) with primary and secondary phosphines PHx (CH2CH2(CF2)n CF3)3–x (x = 1, 2; n = 5, 7, 9). The addition was completed in the presence of the less active initiator 1,1'-azobis(cyclohexylcarbonitrile) to prevent polymerization. Phosphines 106a – d exhibited very high fluorophilicity with a partition coefficient between toluene and PFMC Pi (FBS) > 80 (Table 3). The catalysts were synthesized by the replacement of the ligand in complex G-III in a solution in trifluoromethylbenzene. 127 Table 3 presents the partition coefficients for phosphines 106a – d and the corresponding catalysts 107a – d.
Scheme 69
Download figure:

Table 3. Partition coefficients (PFMC/PhMe) of phosphines 106a – d and complexes 107a – d.
| Phosphine | Pi (FBS) | [Ru] | Pi (FBS) |
|---|---|---|---|
| 106a | 82.3 | 107a | 0.15 |
| 106b | >300 | 107b | 0.66 |
| 106c | >300 | 107c | 3.46 |
| 106d | 82.3 | 107d | 0.13 |
Due to high fluorophilicity, catalyst 107c was almost inactive in dichloromethane. Catalysts 107a,b were tested in a series of benchmark RCM reactions. The initiation rate in dichloromethane – fluorous solvent biphasic systems was much higher than that in pure dichloromethane. This acceleration is associated with the phase transfer of the fluorous phosphine ligand to a fluorous solvent. The catalyst activation via phase transfer of the ligand (phase-transfer activation) was also found in ROMP of norbornene in the presence of catalyst 107b. 129 It was shown that complex 107b can be reused for three cycles in RCM of DEDAM. 130
Indenylidene catalysts 108a,b, analogues of complexes 104 and 105, were described by Nolan and co-workers. 131 Complexes 108 were also synthesized from pyridine-containing indenylidene precursor M31 (Scheme 70). Their catalytic activity was tested in a wide range of CM, RCM, RCEYM and RRM (ring-rearrangement metathesis) reactions. In most cases, complex 108b showed higher activity compared to the non-fluorinated analogue M20 (see Scheme 70).
Scheme 70
Download figure:

Gladysz and co-workers 132 proposed another approach to the synthesis of ruthenium catalysts bearing fluorous L-type ligands. They prepared third-generation catalyst 110 containing two fluorous pyridine ligands (Scheme 71). Fluorous pyridine 109 was synthesized by the Heck reaction of 3,5-perfluoroalkylethylene with dibromopyridine followed by the double bond hydrogenation under mild conditions. The reaction of 109 with complex G-III afforded complex 110. 133 Pyridine was completely removed from the reaction mixture by a series of freezing, evacuating and thawing cycles.
Scheme 71
Download figure:
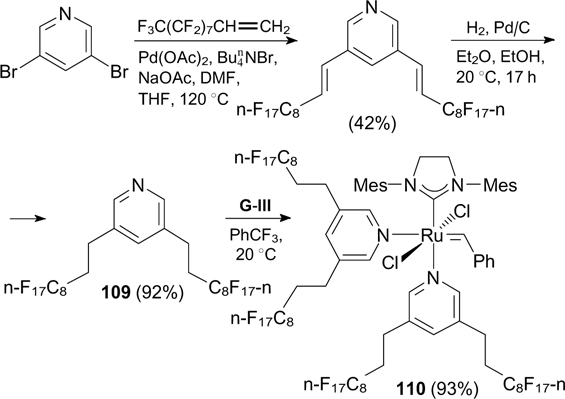
As in the case of catalysts 107a – d, the activity of catalyst 110 in RCM was significantly higher in dichloromethane – fluorous solvent biphasic systems.
Scheme 72
Download figure:
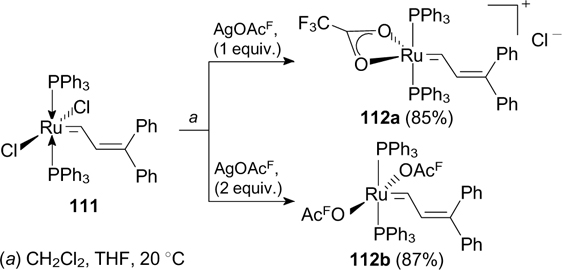
6. Fluorine in X-type ligands
First ruthenium-based metathesis catalysts containing fluorine in an X-type ligand were described in 1993. 134 The synthesis involved the replacement of chlorine in Grubbs complex 111 with trifluoroacetate mediated by the appropriate silver salt (Scheme 72). Cationic catalyst 112a showed high activity in ROMP of norbornene. 135,136
It is worth noting that this reaction in hexane affords water-bridged dinuclear complex 113a bearing two trifluoroacetate groups (Scheme 73). Related compounds were synthesized using complex G-I as the starting compound. 137, 138
Scheme 73
Download figure:

Catalysts 113a,b,d were tested in self-metathesis reactions of internal olefins, in which they exhibited moderate activity. Catalysts 113c,e with pentafluorobenzoate ligands proved to be inactive in these reactions.
Stable four-coordinate complexes 114 and 115a,b were synthesized using bulky X-type ligands (Scheme 74). 139 Non-fluorinated complex 114 was prepared by the reaction of potassium tert-butoxide with complex G-I and then it was treated with an excess of fluorinated alcohol.
Scheme 74
Download figure:

Complexes 115a,b showed moderate activity even upon heating (60 °C), but they were highly active upon the addition of hydrochloric acid to the reaction mixture.
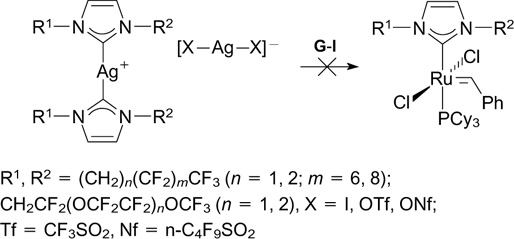
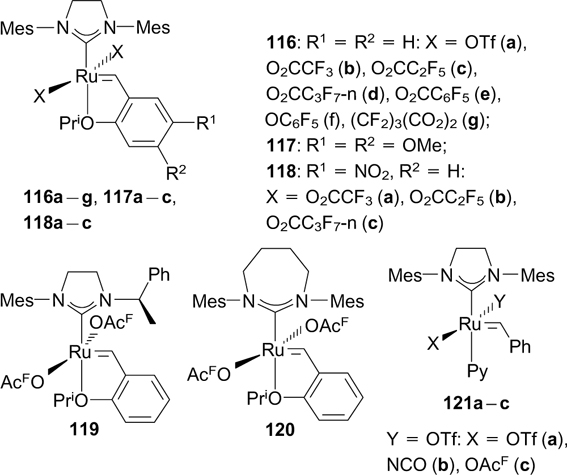
The method developed previously for the replacement of X-type ligands mediated by silver salts was used by Buchmeiser and co-workers 140 – 148 in 2004 – 2011. In this period of time, a series of catalysts (116 – 121), mainly Hoveyda-type catalysts, bearing different fluorinated X-type ligands were synthesized.
Complex 116b exhibited high activity in a wide range of metathesis transformations. Catalyst 116a was less active. 140 Complexes 116b 141 and 116c,d 143 catalyzed the cyclopolymerization of diethyl dipropargylmalonate (DEDPM, Scheme 75) but did not induce polymerization in a living manner.
Structures 116 – 121
Download figure:
Scheme 75
Download figure:
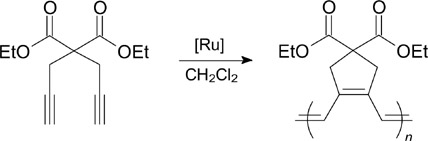
The cyclopolymerization of 1,6-heptadiynes in a living fashion can be performed in the presence of catalysts 117a – c and 118a – c. 141, 142 An elongation of the perfluoroalkyl chain of the catalyst led to a narrower molecular mass distribution of the resulting polyene. 143 Catalyst 117a was found to be efficient in the cyclopolymerization of heptadiynes containing free hydroxy groups, in contrast to molybdenum-derived Schrock catalysts. 144 Besides, compounds 116b, 117a and 118a were tested in the cyclopolymerization of N,N-dipropargylamines, where they showed high efficiency in the catalysis of polymerization processes. 145 Catalysts 116e and 119 were synthesized with the goal to determine the effect of the ligand bulkiness on the catalytic properties, in particular on the regioselectivity of the cyclopolymerization of dipropargyl substrates. 146 Complex 120 demonstrated high catalytic activity in the cyclopolymerization of DEDPM, but was found to be inactive in RCM of DEDMM. 147 Complexes 121a – c and 116g showed high efficiency in the copolymerization of cyclopentene and norbornene. 148
The hexafluoroglutaric group can be used as a linkage unit for the immobilization of ruthenium catalysts on a support. The first attempt to immobilize a Grubbs catalyst through a fluorine-containing ligand was reported by Mol and co-workers. 149 For this purpose, hydroxyethyl-functionalized styrene – divinylbenzene copolymer was treated with hexafluoroglutaric anhydride followed by the preparation of the corresponding silver salt and the subsequent addition of Grubbs catalyst G-I (Scheme 76). Catalytic system 122 was highly active in RCM of DEDAM (the catalyst retained the activity after five cycles of reuse) and in self-metathesis of methyl oleate and trans-4-decene.
Scheme 76
Download figure:

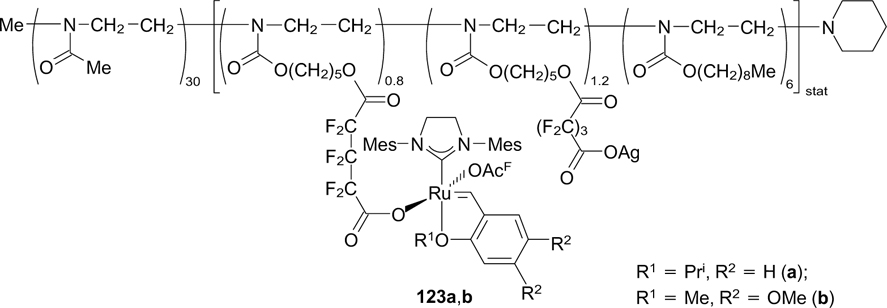
Buchmaiser and co-workers 140, 150, 151 and Braddock et al. 152 immobilized HG-II-type catalysts on polymeric supports using methods similar to that described above. Catalysts 123a and 123b immobilized on amphiphilic oligomeric poly(2-oxazoline) allowed the cyclopolymerization of DEDPM in aqueous medium. 141
The hexafluoroglutaric linkage was utilized to graft Hoveyda-Grubbs-type catalysts on a monolith poly(ether ether ketone) column 150, 151, 153 or silica gel 154 – 156 for flow reactors. Besides, the hexafluoroglutaric moiety was used to produce catalysts bearing amphiphilic (124) and charged (125) ligands. 157, 158
Structures 123
Download figure:
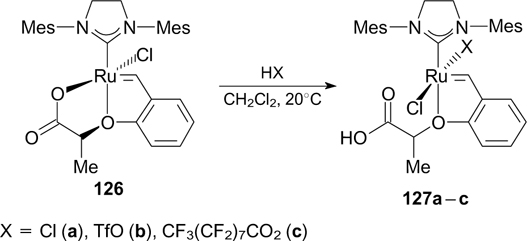
Grela and co-workers 159 synthesized catalyst 126 bearing a chelating carboxylate group. This catalyst showed latent behaviour, being inactive in RCM at room temperature but highly efficient in the presence of an acid or at elevated temperatures. 159 For instance, complexes 127a – c were synthesized in situ; however, fluorine-containing compounds 127b,c were found to be less active than chlorinated compound 127a (Scheme 77). The addition of aqueous hydrofluoric acid or the hydrofluoric acid pyridine complex (HF ⋅ Py) to a solution of 126 affords unstable complex 127d with a Ru–F bond. 160
Structures 124, 125
Download figure:

Scheme 77
Download figure:
Subsequently, Gawin and Grela 160 reported the synthesis of isopropyl-containing complexes 128a,b (Scheme 78). Catalyst 128b bearing the trifluoroacetate ligand proved to be much more active in RCM of DEDAM compared to its chlorinated analogue 128a. Besides, catalyst 128b was highly stable and demonstrated excellent conversion at elevated temperatures or at longer reaction times.
Scheme 78
Download figure:

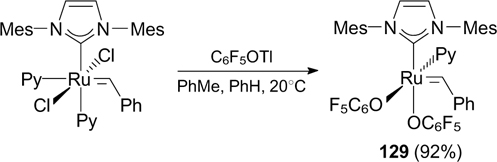
Fogg and co-workers 161 described the reaction of thallium perfluorophenoxide with an unsaturated analogue of catalyst G-III (Scheme 79). The resulting complex 129 exhibited high activity in RCM of DEDAM even at low catalyst loading (0.05 mol.%) and in RCM giving medium-sized rings. 162
Scheme 79
Download figure:
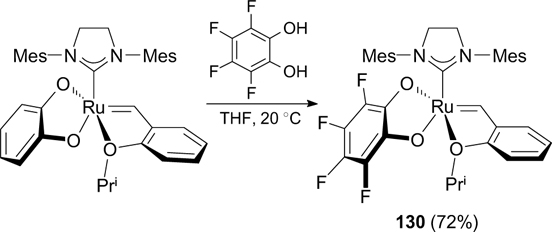
In 2014, Hoveyda and co-workers 163 developed a method for the synthesis of complex 130 based on the fast exchange of the catecholate ligand for its tetrafluorine-substituted analogue (Scheme 80). Complex 130 was found to be highly stable in dichloromethane and chloroform however, its catalytic activity was not evaluated.
Scheme 80
Download figure:
A similar complex 131 containing the tetrafluorodithiobenzene ligand (Scheme 81) 41 exhibited high activity in CM of allylbenzene with Z-2-butene-1,4-diol, which occurs with the retention of the Z configuration. The catalyst was synthesized starting from 1,2,3,4-tetrafluorobenzene via the transmetalation with zinc tetrafluorobenzene-1,2-thiolate.
Scheme 81
Download figure:

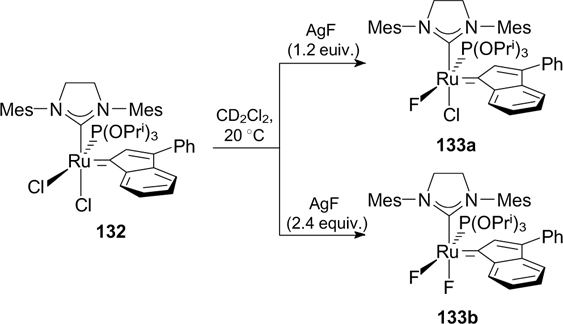
In 2015, Cazin and co-workers 164 reported the first synthesis of alkene metathesis catalysts 133a,b containing a fluorine atom as the X ligand (Scheme 82). These complexes were prepared by the treatment of compound 132 with silver fluoride. Monofluorinated derivative 133a was more active than difluorine-substituted analogue 133b and was tested in CM, RCM and RCEYM reactions, in which it proved to be efficient.
Scheme 82
Download figure:
A series of ruthenium catalysts bearing perfluoro(polyoxa)alkanoate ligands 134 – 136 were synthesized by Kvíčala and co-workers 165 in 2014. The fluorous partition coefficients Pi (FBS) were determined for all complexes, the values of which vary from 0.03 (134a) to 2.9 (135d). In the series of the synthesized catalysts, only compounds 135a – d and 136b – d behave as heavy fluorous catalysts [Pi (FBS) > 1]. Complexes of the series 134 and 136 showed the highest activity in RCM reactions. Complex 136d used in RCM of DEAMM and DEDMM can be recovered with PFMC and reused.
Structures 134 – 136
Download figure:

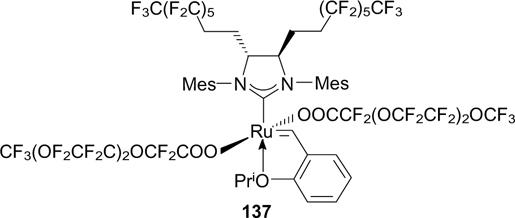
A year later, the same research group 33 synthesized complex 137 having very high fluorophilicity [Pi (FBS) = 27]; however, the data on its activity in metathesis reactions were not reported. 33
Structure 137
Download figure:
A series of fluorous catalysts 138a – c and 139a,b with polyfluorooxaalkanoate ligands containing an asymmetric carbon centre (chiral carbon) were synthesized in 2016. 166 These complexes were tested in RCM reactions with benchmark substrates. Catalysts 138b and 139b were found to be the most active due to low steric hindrance and high electrophilicity of 2-fluoro-2-(perfluoropropoxy)acetate ligands.
Structures 138, 139
Download figure:

Stephan and co-workers 167 reported the synthesis of a series of ruthenium alkylidene complexes with two different NHC ligands and evaluated their catalytic activity. Among the characterized complexes were those with fluorinated thiophenolate ligands 141a – c. These catalysts were synthesized by the reaction of hydride complex 140 with fluorinated aryl vinyl sulfides under mild conditions (Scheme 83). The resulting complexes showed moderate activity in ROMP of cyclooctadiene and in RCM of DEDAM, the activity being significantly higher in the presence of Lewis acid (BCl3). 168
Scheme 83
Download figure:

Due to the electron-withdrawing properties, fluorine-containing X-type ligands are most often utilized in ruthenium metathesis catalyst to increase the catalytic activity of the latter. Besides, fluorine-containing X-type ligands, most often hexafluoroglutarate, are employed for catalyst immobilization on solid or polymeric supports. Such supported catalytic systems are mentioned in dedicated reviews. 169 – 172
7. Conclusion
The alkene metathesis is one of the most efficient tools applied by synthetic chemists to construct various unsaturated systems, from simple olefins to complex biologically active compounds and polymers. Ruthenium alkylidene complexes, particularly those bearing NHC ligands, which are characterized by high air- and moisture-stability combined with tolerance to a wide range of functional groups, are most commonly used for these purposes. Since the design of an ideal catalyst for all types of metathesis reactions is unlikely, the important strategy for the development of the most efficient and selective catalytic systems to solve a particular synthetic or engineering problem is based on the alteration of the structure of ligands and substituents at the ruthenium centre. Significant advances in organofluorine chemistry achieved in the past two decades provide additional opportunities for performing targeted modifications of alkene metathesis catalysts taking into account the unique physicochemical properties of the fluorine atom and fluoroalkyl groups, e.g., the small atomic radius, high electronegativity, lipophilicity and the ability to form hydrogen and coordination bonds. This review summarizes literature data on the methods for the introduction of fluorine atoms and fluoralkyl groups into different ligands to construct metathesis-active ruthenium carbene complexes. It also analyzes the influence of fluorine-containing ligands on the catalytic activity of the complexes.
In most cases, the choice of fluorinated substituents and their attachment sites in NHC ligands are dictated by the desire to increase the electrophilicity of the ruthenium atom due to the electron-withdrawing effect of fluorine atoms and fluoroalkyl groups. These modifications facilitate the interaction of an alkylidene complex with an olefin substrate in the key step of the catalytic cycle, thereby increasing its activity. Besides, in some cases, catalysts bearing unsymmetrical fluorinated NHC ligands were found to have additional Ru–F coordination, which significantly stabilizes the complex. The presence of fluorine atoms in chelating alkylidene ligands provides an increase in the catalyst initiation rate due to a weakening of the ruthenium – heteroatom bond. A decrease in the electron density on L- and X-type ligands also often leads to an increase in the catalytic activity of the complexes in metathesis reactions. The introduction of polyfluoroalkyl groups into ligands allows for the solution of the problem of catalyst recovery with fluorous biphasic systems for reuse.
Despite considerable progress in the field of fluorous modifications of ruthenium alkylidene complexes aimed at increasing their selectivity and efficiency in metathesis reactions, developing tools for catalyst immobilization on solid supports, improving recyclability and recoverability of catalysts and increasing their solubility in industrially important hydrocarbon solvents, these issues are still challenging.
The review was prepared with the financial support of the Russian Science Foundation (Project No. 16-13-10364) and the Peoples' Friendship University of Russia (RUDN University, Programme 5-100). The access to literature was provided by the A.N.Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences with the financial support from the Ministry of Science and Higher Education of the Russian Federation.
8. List of abbreviations and designations
| ADMET | — acyclic diene metathesis polymerization, |
| AIBN | — azobis(isobutyronitrile), |
| Amt | — tert-amyl, |
| ARCM | — asymmetric ring-closing metathesis, |
| AROCM | — asymmetric ring-opening cross-metathesis, |
| CM | — cross-metathesis, |
| COE | — cyclooctene, |
| Cy | — cyclohexyl, |
| DCE | — 1,2-dichloroethane, |
| DEAMM | — diethyl allylmethallylmalonate, |
| DEDAM | — diethyl diallylmalonate, |
| DEDMM | — diethyl dimethallylmalonate, |
| DEDPM | — diethyl dipropargylmalonate, |
| DIPEA | — diisopropylethylamine, |
| DIPP | — 2,6-diisopropylphenyl, |
| DMAP | — 4-dimethylaminopyridine, |
| FBS | — fluorous biphasic system, |
| F-SPE | — fluorous solid-phase extraction, |
| KHMDS | — potassium hexamethyldisilazide, |
| Mes | — mesityl, |
| Naph | — naphthyl, |
| Nf | — perfluorobutyl (nonaflyl), |
| NHC | — N-heterocyclic carbene, |
| PFMC | — perfluoro(methylcyclohexane), |
| Piv | — pivaloyl (tert-butylcarbonyl), |
| polyCOE | — poly(cyclooctene), |
| Py | — pyridine, |
| RCEYM | — ring-closing enyne metathesis, |
| RCM | — ring-closing metathesis, |
| RGO | — reduced graphene oxide, |
| ROMP | — ring-opening metathesis polymerization, |
| RRM | — ring-rearrangement metathesis, |
| SM | — self-metathesis, |
| Tf | — trifluoromethanesulfonyl (triflyl), |
| TON | — turnover number. |
Footnotes
Dedicated to Professor Pierre H. Dixneuf from the University of Rennes (France) for his outstanding contribution to organometallic chemistry and catalysis.