

Abstract
Syntheses of polyfunctional benzene derivatives from both cyclic and acyclic non-aromatic precursors are reviewed. Such aromatizations include transformations that use Diels – Alder reactions, transition metal-catalyzed reactions, base mediated aldol or Michael addition reactions or electrocyclic and pericyclic reactions. This review is limited to research that has been published since the last reviews in 2006, 2008, 2015 and 2018 and is specifically focused on aromatic and heteroaromatic compounds synthesized by construction of the benzene ring. It excludes heterocyclic compounds that lack a monocyclic benzene core. Applications of such aromatization reactions for the total syntheses of natural products as well as potential medicinal chemical entities are included.
The bibliography includes 155 references.
| A.G.M. Barrett. PhD, Sir Derek Barton Professor of Synthesis in the Department of Chemistry at the Imperial College London. |
| E-mail: agm.barrett@imperial.ac.uk |
| T.-K.Ma. Postdoctoral research associate at the same College. |
| E-mail: Freeman.ma@ucl.ac.uk |
| T.Mies. PhD student at the same College. |
| E-mail: thomas.mies14@imperial.ac.uk |
| Current research interest of the authors: synthesis of bioactive natural, heterocyclic, organometallic compounds, macrocyclic ethers, lactones and porphyrazines, design of novel therapeutic agents for detection and treatment of cancers, catalytic sustainable organic synthesis, application of combinatorial chemistry and parallel synthesis for the development of pharmaceuticals, applications of microfluidic flow reactors in synthesis. |
1. Introduction
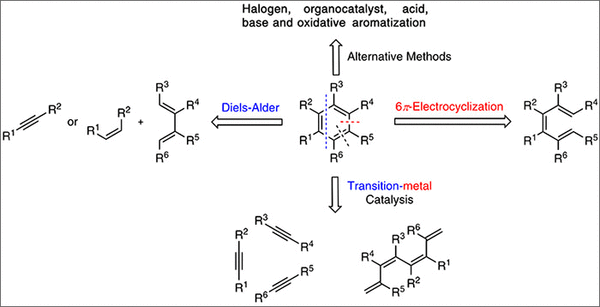
The classical methods for the synthesis of polyfunctional aromatic compounds depend on the use of a range of reactions including sequential electrophilic aromatic substitution reactions and o-metalation with addition or substitution reactions using electrophilic reagents. Additionally, transition metal-catalyzed substitution and C–H activation are also of considerable value in this regard. All these transformations depend on the conversion of an aromatic precursor to another aromatic compound of enhanced complexity. There is an alternative process: the conversion of a non-aromatic precursor, which may be cyclic or acyclic, into an aromatic compound; such transformations are complementary to classical approaches and are often used for the synthesis of arenes with ring substitution patterns not readily available by classical arene derivatizations.
An early example for this type of aromatization is the conversion of testosterone or androstadienedione into estradiol, both enzyme-catalyzed processes in vivo (Scheme 1, conditions a). Related bioinspired dehydrogenative aromatizations via transition metal-catalyzed oxidations have been reported in the first half of the 20th century. 1, 2 Alternative reactions using oxidants such as iodine have since been extensively studied for the synthesis of many aromatic compounds mainly from functionalized cyclohexane or cyclohexanone precursors (see Scheme 1, conditions b – d ). 3 – 8

Download figure:
Standard imageThis review aims to complement an overview of these reactions by highlighting additional methods for ring formation and subsequent aromatization processes, which allows for the synthesis of arenes with various functionalities otherwise not readily available from the derivatization of aromatic precursors. These alternative reactions possess a high degree of flexibility for the synthesis of polyfunctional arenes.
Such advantages are reported for the base-catalyzed benzannulation reactions, 9, 10 Diels – Alder cycloadditions 11 – 16 and electrocyclic and pericyclic reactions followed by aromatization. 17 – 19 These strategies are often employed, since in addition to the above advantages, they can also reduce the costs of scale-up synthesis with the use of inexpensive building blocks and transformations with high atom economy.
The synthesis of aromatic compounds through transition metal-catalyzed reactions, such as ring-closing alkene metathesis (RCM) or cyclotrimerization of alkynes or alkenes and subsequent aromatization, often allows for the synthesis of aromatic compounds with a range of bespoke substituents that would not be easily accessible through other methods. 20 – 22 RCM shows remarkable functional group tolerance and can often be carried out under mild reaction conditions. Cyclotrimerization reactions are also often conducted under mild conditions and show broad functional group compatibility. 23 A highlight of this methodology is the Funk and Vollhardt's 24 estrone synthesis via a [2+2+2]-cyclotrimerization strategy to elaborate the aromatic ring complementing the biomimetic dehydrogenative processes described above (Scheme 2).

Download figure:
Standard imageThe reaction strategies herein described have distinct advantages and disadvantages and are complementary to each other for the synthesis of aromatic compounds. The review covers a selection of methods for cyclization reactions and subsequent aromatizations published between 2016 and early 2020. For a more comprehensive and exhaustive selection of aromatization methods, the authors draw attention to the recently updated Chapter: Formation of Arenes by Aromatization in Comprehensive Organic Transformations by Larock and Pletnev † as well as various prior reviews. 25 – 33
The following designations and abbreviations are used:
| Ad | — adamantyl, |
| All | — allyl, |
| Atom Economy | — ratio of atoms transferred from starting material to products, |
| BINAP | — 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl, |
| Boc | — tert-butyloxycarbonyl, |
| [bmim][Cl] | — 1-butyl-3-methylimidazolium chloride, |
| BMIDA | — boronic acid N-methyliminodiacetic acid ester, |
| Bn | — benzyl, |
| Cbz | — carboxybenzyl, |
| CDI | — 1,1'-carbonyldiimidazole, |
| Cy | — cyclohexyl, |
| DABCO | — 1,4-diazabicyclo[2.2.2]octane, |
| DCE | — 1,2-dichloroethane, |
| DBU | — 1,8-diazabicyclo[5.4.0]undec-7-ene, |
| DDQ | — 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, |
| DEC | — N,N-diethylcarbamoyl, |
| DMAD | — dimethyl acetylenedicarboxylate, |
| DMP | — Dess–Martin periodinane, |
| E1CB | — elimination unimolecular conjugate base, |
| FN | — functionalized nanoparticle, |
| Fu | — furyl, |
| FVP | — flash vacuum pyrolysis, |
| HIV | — human immunodeficiency virus, |
| IC | — inhibitory concentration, |
| LC | — median lethal dose, |
| LED | — light-emitting diode, |
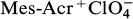
| — 9-mesityl-10-methylacridinium perchlorate, |
| MIC | — minimum inhibition concentration, |
| MOM | — methoxymethyl, |
| Ms | — methylsulfonyl (mesyl), |
| MTBE | — methyl-tert-butyl ether, |
| MW | — microwave, |
| NaBARF | — sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, |
| Naph | — naphthyl, |
| p-NBA | — 4-O2NC6H4COOH, |
| NBS | — N-bromosuccinimide, |
| NFSI | — N-fluorobenzenesulfonimide, |
| NHC | — N-heterocyclic carbene, |
| Ns | — 4-nitrobenzenesulfonyl (nosyl), |
| o/n | — overnight, |
| PG | — protecting group, |
| PMB | — para-methoxybenzyl, |
| Py | — pyridyl, |
| RCM | — ring closing alkene metathesis, |
| SET | — single electron transfer, |
| TBAF | — tetra-n-butylammonium fluoride, |
| TBD | — 1,5,7-triazabicyclo[4.4.0]dec-5-ene, |
| TBS | — tert-butyldimethylsilyl, |
| TEMPO | — (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, |
| TES | — triethylsilyl, |
| Tf | — trifluoromethanesulfonate (triflate), |
| TFA | — trifluoroacetic acid, |
| TFP | — tri-2-furylphosphine, |
| Th | — thienyl, |
| THP | — tetrahydropyranyl, |
| TIPS | — triisopropylsilyl, |
| TMS | — trimethylsilyl, |
| TMTU | — tetramethylthiourea, |
| Ts | — 4-toluenesulfonyl (tosyl), |
| w/u | — work-up. |
2. Diels – Alder reactions and subsequent aromatization
The Diels – Alder cycloaddition is an exceptionally powerful reaction for the synthesis of carbocycles, given the chemo-, regio- and stereoselectivity which is retained from the starting materials in addition to excellent atom economy. By employing a post-cycloaddition oxidation or an elimination reaction, aromatization can be carried out, thereby providing a range of heavily substituted aromatic compounds efficiently. 29, 34 These reactions may be sub-divided according to the diene or dienophile precursors.
2.1. Quinone precursors
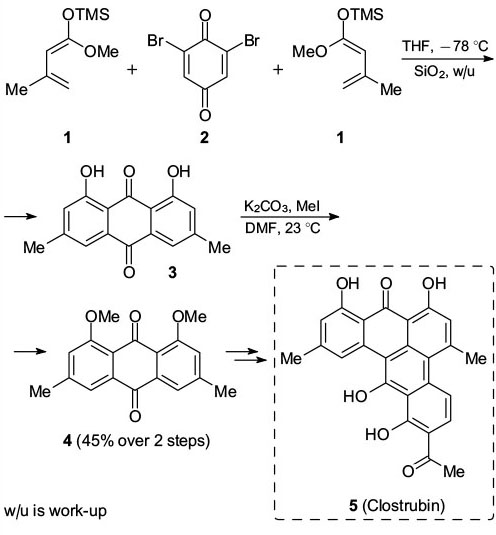
Since previous reviews, Diels – Alder reactions of quinones and quinonimine ketals have been reported to be useful for the syntheses of aromatic compounds. Additionally, Brassard's, Danishefsky's and other related dienes remain particularly useful building blocks for the synthesis of α,β-unsaturated cyclohexenones, which are readily converted into phenols by oxidation using oxygen, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), etc. For example, Li and co-workers 35 reported that the reaction of dibromobenzoquinone 2 with diene 1 and subsequent dehydrobromination gave the northern anthraquinone fragment 3 of clostrubin (5) on a multi-gram scale (Scheme 3). The natural product was shown to have strong antibiotic activities with minimum inhibition concentrations (MIC's) at 0.12 μmol L−1 against methicillin-resistant Staphylococcus aureus and 0.97 μmol L−1 against vancomycin-resistant enterocci.
Download figure:
Standard imageIn 2017, Sayyad and Kaliappan 36 reported a Diels – Alder reaction and subsequent aromatization for the synthesis of the polycyclic anthraquinones 8 functionalized with delicate sugar moieties (Scheme 4). Other dienophiles that were employed in this sequence included various quinones as well as dimethyl acetylenedicarboxylate (DMAD) together with various sugar functionalized dienes followed by aromatization under aerobic, acidic conditions.
Khan and co-workers 37 recently reported the use of the Diels – Alder and aromatization strategy in their synthesis of magterpenoid C (11) via a key silica-gel mediated [4+2]-cycloaddition reaction followed by oxidative aromatization with manganese dioxide (Scheme 5). The magterpenoids A–C inhibit protein tyrosine phosphatase 1B, and thereby enhance insulin sensitivity in cells. An attempted synthesis of the more complex tricyclic magterpenoid B by the same methodology, however, was not successful.

Download figure:
Standard image
Download figure:
Standard imageTong and co-workers 38 reported the first atropoisomerically-selective synthesis of conformationally stable 1,1'-naphthyl-anthraquinones 16 from the Diels – Alder reaction of (1-acetoxy-1-(1-naphthyl)methyl)allenoates 12 and 2-hydroxy-1,4-naphthoquinones 14 catalyzed by an enantiomerically pure ferrocene-based phosphine (Scheme 6). These reactions were extended to those of a range of substituted (1-acetoxy-1-(2-mono- and 2,6-disubstituted-phenyl)methyl)allenoates, mono- and disubstituted-2-hydroxy-1,4-benzoquinones and a hydroxybenzenoanthracenequinone to provide the corresponding adducts (16). The postulated mechanism involved phosphine addition to the allene and acetoxy elimination to form the diene 13, which readily reacted with the dienophile 14 to produce the adduct 15. Subsequent base mediated ylide formation, phosphine elimination and dehydration induced aromatization.

Download figure:
Standard image
Download figure:
Standard imageIn his most recent meroterpenoid studies, George and co-workers 39 employed a Diels – Alder and aromatization sequence for the synthesis of the racemic aromatic core of the naphterpin and marinone natural products (Scheme 7). Quinones 17 and Brassard's diene (or derivative of Brassard's diene) and subsequent aerobic oxidation gave the tricyclic arenes 19. Within one to three steps, these products 19 were converted into four different naphterpin (20 and 21) and marinone (20) natural products.
Hsu and Huang 40 employed a Diels – Alder reaction of naphthoquinone 22 with diene 23 in the presence of the Lewis acid boron triacetate, followed by methanol elimination, TMS-ether hydrolysis and aerobic oxidation to give anthraquinone 25, furnishing three of the four rings of parimycin (26) (Scheme 8). The use of boron triacetate was critically important for the observed regioselectivity as well as enabling the reaction to proceed at room temperature. Anthraquinone 25 was subsequently converted into racemic parimycin (26) and its (R)-(–)-enantiomer (27) thereby defining the absolute stereochemistry of the natural product as the (S)-(+)-enantiomer, a potent antitumour compound with activities against stomach, lung, breast and skin cancers with IC70 values between 0.9 to 6.7 μg mL−1.
Chen and co-workers 41 showed that the formal Diels – Alder reaction of quinone imines 28 with dienals 29 catalyzed by the prolinol derivative 30 provided substituted aromatic compounds 33 with excellent relative and absolute stereochemical control following trimethylsilane mediated dehydroxylation of the carbinolamines 32 (Scheme 9). These products 33 included examples with a broad range of electron-withdrawing and -donating substituents. The method additionally showed good compatibility with a range of protecting groups (PG = Boc, Cbz, Ts) and proceeded with high enantioselectivities.

Download figure:
Standard image
Download figure:
Standard image2.2. Thiophene and furan precursors
Kamimura and co-workers 42 reported the synthesis of phthalimides 38 from maleimides 35 and furfural hydrazones 34 under microwave irradiation (MW) in ionic liquids (Scheme 10). The only by-product of this reaction is water and the ionic liquids were shown to be repeatedly recyclable with no loss of functionality.
Jérôme and coworkers 43 described the synthesis of the o- and m-xylene derivatives 42 and 43 by allowing the furfural acetal 39 to react with acrylonitrile 40 under Lewis acid catalysis to give the Diels – Alder adduct 41 (Scheme 11). 43 This Diels – Alder reaction showed modest selectivity towards the o-endo-adducts. Various bases were screened for the conversion of the Diels – Alder adducts into the aromatic compounds 42 and 43, with regioselectivities ranging from 1.5 – 3.4 : 1 in favour of the m-product 43. By prematurely quenching the base induced aromatization reaction of adduct 41, the selectivity towards the m-xylene derivative 43 was further enhanced. Subsequent derivatization was used in a new route to m-xylenediamine, an important monomer in polymer chemistry.
Recently, Lei and co-workers
44
reported the conversion of thiophenes 44, 45 and acetylenes 46, 48 to aromatic compounds 47, 49 under exceptionally mild photocatalytic conditions using 9-mesityl-10-methylacridinium perchlorate (Mes-Acr+
 ), with the generation of elemental sulfur as a byproduct (Scheme 12). Related Diels – Alder reactions of thiophene were previously reported to proceed at very high temperatures (typically >220 °C)
45
but this was reduced to room temperature with photocatalysis. It was postulated that the formal [4+2] addition was initiated by the oxidation of thiophene (44) to the corresponding radical cation 50 by the excited state of the photocatalyst. Addition of this species to alkynes 46 gave the radical cation adduct 51, which was reduced by a single electron transfer (SET) from the reduced photocatalyst, thereby regenerating the photocatalyst and forming the Diels – Alder adduct 52, which subsequently underwent elimination of sulfur accompanied by aromatization. The authors reported related syntheses of aromatic compounds from the corresponding selenophenes.
), with the generation of elemental sulfur as a byproduct (Scheme 12). Related Diels – Alder reactions of thiophene were previously reported to proceed at very high temperatures (typically >220 °C)
45
but this was reduced to room temperature with photocatalysis. It was postulated that the formal [4+2] addition was initiated by the oxidation of thiophene (44) to the corresponding radical cation 50 by the excited state of the photocatalyst. Addition of this species to alkynes 46 gave the radical cation adduct 51, which was reduced by a single electron transfer (SET) from the reduced photocatalyst, thereby regenerating the photocatalyst and forming the Diels – Alder adduct 52, which subsequently underwent elimination of sulfur accompanied by aromatization. The authors reported related syntheses of aromatic compounds from the corresponding selenophenes.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageIn 2019, Gundersen et al. 46 reported the conversion of N-allyl- or N-propargyl substituted indole or indoline derivatives 53 into the core of bioactive lycorine alkaloids 54 via a Diels – Alder reaction followed by acid-catalyzed dehydration and aromatization (Scheme 13). When the precursor 53 was functionalized with N-allyl groups, the reaction gave rise to arenes (54, R = H), whereas the use of precursors 53 with N-propargyl groups provided phenols (54, R = OH). The authors also reported related intramolecular Diels – Alder and aromatization sequences for o-(2-furyl)-N-allyl- and N-propargylaniline derivatives.

Download figure:
Standard imageIn 2020, Zubkov and co-workers 47 reported an aromatization method that involves the formation of arenes 67 and 75 from 3-(2-furyl)-allylamines 56 and 73 and bromomaleic anhydride 55 (Scheme 14). The authors proposed the initial formation of amides 57 and 58, which subsequently underwent an intramolecular Diels – Alder reaction, with exo-control. Amides 57 plausibly undergo reaction via intermediates 61 – 66 to produce the final lactams 67. In a comparable way, amides 58 plausibly undergo reaction via intermediates 68 – 72 to produce the same final lactams 67. The lactams 67 were obtained in moderate to good yields after recrystallization. When the authors used 3-(3-furyl)allylamines 73, the corresponding aromatization product 74 was shown to be sensitive to acidic conditions, which resulted in the opening of the dihydrofuran ring to yield phenols 75 in moderate yields after recrystallization.

Download figure:
Standard image2.3. Pyrone starting material
Pyrones are exceptionally good dienes for the synthesis of aromatic compounds since the initial cycloadducts 77 readily undergo loss of carbon dioxide in the aromatization step (Scheme 15). Snyder and co-workers 48 synthesized a range of indoline derivatives 78 in excellent yields using this method.
In 2017, Kraus and Wang 49 synthesized a range of differentially substituted aromatic compounds 82, 84 by simple variation of the dienophile 80 in the Diels – Alder reaction step and subsequent aromatization by loss of carbon dioxide (Scheme 16). Interestingly in these examples, intermediate Diels – Alder-lactones 81 and 83 were stable and could be isolated.
Additionally, Yu and Kraus 50 expanded the substrate scope of this reaction by including various enamines 85, 90, 91 as dienophiles to provide the arenes 88, 93 and 94 (Scheme 17). A range of enamine substrates bearing various alkyl and aryl groups as well as cyclic enamines were used in this reaction. Interestingly, the pyrrolidine enamine adducts 87 rapidly underwent elimination of both carbon dioxide and pyrrolidine and aromatization to provide the tetrahydronaphthalene 88, whereas the morpholine adducts 92 were stable enough for purification but were aromatized using base-mediated elimination. When the cyclopentene derivative of enamine 86 was used, the reaction gave ketone 89 via a conjugate addition, carbon dioxide elimination and hydrolysis of the enamine rather than the anticipated Diels – Alder product.

Download figure:
Standard image
Download figure:
Standard image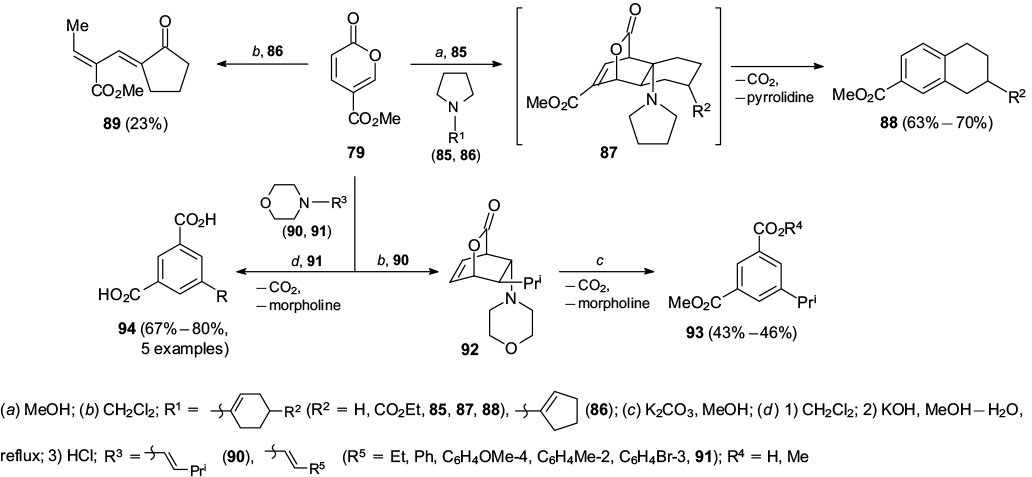
Download figure:
Standard imageMethyl coumalates 79 (see Scheme 16) and coumalic acid 95 are feedstock chemicals accessible from malic acid through dimerization. Shanks and co-workers 51 investigated the transformation of acid 95 into the bulk chemicals benzoic (99a) and isophthalic acids (99b) (Scheme 18) via a Diels – Alder reaction of 95 with ethylene (96) which initially gave lactone 97. The advanced cyclohexadiene moiety 98 was accessible through either Lewis acid mediated (R = H) or Brønsted acid (R = CO2H) mediated lactone decarboxylation. Finally, catalytic dehydrogenation gave benzoic acid 99a effectively from feedstock biomass.

Download figure:
Standard imageSubsequently, Shanks and co-workers 52 were able to expand this methodology to synthesize p- or m-toluic acids (103a, 104a) or their esters (103b, 104b) in excellent yields starting from methyl coumalate 79 or coumalic acid 95 and propylene (100) (Scheme 19). By raising the temperature, the reaction proceeded smoothly to the cyclohexadiene intermediates 102, which upon addition of Pd/C could be dehydrogenated to compounds 103a,b, 104a,b.

Download figure:
Standard imageIn 2018, Cui and co-workers 53 synthesized heavily substituted indole derivatives 109 as well as aniline derivatives 113 via a one-pot cascade sequence of alkyne (106, 111, 112a) and alkynyl-acid 110 coupling to produce pyrones 107, which underwent a subsequent Diels – Alder and aromatization sequence (Scheme 20). A broad scope of various aliphatic and aromatic substituents were tolerated in these transformations.
In 2019, Nakazaki and co-workers 54 reported the synthesis of an estrogen analogue 116 using an intramolecular pyrone – Diels – Alder and aromatization sequence of alkynyl-pyrone 114 in their studies on the potential medical chemistry applications of highly oxygenated steroids (Scheme 21).
Garg and co-workers 55 reported that electron-rich pyrones 121, which can be synthesized from 6H-1,3,4-oxadiazin-6-one (119), reacted with the strained azacycloalkyne 118, generated in situ from the silyl-triflate precursors 117 with caesium fluoride, to provide the heterocyclic compounds 123 and 124 after carbon dioxide elimination with concomitant aromatization. The initial cycloaddition with a ratio of precursors 117 : 119 of 1 : 2 and dinitrogen loss gave both regioisomers 121a,b but the second adduct underwent selective decomposition with the excess caesium fluoride present to leave only the azapyrone 121a (74%). When the reaction was carried out with a ratio of precursors 117 : 119 of 2 : 1, a double cycloaddition and carbon dioxide loss occurred giving both arenes 123 and 124. The aza-pyrone 121a was also converted into the corresponding derivatives by reaction with other strained cycloalkynes (cyclohexyne 125 and 4-oxacyclohexyne 126). These reactions proceeded under mild conditions and in good yields [71% and 62% (123 : 124 ratio is 1.5 : 1), respectively] (Scheme 22). Additional examples from the Garg group included related trapping reactions with arynes and heteroarynes but these are outside the scope of this review.

Download figure:
Standard image
Download figure:
Standard imageRecently, Wilson et al. 56 reported a cascade sequence of an intramolecular Diels – Alder reaction of the pyridones 127 followed by the elimination of isocyanic acid (129) instead of carbon dioxide to synthesize isoindolinones 130; such heterocyclic compounds are common scaffolds in medicinal chemistry (Scheme 23).
Wang and co-workers 57 reported the synthesis of benzoate esters 135 (Scheme 24). Initially, the synthesis provided lactones 133 at 120 °C, however raising the temperature to 150 °C resulted in smooth conversion to the cyclohexadienes 134. Subsequently, this route was extended by addition of DDQ, thereby providing benzoate esters 135. Finally, employing alkynes 136 instead of alkenes 132 gave the similar arenes 135.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image2.4. Miscellaneous substrates
Abaee et al. 58 reported a one pot procedure for the synthesis of bicyclic aromatic compounds 142 from enones 137, aldehydes 138 and acetylenedicarboxylate esters 112a,b using recyclable, silica-coated magnetite nanoparticle-based catalyst FN-Fe3O4 (139) followed by aerobic oxidative aromatization (Scheme 25).
In 2017, Philkhana and Reddy 59 reported the synthesis of fregenedadiol (146) and fregenedadiol acetate (147) using a Diels – Alder cycloaddition and oxidative (DDQ) aromatization sequence as key steps to elaborate the arene core 145 (Scheme 26). Reaction of diene 143 with dimethylacetylenedicarboxylate 112a gave the cycloadduct 144 and this was oxidatively aromatized with DDQ to provide the arene 145, which was converted to the natural products 146 and 147 respectively in two (reduction, hydrogenolysis) and three (reduction, hydrogenolysis, acylation) steps.
Thamattoor and co-workers 60 reported the synthesis of the reactive intermediate, 3-oxacyclohexyne (126), via an unusual photochemical rearrangement reaction (Scheme 27). 60 In this study, the phenanthrene derived precursor 148 underwent photolytic cleavage and thereby formed carbene 149, which, in turn, rearranged to the reactive species 126. Its generation in situ in the presence of cyclopentadienones 150 gave the Diels – Alder adduct 151 and this underwent a retro [4+1] reaction with loss of carbon monoxide and aromatization to provide the isochromanes 152 in moderate yields.

Download figure:
Standard imageIn 2019, Carreira and co-workers 61 reported the completion of the total synthesis of (–)-merochlorin A (160) by a late stage introduction of the aromatic core (Scheme 28). Dehydrogenation of ketone 153 by reaction of the derived enolate with imino-sulfenyl chloride 154 gave the enone 155, which underwent a Diels – Alder reaction with Brassard's diene 156 with hydrolytic cleavage to produce the core cyclohexenone unit 157. This was converted into the enol trimethylsilyl ether 158, which underwent Saegusa – Ito oxidation to form the resorcylate 159. Subsequent de-O-methylation of ether 159 completed the synthesis of (–)-merochlorin A (160), a potent antibiotic with MIC's of 0.15 – 0.3 μmol L−1 against C. dificile and 2 – 4 μmol L−1 against S. aureus.
In 2019, Liang and co-workers 62 synthesized the ABCE ring system of daphenylline 165 using a Diels – Alder reaction of amine-borane complex 161 with dimethyl acetylenedicarboxylate (112a) and subsequent aerobic oxidation to produce the arene 163, which was purified as the amine-borane salt (Scheme 29). Without amine coordination with borane as in the complex 161, the critical Diels – Alder reaction did not occur. Additionally, diverse alternative oxidants for the aromatization reaction of diene 162 to arene 163 were unsuccessful.

Download figure:
Standard image
Download figure:
Standard imageRecently, Yu and co-workers 63 synthesized the aromatic core of psymberin (170, irciniastatin A) by the reaction of alkynyl-ester 166 with diene 167 and subsequent aromatization by the expulsion of isobutylene following earlier Danishefsky and co-workers 64 – 66 precedent (Scheme 30). Psymberin was shown to have high cytotoxic activities against breast cancer, colon cancer and melanoma (LC50 values < 2.5×10−9 mol L−1).
Moreover, Yuan and co-workers 67 synthesized a range of bioactive sesquiterpenes 173 through the key intramolecular Diels – Alder and oxidative aromatization sequence of alkyne 171 (Scheme 31) via diene 172. Further functionalization of the ketone at the α-position was used to readily synthesize a range of natural products 174 – 178.
Some benzochromenes exhibit interesting biological properties including antibacterial, antioxidative and antimicrobial activities and possess material properties useful for the fabrication of photovoltaic devices. Xu and co-workers 68 reported a synthesis of these heterocyclic compounds through a Wolf-rearrangement of a diazoacetoacetate derivative 179 to the corresponding ketene-based diene 180 (Scheme 32). This reactive intermediate gave 6H-benzo[c]chromene derivatives 182 following an intramolecular Diels – Alder reaction and subsequent aromatization through tautomerization.

Download figure:
Standard image
Download figure:
Standard imageZhang and co-workers 69 reported that bifunctional ligands L, with a Lewis-basic phosphine coordinating to the metal catalyst as well as a Brønsted-basic amine to act as a proton shuttle, together with a π-Lewis acid such as gold(I) chloride mediated the rearrangement of acetylenic amides 183 into amino-furans 185 (Scheme 33). These highly electron-rich intermediates were trapped, in situ, with a dienophile in an inter- or intra-molecular Diels – Alder reaction. Ring opening of the resultant dihydrofuran intermediates 187 and elimination of water gave the tetrasubstituted arenes 188 and 190. The intramolecular reaction affording the latter arene was extended with variation in the tether.
Kirschning and co-workers 70 reported three different routes to synthesize chromanes, a heterocyclic unit found in various biologically active natural products (Scheme 34). Reaction of vinyl-dihydropyran 191 with nitroalkenyl sulfoxide 192 gave cyclohexadiene 194 by Diels – Alder reaction and subsequent elimination of phenylsulfenic acid. Without the need of further oxidants, cyclohexadiene 194 underwent aromatization by elimination to yield alcohol 195. Diels – Alder reaction of vinyl-dihydropyran 196 with keto-acetylene 197 gave cyclohexadienes 198 and 199, which were subsequently converted into chromane 200 via DDQ mediated oxidation. Reaction using excess DDQ resulted in cleavage of the silyl-ether and oxidation of the benzylic alcohol giving the corresponding ketone. Other acetylenes and additives were examined, but the yield of the Diels – Alder reaction was not significantly better. Subsequently, the use of high pressure was investigated, which gave dihydrobenzenes 198, 199 in combined 35% yield. In addition, chromane 202 was formed under these conditions, presumably due to a second Diels – Alder reaction of dihydrobenzene 199 with keto-acetylene 197 and subsequent elimination of ethylene from 201 to produce chromane 202.
The pyrone Diels – Alder reaction was investigated by Kirschning and co-workers 70 initially on a model system, which gave lactones 204 and 205. Subsequent loss of carbon dioxide gave cyclohexadiene 206 and this was aromatized with DDQ to produce chromane 207 (Scheme 35). Optimum yields were obtained at room temperature and high pressure over prolonged reaction times. This method was successfully applied to dihydropyran 208 and methyl coumalate 79 in an inverse electron demand Diels – Alder reaction followed by the loss of carbon dioxide and DDQ mediated oxidative aromatization.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageIn recent studies on the synthesis of biphenyl and heterobiaryls, Temperini et al. 71 employed a Diels – Alder reaction of the enol acetate derived from 211, via transacylation with iso-propenyl acetate (213), with acetylene 212 (Scheme 36). 71 Yields were better at 145 °C than 110 °C, and elevated pressure in a closed metal vessel gave dihydrobenzenes 214, which upon DDQ mediated oxidative aromatization furnished biaryls and heterobiaryls 215 in moderate to good yields.

Download figure:
Standard image
Download figure:
Standard imageRecently, Hu and co-workers 72 reported that tetra-ynes 216 underwent cyclization via the arynes 217, 218, which were trapped by reaction with oxazolines 219 to give arenes 223. The authors suggested a plausible mechanism via intermediates 220 to 222 (Scheme 37). Equivalent reactions using oxazolines 224 gave the indoles 228, which were formed presumably via the intermediates 225 – 227.
Also, it was reported that reaction using oxazolines 229 without a substituent at the imine carbon atom gave adducts 232 in good yields (Scheme 38). These reactions were again suggested to proceed via intermediates 230 and 231.

Download figure:
Standard image
Download figure:
Standard image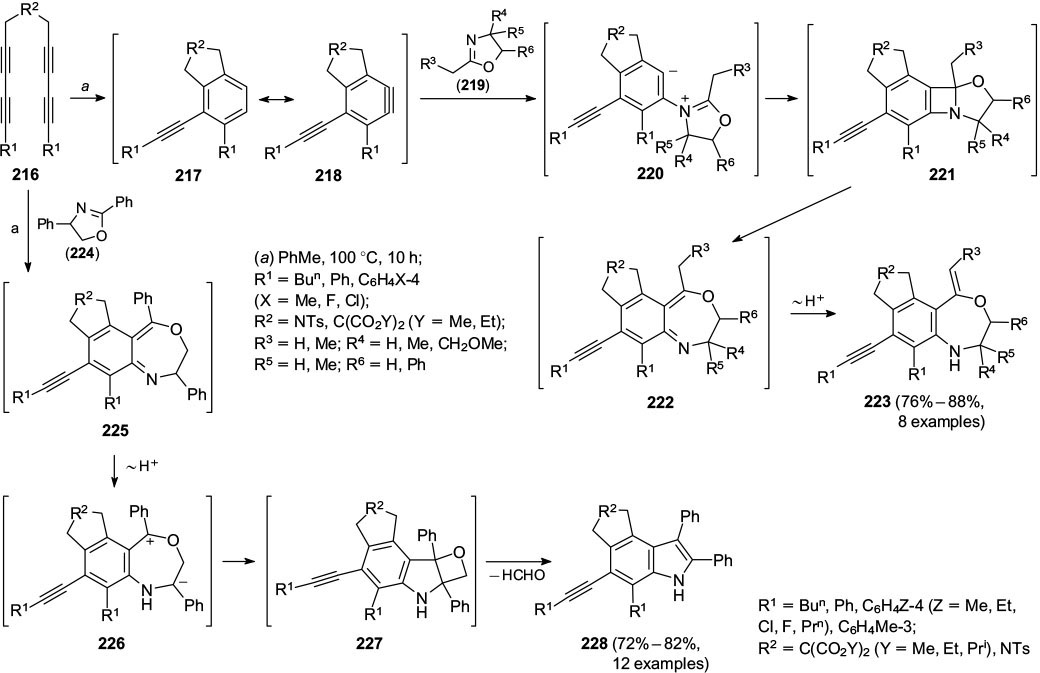
Download figure:
Standard image
Download figure:
Standard image3. Electrocyclic(pericyclic) reactions and aromatization
6π-Electrocyclic and pericyclic reactions are governed by orbital-symmetry, which controls their mode of cyclization (disrotatory or conrotatory). This stereoelectronic control has been used in highly regioselective and stereoselective syntheses of carbocycles or heterocycles through the simple use of heat or light. Aerobic oxidation or the addition of oxidants was used for aromatization to provide arenes with various substitution patterns with broad functional group compatibility and atom economy.
1,3-Cyclohexadienes are the initial product of 6π-electrocyclization reactions and were found to be convenient precursors for the synthesis of benzene derivatives by oxidation. Additionally, 6π-electrocyclizations have been proposed as possible pathways in the biosynthesis of natural products. In conjunction with other nπ-electrocyclic (n = 4, 8) or pericyclic reactions, these cascade reactions have been applied photochemically for the rapid assembly of complex products. 73 UV irradiation or thermally initiated 6π-electrocyclic and pericyclic reactions are therefore useful for the conversion of acyclic precursors into benzene derivatives. This chapter will exclusively focus on the synthesis of all carbon aromatic compounds; equivalent methods for heteroarene syntheses have been reviewed by Ding and Kaufman. 74, 75
Chuang and co-workers 76 reported that the (formal) electrocyclization of polyfunctional triene ylides 236, which were synthesized from enyne-esters 234, triarylphosphines 235 and imines or aldehydes 233, gave stabilized ylides 237 and subsequently oxindole and benzofuranone ylides 238 (X = NR, O, respectively). These were further derivatized to produce isatins 239 and isooxazolinones 240 (Scheme 39).

Download figure:
Standard imageIn 2016, Wilson and co-workers 77 reported that reaction of bromodienes 241 and trifluoroborate 242 under Molander – Suzuki – Miyaura conditions gave trienes 243, which underwent sequential thermal 6π-electrocyclization and dehydrofluorination reactions to give indanes, tetralins and benzosuberans 244 (Scheme 40). 77
Huang and co-workers 78 applied a 6π-electrocyclization and aromatization sequence by the addition of amines 246 to ynedienyl-amines 245 and 248 followed by siloxane elimination to synthesize variously substituted 1,3-diaminobenzene derivatives 247, 249 (Scheme 41). The reaction conditions tolerated a range of amine substituents with both cyclic and acyclic amines as defined below.
Delavatine A (254) and (–)-incarviatone A (255) are two secondary metabolites from Incarvillea delavayi. These plants are found in Southwest China and the Himalayas and have been used in traditional Chinese medicine to treat anemia and dizziness. Both have intriguing biological activities with delavatine A (254) possessing anti-cancer activity against several human cancer cell lines. Sarpong and co-workers 79 have developed a unified approach for the synthesis of these two natural products as they share a common molecular scaffold (Scheme 42). The polyene precursor 250 upon silylation with TBSOTf or TIPSOTf gave penta-ene 251 with a core triene with the (Z,Z,E)-geometry. This polyene 251 underwent a 6π-electrocyclization reaction and subsequent DBU-mediated aromatization of intermediate 252 to give the polyfunctional indane 253. Subsequently, it was found that this transformation worked well in a one-pot procedure with precise base stoichiometry (10 equiv.) to provide indane 253 as a 12 : 1 mixture of diastereoisomers. Within three steps this indane 253 was subsequently converted into delavatine A (254) and also used to complete a formal total synthesis of (–)-incarviatone A (255).

Download figure:
Standard image
Download figure:
Standard imageCoffin and Ready 80 reported the preparation of the advanced intermediate 262 in their enantioselective synthesis of (+)-dysoline (263) (Scheme 43). A highly regioselective Danheiser benzannulation between diazoketone 257 and alkyne 256 was used to synthesize the core chromenone 262. Irradiation with blue light induced a Wolf-rearrangement of diazo-ketone 257 to provide ketene 258, which, in turn, underwent a selective [2+2] addition with alkyne 256 to produce cyclobutenone 259, followed by a retro-4π-electrocyclic opening and subsequent 6π-electrocyclization of triene 260 and aromatization through tautomerization to give the advanced intermediate 262. The natural product 263 was shown to have cytotoxic activity against HT1080 fibrosarcoma cell lines (IC50 0.21 μmol L−1).
In 2015 and 2018, the group of Magauer 81, 82 reported a unified approach for the synthesis of variously substituted phenols 268, anilines and phenol ethers 269 and polycyclic compounds 271 and 272 through the 2π-disrotatory electrocyclic ring opening of bicyclo[3.1.0]hexan-2-ones 264 and 270 (Scheme 44). The optimal reaction conditions were applied to the synthesis of phenols 268 tolerating various electron-donating and electron-withdrawing substituents. In addition, a penta-substituted derivative of phenol 268 was synthesized in reasonable yield (53%) after a [1,2]-shift.
In 2017, Zhu and co-workers 83 developed a gold-catalyzed ring expansion including a 6π-electrocyclization-aromatization sequence to prepare a selection of mono- and bicyclic aromatic compounds 278 (Scheme 45). The authors' proposed mechanism of reaction involved enyne-activation and ring expansion to provide nonacycle 275, followed by a formal 6π-electrocyclization of the intermediate hexatriene 276 and subsequent aromatization.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageIn 2018, Li and co-workers 84 reported a divergent total synthesis of three Daphniphyllum alkaloids, namely, (–)-daphenylline (285), daphnipaxianine A (286) and himalenine D (287) using a late stage aromatization of the common intermediate 279 (Scheme 46). The natural products have been used in traditional Chinese medicine and have a range of biological activities, including antitumour and antiviral properties. Their synthesis of the central aromatic ring of the daphenylline precursor 284 applied a formal 6π-electrocyclization and aromatization sequence. Pentacyclic triketone 279 was allowed to react with guanidine 280 (triazabicyclodecene, TBD), which induced a vinylogous aldol reaction with the adjacent ketone to produce the cyclopentene-anion 281. This set the stage for a 6π-electrocyclic ring expansion, which upon elimination of hydroxide from 282 produced the aromatic core of daphenylline 283, followed by an additional aldol reaction of the acetophenone moiety with the proximal ketone. This elegant reaction cascade sequence led to the formation of one aromatic and 2 different ring systems in one procedure. Reductive deoxygenation completed the synthesis of the natural product (285).

Download figure:
Standard image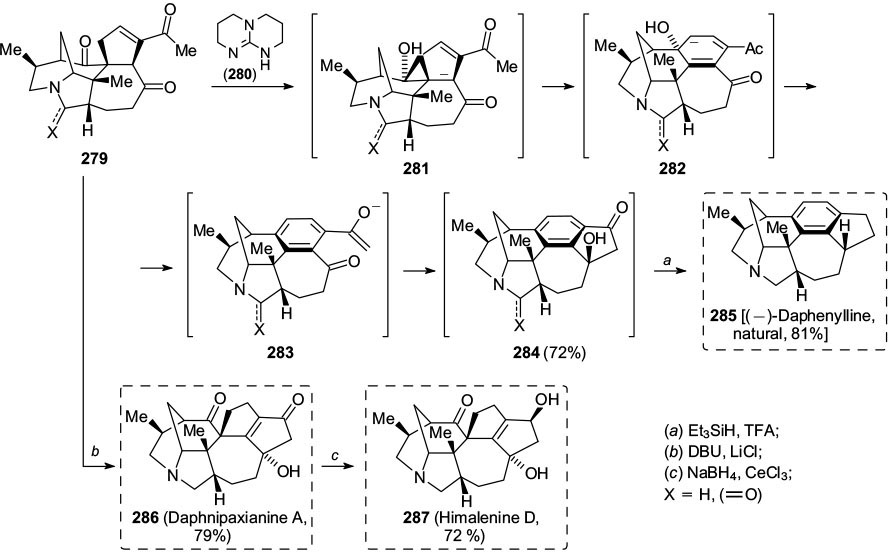
Download figure:
Standard image4. Transition metal-catalyzed reactions and aromatization
A series of ortho-terphenyl derivatives 291 were prepared by Dash and co-workers 85 utilizing a ring closing alkene metathesis (RCM) reaction as a key step (Scheme 47). Treatment of diketones 288 with allylmagnesium bromide or sequentially with (allyl)1-2–XBr (X = Zn, In) and allylmagnesium bromide provided the diallylated compounds 289, which were subjected to RCM with Grubb's 2nd generation catalyst thereby providing tricyclic diols 290. Subsequent reaction of these diols 290 with p-toluenesulfonic acid resulted in aromatization via double dehydration to provide the arenes 291.
In 2016, Xie and co-workers 86 reported studies on the total synthesis of (–)-rubriflordilactone B (295), a natural product which showed bioactivity against HIV-1 replication (Scheme 48). The key aromatization step in their approach involved a rhodium-catalyzed formal [2+2+2] cycloaddition of triynes 292 to produce arene 293, which was subsequently allowed to react with p-toluenesulfonic acid to give an advanced lactone intermediate 294.
Matsuda and Matsumoto 87 reported a rhodium(I) catalyzed approach for the synthesis of arenes 301 (Scheme 49). It was found that Wilkinson's catalyst [(Ph3P)3RhCl] mediated the insertion of alkyne 46 into the C–C bond of cyclobutenes 296 to give a range of tetra- and pentasubstituted arenes 301.

Download figure:
Standard image
Download figure:
Standard imageBenzocyclobutenes are valuable synthetic intermediates in organic chemistry. Recently Shi and co-workers 88 reported a gold(I)-catalyzed dehydrogenative cycloisomerization of 1,5-enynes 302 to produce disubstituted benzocyclobutenes 305 after oxidative aromatization using DDQ (Scheme 50). Furthermore, cyclobutane and cyclohexane but not cyclopentane tethered 1,5-enynes 306 were also aromatized under the reaction conditions to produce arenes 307 as well as enynes with the R1R2C = CH2 tethers (where R1 = R2 = Me, Et; R1 = Me; R2 = Ph, C6H4Cl-4).
A new method for the synthesis of aryl triazoles was reported by Fan and co-workers 89 (Scheme 51). This involved one pot cascade reactions of alkynes 308 with aliphatic keto-azides 309 and allenic ketones 311. Firstly, the alkynes 308 and azides 309 underwent copper-catalyzed Huisgen cycloaddition reactions to give triazole intermediates 310, which were subsequently allowed to react with allenic ketones 311 via Michael addition and intramolecular condensation to yield the aromatic compounds 313.
Chan and co-workers 90 reported that in the presence of DDQ or N-fluorobenzenesulfonimide (NFSI) as an oxidant, the known cycloisomerization of 1,4-enyne esters 314 to cyclopropanated cyclopentene-derivatives 315 occurred with oxidative ring expansion to provide phenol derivatives 317 in moderate to good yields (Scheme 52). The reaction tolerated various aliphatic and aromatic substituents in the meta-position thereby highlighting the value of synthesizing aromatic compounds from acyclic precursors given the preference for ortho- and para-substitution of phenols on reaction with electrophiles or via ortho-lithiation based processes.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageZi and co-workers 91 reported a de novo synthesis of substituted phenols 320 via oxidative cycloaromatizations of dienynes 318 (Scheme 53). 91 The key step involved their rhodium-catalyzed conversion into the corresponding metal vinylidenes, which were subsequently oxidized to provide metal ketenes 319. These reactive intermediates 319 underwent cycloaromatization to provide phenols 320 in fair to good yields.

Download figure:
Standard image
Download figure:
Standard imageIn 2018, Li and co-workers 92 reported a palladium-catalyzed carbonylative benzannulation approach to synthesize substituted phenols 324 (Scheme 54). Hydrosilane 322 was used to mediate the palladium-catalyzed reductive [5+1] cycloaddition of enynes 321 with carbon monoxide to provide a range of substituted phenols 324 with excellent functional group tolerance.
The gibberellins are a family of tetracyclic ent-kaurenoid diterpenes that exhibit multiple biological activities including promoting plant germination and growth. When Zhang and co-workers 93 prepared triazole derivatives of allogibberic acid 326, they utilized two different aromatization approaches to elaborate the arene core (Scheme 55). Aromatization of gibberellin 325 with hydrochloric acid provided arene 326, which was subsequently esterified to produce methyl ester 327. On the other hand, sequential esterification and Dess Martin periodinane (DMP)-mediated oxidation of gibberellin 325 gave enone 328, which was subsequently aromatized using quinone 329 and Pd(PPh3)4 in aqueous DMSO to give phenol 330.
In 2018, Loh and co-workers 94 reported the synthesis of highly substituted salicylaldehydes 336 starting from dialkenyl ketones 331 and diazoketones 332 using rhodium catalysis (Scheme 56, conditions a). The authors proposed that the reaction proceeded via intermediates 333 – 335. This reaction tolerated various alkyl, aryl and alkenyl substituents and was also useful for the synthesis of a series of arylene-spaced bic(salicylaldehydes) in moderate yields.

Download figure:
Standard image
Download figure:
Standard image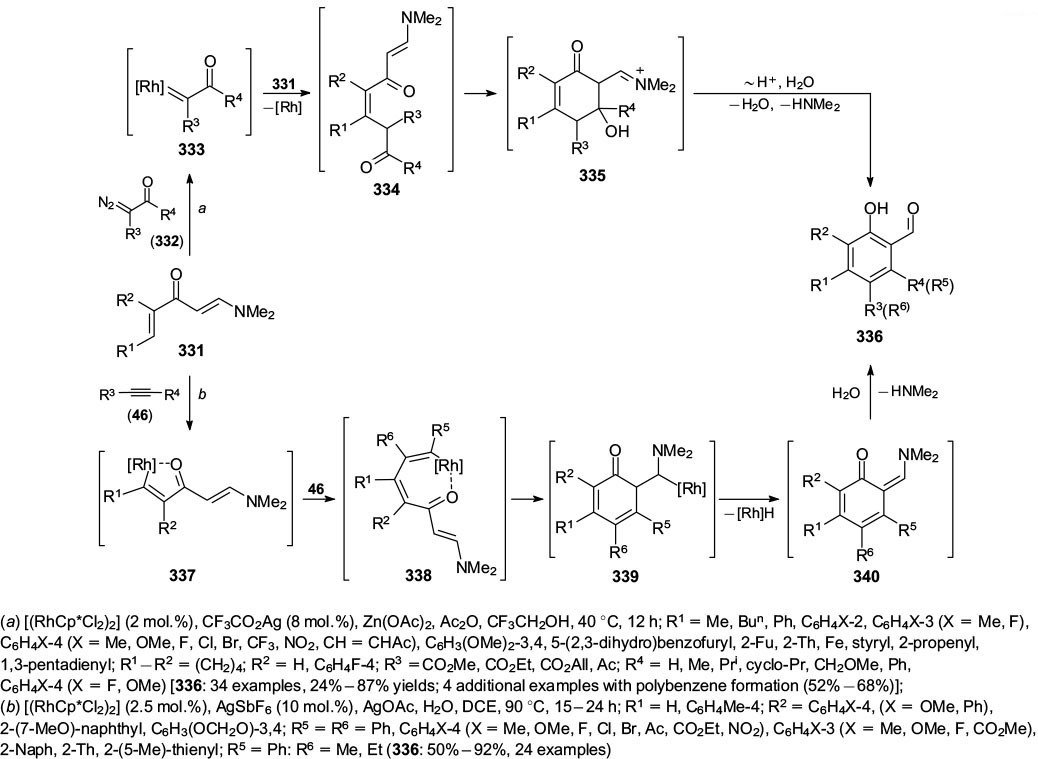
Download figure:
Standard imageGao and co-workers 95 reported the synthesis of the same structures 336 via rhodium catalyzed reaction of similar dienamides 331 with acetylenes 46 (Scheme 56, conditions b). The authors suggested that the process proceeded via the organo-rhodium intermediates 337 to 339, quinomethide 340 and its hydrolysis.
Gawali and Gunanathan 96 reported that iron-pincer complex 341 catalyzed a regioselective cyclotrimerization to yield trisubstituted benzene derivatives 345, 346 (Scheme 57). Activation of the iron dichloride in the 341 with ethylmagnesium bromide produced iron(I)-hydride, which was the active catalytic species for the reaction and mediated the stepwise formal [2+2+2] cycloaddition of aryl alkynes 308 via intermediates 342 – 344 to provide arenes 345 and 346. The authors also reported that aliphatic alkynes reacted in a similar fashion to yield the corresponding arenes.

Download figure:
Standard imageToyota and co-workers 97 reported a palladium-mediated aromatization of various alkenyl-β-keto-esters 347 to produce substituted phenols 324 (Scheme 58). The authors proposed that the reaction proceeded with the enolate attack of the terminal carbon to form an acetoxy palladium intermediate 348. Subsequent β-elimination and oxidative aromatization gave phenols 324.
In 2019, Gong and co-workers 98 described the total synthesis of dehydrobotrydienal (355), dehydrobotrydienol (356) and 10-oxodehydrodihydrobotrydial (357). These botryanes are structurally diverse sesquiterpenoids that are isolated from the phytopathogenic fungus Botrytis cinerea (Scheme 59). Co-tetramethylthiourea (351, TMTU)-catalyzed tandem Pauson – Khand and 6π-electrocyclization reactions of trienyne 350 gave the tricyclic ketone 353. Subsequent oxidative aromatization mediated by DDQ gave the aromatic ketone 354, which was subsequently transformed into the above-mentioned botryanes 355 – 357.

Download figure:
Standard imageAryl triazenes are versatile synthetic intermediates since they can be transformed into many different compounds on reaction of the derived arenediazonium salts with different oxygen- and nitrogen-containing nucleophiles. Recently, Cramer and co-workers 99 reported the synthesis of heavily substituted aromatic triazenes 363 and 365, which utilized a ruthenium-catalyzed cyclization reaction (Scheme 60). Reaction of tethered diynes 358 with alkynyl triazenes 359 in the presence of a RuII complex resulted in formal [2+2+2] cyclotrimerization reactions, producing aryl triazenes 363. Similarly, 1-diynyl triazenes 364 and alkynes 46 underwent a related [2+2+2] cycloadditions to produce aryl triazenes 365. The retention of the triazine unit during these aromatization reactions is noteworthy.
Ma and co-workers 100 reported a palladium-catalyzed synthesis of 1,3-dihydroisobenzofurans 371, 372 and 374, 375 (Z = O) and isoindolines 374 and 375 (Z = NTs) (Scheme 61). The authors found that palladium acetate and tri-2-furylphosphine (TFP) promoted the cyclization of dienyl carbonates 367 with terminal alkynes 366 to produce dihydroisobenzofurans 371 and 372, favouring regioisomers 371 due to regioselective double alkyne insertion reactions. Furthermore, a range of oxygen- or nitrogen-tethered carbonates 373 were also cyclized with terminal alkynes 366 to produce arenes 374 and 375.
Yang and co-workers 101 reported a simple formal [3+3] cycloaddition approach to produce polysubstituted terphenyls 382 from vinylethylene carbonates 376 (Scheme 62). With palladium catalysis, the latter compounds were converted into the corresponding α,β-unsaturated aldehydes 377, which were subsequently allowed to react with allylic sulfones 378 to induce a regioselective [3+3] oxidative cycloaddition in the presence of DBU to give terphenyl compounds 382.
Kale and Liu 102 reported that the cyclohexene-bridged diyne 383 underwent gold-catalyzed aromatization reactions with para-substituted nitrosobenzenes 384 to give tetrahydronaphthaldehyde derivatives 388 (Scheme 63). It was found that the propargylic oxygen substituent of the 1,6-diyne substrate influenced the reaction outcome with larger silyl groups leading to higher reactivity due to the operation of the Ingold – Thorpe effect.

Download figure:
Standard image
Download figure:
Standard imageDeng and co-workers 103 reported a palladium-catalyzed aerobic benzannulation approach for the preparation of highly functionalized anilines 396 (Scheme 64). It was found that these anilines were formed via palladium-catalyzed formal [3+2+1] cycloadditions between amines 389, aldehydes 138 and β-dicarbonyl compounds 390. It is important to note that the addition of acetic acid was essential to promote both the condensation process as well as in the oxidative dehydrogenation step.
Ghorai and Lee 104 reported aryne-based multicomponent coupling reactions for the preparation of highly functionalized aromatic compounds 402 – 406 (Schemes 65, 66). They reported the hexadehydro Diels – Alder reaction of the tetra-ynes 214, 397 to produce aryne intermediates 398, which readily underwent reaction with aryl isocyanides to form nitrilium intermediates 399. Subsequent trapping of these species with carboxylic acids gave the corresponding imides 401 after Mumm rearrangement. This method was further extended for the syntheses of imidate 404, amidine 405 and amide 406 by respectively replacing the carboxylic acid with an alcohol, sulfonamide or water (see Scheme 66). All these 3-component coupling reactions were generalized by variation in the tetra-yne, alcohol and sulfonamide. Also, the processes could be catalyzed using silver(I) hexafluoroantimonate but yields were better in its absence.
(–)-Rubriflordilactone B (295) is a highly functionalized polycyclic nortriterpenoid extracted from the plants of the Schisandra sp. and, as mentioned above, displays antiviral and anticancer activities. Recently, Anderson and co-workers 105 reported a convergent strategy for the total synthesis of this natural product in which the aromatic core was elaborated from a non-aromatic precursor (Scheme 67). Triyne 408, derived from triyne 407 by desilylation, underwent a rhodium-catalyzed cyclotrimerization reaction in the presence of Wilkinson's catalyst [(Ph3P)3RhCl] to give the arene core of alcohol 409, an advanced intermediate which was further converted into (–)-rubriflordilactone B (295) and (–)-pseudo-rubriflordilactone B (410).

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageHyland and co-workers 106 disclosed the synthesis of isoindolinones 418 via a gold catalyzed cycloisomerization and aromatization of diyne 411 (Scheme 68). The authors suggested that the reaction proceeded via gold acetylides 412 and 413, a second gold incorporation as acetylene complex 414, and cyclization via intermediates 415 – 417.

Download figure:
Standard image
Download figure:
Standard imageAs a follow up publication from the earlier dihydroisobenzofuran synthesis (see Scheme 61), Ma and co-workers 107 reported that the use of α-disubstituted propargylic alcohols 420 gave tricyclic arenes 424 with only minor amounts of the β-elimination products (Scheme 69). The authors suggested a mechanism where Pd-coordination induced the elimination of the carbonate and gave the reactive intermediate 421, which reacted with propargylic alcohol 420 to give the allene 422. Arene 423 formation was followed by nucleophilic displacement of the catalyst to release the benzoisofuran derivatives 424 in fair to good yields. The use of enantiomerically pure diynes 367a,b gave the arenes 424a,b with retention of configuration at the propargylic position.
Recently, Zhu and co-workers 108 showed that diynes 425 on reaction with a catalytic amount of cobalt dichloride and pinacolborane 426 in the presence of phenanthroline gave vinyl boranes 427 in moderate to good yields (Scheme 70). Vinyl borane 427a was subsequently converted into cyclohexadienes 428, employing a Suzuki – Miyaura coupling reaction followed by a thermal 6π-electrocyclization. Oxidative aromatization mediated by DDQ gave indanes 429 in good yields.
Lee and co-workers 109 expanded their earlier work 104 on imides 401, imidates 404, amidines 405 and amides 406 (see Schemes 65, 66) by trapping the aryne intermediate 432 from triynes 430 by nitriles in the presence of a silver catalyst (Scheme 71). Silver complexed aryne 433 gave rise to two regioisomeric nitrilium ions 434 and 437, which underwent conversion into the two isomeric quinazolines 436 and 439 on treatment with a second equivalent of nitrile. The observed regioselectivity was controlled by the steric bulk of the R5 group with tert-butyl favouring nitrilium ion 434 whereas nitrilium ion 437 was favoured with the smaller TMS group. Aromatic substituents resulted in a slight preference of 434 over 437.
Additionally, the same group 110 reported that arynes of type 434 derived from 440 were trapped with isonitriles to give benzocyclobutene-diimines 441 in moderate yields (Scheme 72). Mixed additions of both nitriles and isonitriles were also performed. In this case, reactions gave indolimines 442 in fair yields, as well as the derived hydrate 443 in moderate yields in some cases.
In 2020, B.Wang and co-workers 111 reported the rhodium catalyzed annulation of heterocyclic amides 444 with diynes 445 (Scheme 73). As a directing group, the amide functionality is known to facilitate Rh–C–H insertion to intermediate 446, which reacted with diyne 445. Subsequently, a five-membered rhodacycle 448 was formed, which in the presence of a nucleophile lost the directing group. Concomitant insertion into the other alkyne gave the metallacycle 449, followed by rhodium elimination to give arenes 450 in low to good yields.

Download figure:
Standard image
Download figure:
Standard imageActivation of diynes 451 with rhodium in the presence of 2 equivalents of quinones 452 gave naphthoquinones 457 (Scheme 74). 112 Initially, the metallacycle 454 was formed, which reacted with quinone 452 in a cycloaddition type fashion to give diene 456 after rhodium elimination. The intermediate cyclohexadiene 456 was aromatized by reaction with a second equivalent of quinone 452 giving naphthoquinones 457 in fair to good yields. Hexasubstituted arenes were accessible via this procedure in moderate yields.
Foubelo and co-workers 113 investigated strategies for the conversion of diynes and enynes into arenes. Firstly, rhodium catalyzed [2+2+2]-cyclotrimerization of diyne 458 and acetylenes 112 gave tetrahydroisoquinolines 463 (Scheme 75). In addition, enynes 464, 465 were cyclized using the Hoveyda – Grubbs catalyst to give dienes 466 and 469. Finally, Diels – Alder reaction of these compounds with dimethyl acetylenedicarboxylate (112a) and subsequent DDQ mediated oxidative aromatization gave tetrahydroisoquinolines 468 and 470.
Li and co-workers 114 reported the oxidative coupling of primary alcohols 472 with differently substituted cyclohexanones 471 on Pd/C for the synthesis of o-substituted phenols 478 (Scheme 76). Oxidation of the alcohols 472 to the corresponding aldehydes 473 followed by base catalyzed aldol reaction gave alcohols 474. Dehydration and alkene isomerization gave cyclohexanone 476, which on aerobic oxidation and tautomerization gave phenols 478 in reasonable yields. Intriguingly, reaction with a steroid triol occurred with regioselective oxidation of the primary alcohol and gave the steroidal phenol 479.

Download figure:
Standard image
Download figure:
Standard imageRecently, Deng and co-workers 115 reported the synthesis of heavily substituted phenols 487 from allenyl diazo ketones 480 (Scheme 77). Upon mixing with a silver salt, allene 481 was cycloisomerized to diazofuran 483. Under rhodium catalysis conditions, this compound was converted into the rhodium-carbene 484, which underwent cyclopropanation and ring expansion to give the cyclohexanone 486. When R1 = Ar, R2 = OEt these underwent tautomerization under the reaction conditions and gave phenols 487 in good yields. In ten other examples when R2 = Ph, OEt the products isolated were the cyclohexenones 486. Addition of a second Lewis acid mediated the aromatization of seven of those examples (R2 = OEt, 50% – 86%) while three (R2 = Ph or R1 = H and R2 = OEt) did not undergo conversion into the phenols 487. Further attempts to transform this reaction into a one-pot procedure were unsuccessful since the reaction stopped at the furan 483 stage.
Toyota and co-workers 116 reported the conversion of enamines 488 into anilines 491 in moderate to good yields by reaction with copper(II) and palladium(II) acetates in DMSO (Scheme 78). The transformation by Pd(OAc)2 mediated cyclization to the palladated cyclohexane derivative 489 and subsequent reductive elimination gave cyclohexadiene 490, which was subsequently converted into the anthranilic acid ester derivatives 491 by further oxidation with Pd(OAc)2. In these cases, Cu(OAc)2 was the best co-oxidant. When the amine 488 was only monosubstituted and the alkene was terminal, a mixture of the cyclohexanone imine 489 and the corresponding pyrrolidine was obtained, which gave rise to a mixture of anilines and pyrroles slightly favouring the aniline. Additionally, the authors showed that the procedure was amenable to a one-pot process starting with the acetoacetate and the amine (with oxygen as the oxidant).

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image5. Other aromatization strategies
This final section of the review will focus on alternative methods that are not classified according to the sections above. This includes aromatization reactions that depend on applications of organo-catalysis, base-mediated reactions (Aldol-reactions, Mannich-reactions, etc.) and oxidative cyclohexane- or cyclohexanone halide – halonium addition – elimination reactions to form phenols. These methods are highly versatile and applicable for the synthesis of heavily substituted phenols from readily available starting materials. As shown in this chapter, new developments in phenol syntheses have facilitated the synthesis of m-substituted derivatives through pre-functionalization and subsequent aromatization. These approaches overcome the limitations of electrophilic aromatic substitution reactions or ortho-lithiation reactions of phenols and phenol derivatives, which strongly favour ortho- or para-substitution reactions and not meta. These reactions may be sub-divided according to their reaction type.
5.1. Halogen-mediated reactions
Luo et al. 117 showed that substituted cyclohexenones 492 reacted with iodine in DMSO under air to provide phenols 493 (Scheme 79). This procedure was applicable to a broad range of different substitution patterns including for the elaboration of tetra-substituted phenols 493.
Kang and co-workers 118 reported the trimethylsilyl triflate-catalyzed aromatization of Hagemman's esters 494 with NBS as the stoichiometric oxidant. Sixteen examples of phenol derivatives 495 were synthesized via this route by simple variation of the aldehyde precursor (RCHO) used in the preparation of the starting cyclohexanone 494 (Scheme 80) from ethyl acetoacetate.
Luo and co-workers 119 reported the conversion of α,β-unsaturated oximes 496 with iodine under air to provide anilines 500 (Semmler – Wolf type reaction) (Scheme 81). Improving on recent findings, this method excluded strong acids or transition metal catalysis. A range of aromatic substituents were tolerated with the R1 and R2 substituents and the reaction provided the desired products with overall moderate to good yields. The authors suggested that the reaction proceeded via the imine radical 497, its tautomerization to radical 498, C-iodination and elimination to the aniline tautomer 499 and aromatization. Whilst the authors have shown by reaction inhibition with 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) that the process involves radicals, the authors of this review consider that the imine radical 497 is an unlikely intermediate and the reaction more likely proceeds via an alternative radical pathway and intermediacy of the dihydroaryl hydroxylamine and dehydrative aromatization.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image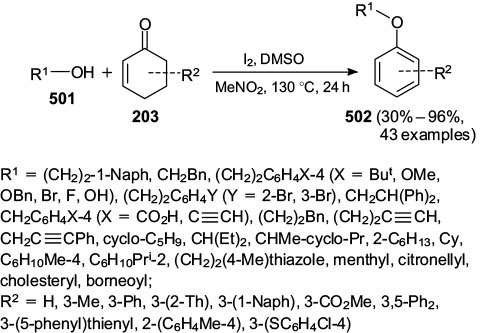
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageJiao and co-workers 120 reported the coupling of cyclohexenones 201 with primary, secondary, terpene based, cyclic, and benzylic alcohols 501 to afford phenol ethers 502 (Scheme 82). The reaction tolerated different substituents on the cyclohexanone scaffold as well, giving tri-substituted phenol ethers in fair to excellent yields.
Sha and Liu 121 discovered a Paal-Knorr based benzofuran synthesis starting from the readily available 2-hydroxy-1,4-diketones 503 (Scheme 83). With the use of trifluoroacetic acid-catalysis, the diketones were converted into the furans; subsequent NBS oxidation resulted in aromatization to produce benzofurans 504.

Download figure:
Standard image
Download figure:
Standard imageYang and co-workers 122 reported a general method for the syntheses of phenols 509 with a varying substitution patterns starting from 1,3-diketone 390a and enones 209 (Scheme 84). Self-condensation of 1,3-diketones 505 mediated by iodine as catalyst followed by transacylation (C to O) of intermediate 506 and subsequent iodine promoted oxidation, presumably via iodides 507, provided a variety of biaryl-phenols 509.
Additionally, the authors showed that 1,3-diketones 390a, 510 were general precursors for the synthesis of biaryl phenols 512, 514, 516 and 517 using α,β-unsaturated ketones 511, 513, 515 and 209a, respectively (Scheme 85). Both symmetrical 513 as well as unsymmetrical ketones (511, 515, 209a) were found to readily react to produce the corresponding phenols in moderate to good yields using this iodine mediated reaction.
In 2019, Wu et al. 123 reported the oxidization of cyclohexanones 471 with iodine in the presence of DMSO for the synthesis of 2-sulfanyl-catechols 518 (Scheme 86). This method proved to be general and worked with a range of alkyl and carboxylate substituents in the 2, 3 and 4 position of the cyclohexanones 471 and the reaction was also applicable to 2-cyclohexenones (e.g., 201a) as starting materials.
5.2. Organocatalyst-mediated reactions
In 2017, Lee and co-workers 124 reported the reaction of the nitroalkene 519 and cinnamaldehyde (520a) with the prolinol catalyst 30 to produce the heavily substituted spirocyclic oxindole derivative 521 as well as the arene 522 (Scheme 87). This reaction proceeded through a catalyst 30 mediated Michael – Michael – Michael – Aldol reaction cascade, a multistep process that proceeded with remarkable enantioselectivity (>96% ee) to provide oxindole 521 and presumably its spirane epimer, which underwent spontaneous aromatization by the elimination of nitrous acid under the reported reaction conditions and directly gave the spiro-arene 522. The authors noted that other organocatalytic reactions of cinnamaldehyde derivatives with nitroalkenes structurally related to nitroalkene 519 gave spirocyclic oxindole derivatives corresponding to oxindole 521 as mixtures of spirane-epimers and no arene products corresponding to arene 522. Presumably these cyclohexenes e.g., 521 could be aromatized by reactions at the nitro-group.

Download figure:
Standard imageIn 2018, Chi and co-workers 125 reported that an N-heterocyclic carbene (NHC) derived from a precatalyst 524 catalyzed the aromatization condensation reaction of α,β-unsaturated p-nitrophenol esters 523 with enediones 392 to produce tetrasubsituted benzene derivatives 530 (Scheme 88). The authors proposed that the reaction proceeded via the Michael and aldol adducts 526 and 527, lactone 528 formation and elimination of carbon dioxide to provide cyclohexadiene 529, with final aromatization carried out with TEMPO as the oxidant. With this method, a broad variety of different biaryl systems 530 were synthesized in moderate to excellent yields.

Download figure:
Standard imageAfter their successful synthesis of tetrasubstituted arenes 530 from α,β-unsaturated esters 523 under NHC catalysis, the Chi group 126 expanded this methodology to prepare stereodefined indane derivatives 537 in good yields and enantiomeric ratios (up to 96 : 4 er) (Scheme 89). The reaction followed a similar pathway as above, however, using the stereodefined NHC-based catalyst 533, gave the products 537 with high enantioselectivities (up to 91% ee). The scope of reaction allowed for variation of the different substituents R1, R2 and R 3 in substrates 531, 532. In addition, several spirocyclic 1,3-dienones 532 gave the corresponding derivatives in good yields and moderate to excellent enantiomeric ratios.

Download figure:
Standard imageIn addition to the synthesis of arenes 530 and stereodefined indanes 537, Chi and co-workers 127 showed that the method was useful for the synthesis of biaryls such as phenyl benzothiazoles 543 (Scheme 90). Reaction of enal 531 and enone 538 gave phenyl benzothiazoles 543, which were formed via a NHC catalyzed Michael-addition and aldol reaction pathway, followed by lactone formation and loss of carbon dioxide to give 542. Oxidation with 3,3',5,5'-tetra-tert-butyldiphenoquinone (534) gave the phenyl benzothiazoles 543 in moderate to excellent yields. Benzothiazoles have been shown to act as directing groups in transition metal-catalyzed C–H activation reactions and are thus useful intermediates for late-stage functionalization.

Download figure:
Standard imageAdditionally, Zhu and co-workers 128, 129 applied the same stereodefined NHC based catalyst for the synthesis of biaryls 551 with good levels of atropoisomeric control (98% ee) (Scheme 91). The reaction proceeded via a similar mechanistic pathway as postulated by Chi and co-workers 125 – 127 and also tolerated various functional groups in the ortho and para positions. The resultant biaryls 551 were isolated in good to excellent yields and enantioselectivities.
In 2019, Wang and co-workers 130 extended the stereoselective NHC catalysed indanone syntheses by employing a stereodefined catalyst to bring about the coupling of various cyclopentene-1,3-diones 532 with α,β-unsaturated enals 531 (Scheme 92). The mechanism, as postulated above, involved a Michael – aldol – aromatization cascade to elaborate the aromatic indanone framework followed by protection of the phenol as the methyl ether. The reaction was shown to tolerate various substituents on the starting cyclopentenediones 532 as well as different aromatic substituents on the enal 531.

Download figure:
Standard image
Download figure:
Standard imageRecently, Han and co-workers 131 published a prolinol catalyzed coupling of acrylonitriles 554 with enals 520 to prepare tetrasubstituted arenes 561 (Scheme 93). Michael-addition of the resonance stabilized anion 555 with the iminium ions 556 gave adducts 557. Proton transfer and Mannich-type addition gave the cyclohexene derivative 559, which upon elimination of the catalyst gave cyclohexadiene 560, which easily underwent oxidation under aerobic conditions to give arene 561. The authors applied the method to synthesize various trisubstituted RF containing arenes in an efficient manner.
In addition, Han and co-workers 131 showed that prolinol 30 catalyzed the reaction of β-trifluoromethyl-enones 562 and enals 520 and gave tetrasubstituted benzaldehydes 568 (Scheme 94). A similar reaction pathway as described above gave intermediate 565, which underwent a prolinol catalyzed aldol reaction to give the cyclohexene core 566. Dehydration and imine hydrolysis gave the pentasubstituted arenes 568 after aerobic oxidation. These two methods starting from a range of enones and enals gave the derived tetra- or penta-functionalized benzoate esters 561 and 568.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageHan and co-workers 132 also reported the synthesis of tetrasubstituted anilines 572 and trisubstituted benzonitriles 561 from acrylonitriles 554 and Michael acceptors 569 and 573 (Scheme 95). The authors suggested that the mechanism involved deprotonation by DABCO and Michael addition to intermediate 570, which upon ring closure, hydrogen cyanide elimination underwent tautomerization to aniline 572. Alternatively, using DMAP and Michael acceptor 573, a Baylis – Hillman type reaction gave intermediate 575, which underwent elimination of DMAP and acetate. A subsequent Michael addition gave the nitronate 577, which underwent elimination of nitrous acid followed by aerobic oxidation to give various RF-substituted benzonitriles in good yields.
5.3. Acid-catalyzed reactions
Similar to Li's approach, Zhai and co-workers 133 retrosynthetically planned their total synthesis of (–)-daphenylline (285) to involve an aromatization step applied to a precursor already bearing the ABCD-ring system of the final natural product (Scheme 96). In their approach, however, they utilized a ring expansion process starting with cyclopentanone 579. After dehydrative cyclization to the furan 580, the cyclopentane unit was found to undergo a [1,2]-alkyl shift to produce furanyl carbenium ion 581, which, in turn, underwent aromatization by the elimination of water and proton loss giving the product 582. In this publication, the results inspired the Zhai group to propose a possible biosynthetic link between the various Daphniphyllum alkaloids.
In 2018, Wan and co-workers 134 reported a method to synthesize m- and p-substituted benzaldehyde derivatives 589 and 591 from enamines 585 and 590 and enals 584 (Scheme 97). An initiatal vinylogous Michael addition of enamine 585 to the protonated enal 584 gave enol 586, which underwent cyclization to the cyclohexene 587. Elimination and aerobic oxidation gave the m- and p-substituted arenes in good yields. In addition, it was shown that the enamines 590 could be formed in situ from acetylenes 210 and dimethylamine, which gave the benzoate esters 591 in good yields following a similar reaction pathway.

Download figure:
Standard image
Download figure:
Standard imageIn 2019, Rubin and co-workers 135 published a synthesis of aniline derivatives 599 via a three-component coupling of 1,3-diketones 592, amines 593 and acetone (594a) (Scheme 98). The authors suggested that the reaction proceeded by enamine formation and subsequent addition to the 1,3-diketones 592 to give the intermediate 596. Cyclization gave the cyclohexane 597, which underwent dehydration to give anilines 599. A large range of substituents were tolerated under the reaction conditions. Furthermore, penta-substituted anilines 601 could be synthesized with this methodology, albeit requiring longer reaction times and further addition of the dehydrating agent 4-toluenesulfonic acid.

Download figure:
Standard imageIn 2020, Xu and co-workers 136 reported the dimerization of various styryl-epoxides 602 to prepare biaryls 609 under microwave accelerated acid-base conditions (Scheme 99). The authors postulated that an initial Meinwald rearrangement took place to give aldehydes 605 or their corresponding enolates. These intermediates underwent dimerization followed by cyclization with an aldol reaction to hydroxyaldehydes 607. Dehydration gave diene 608, which underwent aromatization presumably by loss of toluene possibly in a concerted process. Biaryl benzaldehyde derivatives 609 were obtained in moderate to excellent yields and the reaction tolerated various functional groups on the aryl core. Noteworthy, substituents on the alkene in the cis-position were not tolerated and the reaction stopped after formation of the Meinwald rearranged product 605.

Download figure:
Standard image5.4. Base-catalyzed reactions
Recently, Ye and co-workers 137 reported a DBU mediated synthesis of tri-substituted arenes 346 from readily available α,β-unsaturated carboxylic acids 610 and α-cyano-enones 611 (Scheme 100). This method was applied for the synthesis of thirty two biaryls 346 in moderate to good yields. Mechanistically, the authors suggested that DBU had a dual role both as base as well as nucleophilic catalyst through a CDI activated acyl transfer of the α,β-unsaturated carboxylic acids to DBU 613. In their control-experiment without DBU, the reaction only formed traces of the product 346 even from the carboxylic acid CDI adducts. The acylated DBU intermediate 613 was found to react with the cyano-enone 611 (or its derived enolate) to produce the cyclohexenes 614, which upon β-lactone 615 formation, decarboxylation 616 and cyanide elimination underwent aromatization to produce the product arenes 346.

Download figure:
Standard image
Download figure:
Standard imageThorimbert and co-workers 138 showed that reaction of methyl coumalate 79 with trifluoromethyl-1,3-diketones 617 gave the corresponding trifluoromethylated arenes 622 (Scheme 101). Various aliphatic and aromatic substituents on the starting diketone were tolerated. Mechanistically, the authors suggested that the reaction proceeded by Michael-addition of the enolate derived from 617 followed by ring opening to the linear dienoate 619. Subsequent ring closure and loss of carbon dioxide and hydroxide in a Grob-type fragmentation gave the arene 622 in good yields. With this study the authors developed a potentially sustainable transformation of a renewable precursor into trifluoromethyl-functionalized arenes in a simple manner.
In 2018, Shi and co-workers 139 published a base promoted synthesis of heavily substituted benzonitriles 629 from diketo compounds 392 and α-cyanoenones 623 (Scheme 102). This method was applied for the preparation of biaryl compounds including hexa-substituted arenes 629 in moderate yields. Mechanistically, the synthesis involved a Michael addition of the enolate derived from cyano enones 623 with enones 392, followed by a tautomerization and aldol ring closure. Subsequent (C to O) acyl transfer and E1cB elimination of benzoic acid gave cyclohexadienes 628. Aerobic oxidation produced arenes 629 in fair to good yields.
Recently, Qiu and co-workers 140 reported the total synthesis of (–)-daphenylline (285) including a key Robinson annulation and aromatization sequence to build up the ABCE ring system (Scheme 103). Diketone 630, prepared from the corresponding ketone using the Stork-Ganem reagent MeCOC(=CH2)SiMe3, was converted into the aromatic compound 633 in good yield through a base mediated aldol-condensation, dehydration and oxidative aromatization in air.

Download figure:
Standard image
Download figure:
Standard imageYadav et al. 141 reported a formal [3+3] benzannulation between bromo-enones 634 and α,β-unsaturated sulfones 635 (Scheme 104). Both of these precursors were readily synthesized from commercially available starting materials through simple functional group interconversions. Subsequent DBU-mediated coupling readily gave tetrasubstituted arenes 639 under mild reaction conditions.
The group of Tummatorn 142 reported the sequential transformation of alkynyl-aryl ketones 640 to provide indenones 641, which were subsequently aromatized to provide 3-hydroxyfluorenes 644 (Scheme 105). This procedure was also used to synthesize 4-azafluorenes by employing ammonium acetate in methanol in the cyclization process (via the corresponding enamine of 641) and aromatization step.
Baidya and co-workers 143 reported the base-mediated coupling of alkylidene malonitriles 645 with cyclopentene-1,3-diones 532 to produce penta- and hexasubstituted 4-amino-1H-indene-1,3(2H)-diones 648 (Scheme 106). The authors suggested that this reaction proceeded via a vinylogous Michael-addition and subsequent cyclization by addition to the nitrile group, followed by oxidative aromatization. The reaction showed a broad functional group tolerance for aliphatic, aromatic and bulky steroid substituents.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageSosnovskikh and co-workers 144 showed that enaminodiones 649 were dimerized by condensation using lithium hydride and tetraethoxysilane to produce trisubstituted phenolic diketones 653 (Scheme 107). The authors postulated that these reactions proceeded via a double-Michael addition to intermediates 651 with subsequent, highly selective retro-Claisen reaction to yield benzophenones after aromatization. This method enabled the authors to rapidly synthesize different phenols 653 from simple precursors in three steps.
Pratap and co-workers 145 reported the synthesis of tetrahydroquinolines 659 from 2-pyrone derivatives 654 or 661 as C(4) building blocks and Boc-protected 3-piperidone 655 as a C(2) building block (Scheme 108). This method, following trifluoroacetic acid Boc-deprotection, gave tetrahydroquinolines 660 from easily accessible precursors in good yields. The authors proposed that Michael addition of the enolate derived from 655 with 2-pyrone 654, followed by ring opening and ring closure to cyclohexadienes 658 and a Grob-type fragmentation with concomitant aromatization gave arenes 659. Trifluoroacetic acid mediated Boc deprotection completed the synthesis of tetrahydroquinolines 660.
In 2019, Kano and co-workers 146 reported the synthesis of benzophenones 666 via base mediated annulation of imino-dienes 662 as C5 building blocks and substituted acetophenones 594 (Scheme 109). The authors proposed reaction mechanism involved Mannich-type additions of the enolates derived from ketones 594 with imines 662 followed by piperidine elimination to give the trienones 664. Cyclization of these gave cyclohexadiene 665. These intermediates underwent aromatization by elimination of a second equivalent of piperidine to give benzophenones 666 in fair to excellent yields. The reaction was extended to 4,4'-diacetyl-biphenyl, 1,3,5-triacetylbenzene and 4,4',4''-triacetyl-triphenylamine derivatives of 594.
In 2019, Wang and Kraus 147 reported the total synthesis of (±)-dihydro eurotiumide B (675), which employed a cascade sequence to build up the arene and lactone ring (Scheme 110). These authors showed that this reaction also worked with other epoxides as well as methyl sorbate, which lacked the epoxide functionality. They proposed that butanolide 667 underwent alkylation via the corresponding enolate with subsequent selective Michael addition onto the α,β-unsaturated ester 668. A subsequent aldol reaction gave the tetrahedral intermediate 670, which underwent fragmentation to the diketone 671 by thiophenolate elimination. Aromatization by tautomerization gave intermediate 672. Opening of the styryl epoxide by participation of the electron rich arene gave enolates 673, which readily underwent aromatization by the diastereoselective addition of thiophenolate. Substitution of the thio ether by sodium methoxide gave dihydro-eurotiumide B (675) in 41% yield.
Yu and Kraus, 148 in further studies on pyrone Diels – Alder chemistry, reported that electron-poor dienophiles including dihydroxy-p-quinones 676 do not undergo Diels – Alder reactions to provide arenes. However, addition of a base such as triethylamine mediated the formal Diels – Alder reaction of 2-pyrones 131 with hydroxyquinones 676 and 681 (Scheme 111). The authors suggested that these reactions involved base mediated enolate formation of the hydroxyquinones and subsequent Michael addition to give adducts 677 thereby allowing the aldol reaction to give intermediate 678. Subsequent decarboxylation and loss of water gave the arenes 680 in fair to excellent yields. In the case of dihydroxybenzoquinone 676 (R 3 = H), a double addition was observed giving rise to anthraquinones 683. Of particular note, acetylene substituents in the R2 position underwent oxidation to the corresponding ketones during the progress of this reaction, presumably after the elimination of water.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageIn 2020, Xu and co-workers 149 reported the synthesis of hydronaphthofurans 685 via rhodium catalyzed coupling of 1,6-enynes 684 with acetylenes 308 (Scheme 112). When (S)-BINAP was used as a ligand in the [2+2+2] cycloaddition reaction, enones 685 were formed with excellent enantioselectivities (>97% ee). Upon addition of sodium hydride, the derived enolate 686 underwent an oxy-accelerated [1,5]-hydride shift with concomitant aromatization to give arenes 688 in good yields with no reduction in enantiomeric excesses (>99% ee). In one case, the authors showed the reaction could be carried out as a one-pot process in good yield (62%) and enantioselectivity (99% ee). Other standard aromatization procedures, such as with DDQ, gave enone 689. Furthermore, aerobic oxidation in the presence of DBU gave arene epoxide 690 in excellent yield and enantioselectivity. In this case, oxidation of diene 685a with oxygen gave rise to hydrogen peroxide, which subsequently underwent reaction with the enone moiety via a stereospecific Scheffer – Weitz epoxidation to give epoxide 690.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageVerma and co-workers 150 reported the synthesis of indane 695 via an aza-Henry reaction between nitrile 691 and nitromethane (Scheme 113). Reaction of the nitronate from nitromethane with nitrile 691 and tautomerization and cyclization gave cyclohexadiene imine 694, which underwent aromatization to give the nitro-indane 695. We consider the mechanism involves these intermediates through nitronate addition to the acetylene and protonation by water to 692, followed by deprotonation to form nitronate 693 and cyclization to 694 and not via a vinyl carbanion as suggested by the authors. In addition, the authors reported 37 additional examples for the synthesis of naphthalene derivatives, which is outside of the scope of this review.
Recently, Tan and co-workers 151 reported a biomimetic total synthesis of the natural products sanctis A (700), iso-sanctis B (701) and sanctis B (702) using a classical orsellinate synthesis (Scheme 114). 152 1,3-Keto-ester 390b was allowed to react under basic conditions with diketene 696 to provide the triketo-ester 697, which readily underwent cyclization and aromatization to give resorcylate 699. This intermediate was converted into the three natural products 700 – 702.

Download figure:
Standard image5.5. Other oxidation reactions
Zhang et al. 153 reported a one-pot procedure for the syntheses of 5,7,8,9-tetrahydrobenzo[b]oxepin-6(2H)-one, which on dihydroxylation using osmium tetraoxide and NMO in aqueous acetone and double methanesulfonylation using methanesulfonyl chloride in pyridine was converted into the dimesylate 703a. Subsequent aromatization was carried out by reaction with tetra-n-butylammonium fluoride (TBAF) to produce the 2,5-dihydrobenzoxepine 704 (Scheme 115). Additionally starting from compound 703b, this reaction was successfully applied for the synthesis of radulanin A (705), a phenol with phytotoxic activity.

Download figure:
Standard image
Download figure:
Standard image
Download figure:
Standard imageIshida and co-workers 154 have recently reported the synthesis of atropisomerically enriched biaryls 708 via manganese dioxide mediated aromatization of cyclohexanones 707, which were readily prepared through an organocatalytic enantioselective (99% ee) Michael – Henry reaction (Scheme 116). A one-pot silyl-enol ether 707 formation followed by manganese dioxide oxidation gave the three biaryls 708 in high atroposelectivities (96 – 98% ee) as well as the 1-naphthyl-analogue (56%, 95% ee).
Lastly, Yanada and co-workers 155 reported the synthesis of benzophenones 716 and 718 from 1,6-diynes 709 and aliphatic and aromatic aldehydes 138 under Lewis acidic conditions (Scheme 117). The authors suggested that the mechanism of reaction involved a carbonyl – acetylene metathesis and was applied to 17 different benzophenones 713. Subsequently, in a one-pot procedure, these compounds were converted into indanes 716 and benzyl acetates 718 the ratio of which depended on the R2 substituent. When R2 was aromatic, upon addition of the oxidant iodobenzene diacetate and indium(III) trifluoromethanesulfonate, the stabilized allylic, benzylic carbenium ion 714 underwent Friedel – Crafts alkylation to give cyclohexadiene 715. Further oxidation with iodobenzene diacetate gave indanes 716 in good yields. Alternatively, when R2 was aliphatic, the same reaction promoted the formation of the allylic, benzylic carbenium ion 714, which was trapped with acetate, and loss of a proton to produce cyclohexadiene 717. Further addition of the oxidant iodobenzene diacetate gave benzyl acetate derivatives 718 in good yields.
6. Conclusion
Inspired in part by nature's methods for the biosynthesis of aromatic compounds, chemists have devised procedures for the synthesis of variously substituted benzenes, phenols, anilines, and other aromatic and heteroaromatic compounds from both cyclic and non-cyclic precursors. The underlying synthetic pathways are robust and well understood processes and depend on Diels – Alder, electrocyclic, pericyclic, cross-coupling, RCM, cyclo-isomerization, aldol or Michael addition reactions. The resulting aromatic compounds have diverse applications from medicinal chemistry to materials. Recent applications are in drug discovery and the total syntheses of meroterpenoids, polyketides and Daphniphyllum alkaloids with both early and late-stage incorporation of the aromatic units. Furthermore, the use of stereodefined catalysts to generate precursors for aromatization, has proven very valuable for the enantio- and diastereoselective synthesis of biaryl atropisomer systems, stereodefined ligands and compounds relevant to the elaboration of molecular machines. It is, therefore, reasonable to expect future developments in this important area of synthetic organic chemistry, given the extensive use of aromatic compounds in pharmaceuticals, agrochemicals, organic electronics and other materials.
We greatly appreciate the GlaxoSmithKline endowment (to A.G.M.B.) and extend our thanks to Dr. Alfred (AH) and Dr. Isabel Bader for their support. We thank Ms. Virginia Manch, Ms. Elfie Cavalli and Ms. Jiaxu Han for proofreading this review article.
Footnotes
- †
R.C.Larock, A.Pletnev. In Comprehensive Organic Transformations. (Eds R.C.Larock). (Wiley, 2018).