


Abstract
Molecular design of magnetically active graphene nanoscale structures is an emerging field of research. The key goal of this research is to produce graphene nanoribbons and graphene quantum dots with specified electronic, optical and magnetic properties. The review considers methods for the synthesis of spin-labelled polycyclic aromatic hydrocarbons, which are homologous precursors of graphene nanostructures, and discusses the advances and prospects of the design of magnetically active graphene materials.
The bibliography includes 134 references.
Export citation and abstract BibTeX RIS
| Yu.A.Ten. Junior Researcher at the NIOC SB RAS. |
| Telephone: +7(383)330 – 6859, e-mail: ten@nioch.nsc.ru |
| Current research interests: chemistry of heterocyclic compounds, stable organic radicals, high-spin systems. |
| N.M.Troshkova. PhD in Chemistry, Researcher at the same Institute. |
| Telephone: +7(383)330 – 6859, e-mail: romanova@nioch.nsc.ru |
| Current research interests: organic synthesis, nucleophilic substitution in aromatic and related compounds, development of methods for the synthesis of new effective biologically active polyfluoro-1,4-naphthoqui-nones. |
| E.V.Tretyakov. Doctor of Chemical Sciences, Deputy Director for Science of the NIOC SB RAS; Senior Lecturer at the Novosibirsk State University. |
| Telephone: +7(383)330 – 9171, e-mail: tretyakov@nioch.nsc.ru |
| Current research interests: functionally oriented synthesis, chemistry of heterocyclic compounds including fluorinated compounds, stable organic radicals, heterospin systems, molecular design of magnets. |
1. Introduction
Development of the modern society is inconceivable without the use of magnetically active systems and materials. First of all, this refers to magnetic materials able to exhibit magnetic ordering throughout their bulk. Such materials were known back in ancient times, they were used to design the first navigation instruments. 1,2 During the scientific and technological revolution, the demand for materials possessing specified magnetic characteristics has spiked, which resulted in the discovery and practical mastering of an extensive group of magnets based on transition and rare earth metals, their diverse alloys, metal oxides and other binary and ternary compounds. 3 A sound achievement is the successful use of molecular approach to design magnetic materials based on organic, organometallic and coordination compounds, the solid phase of which is able to switch to the ferro- or ferrimagnetic or weakly ferromagnetic state below the critical temperature. 4 – 12
Another group of magnetically active materials is comprised of paramagnetic compounds containing isolated exchange-coupled two- (and more) spin systems. Under the action of external factors, these systems can change their state, which results in either drift or abrupt change of physical characteristics at the macroscopic level. 13 These materials can actually transcode an external signal or an external stimulus at the molecular level into an easily detectable macroparameter; therefore, they are considered as working elements of ultrasensitive sensors, non-volatile magnetoresistive random access memory cells and quantum computer components.
There also exists a special group of single-spin systems with a specific quantum nature causing them to behave as permanent magnets even at the level of a single molecule or atom (single-molecule and single-atom magnets, respectively). 12 – 16 The magnetic bistability of these materials makes them applicable in spintronics, first of all, for the design of ultradense data recording and storage devices and ultrasmall spin diodes and transistors.
For many years, compounds with d- and f-element atoms as spin carriers predominated in magnetically active systems and materials. The continuous search for new magnets carried out in the last decade resulted in the discovery of ferromagnetic materials containing only p- and s-elements. 17 These compounds may possess a number of attractive properties such as low density, biocompatibility, plasticity, etc., which stimulates the search for magnetism in systems based on light elements. 18 One more encouraging fact is that Curie points of up to 65 K (under pressure) have been attained among ferromagnets of this type, which is currently the record high value. 19,20
While speaking of record high values of various sorts, it should be noted (without citing the relevant publications) that syntheses of organic compounds exhibiting magnetic hysteresis that is retained up to hundreds of Kelvin degrees have been regularly reported in the literature. However, subsequently the reported results could not be reproduced, which initially raised skepticism and distrust in the possibility to prepare magnets based on s- and p-elements. The development of physicochemical methods for studying processes behind the functioning of magnetic materials crucially changed the situation. A turning point in the attitude to the magnetism of light elements, in particular the magnetism of carbon-based systems, was associated with the appearance of reproducible results obtained for polymerized fullerene 21,22 and proton-irradiated graphite. 23 It was realized that under extreme conditions (high temperature, high pressure, or exposure to radiation or ion bombardment), numerous defects of paramagnetic nature, the topological structure of which is favourable for ferrimagnetic ordering of π-delocalized spins, may arise in carbon or organic materials via stochastic structural rearrangements. The total number of spins in separate ferromagnetic clusters is relatively low; therefore, the full magnetization of these purely carbon structures is always low. 18 However, the most important issue is that, upon drastic impacts, organic compounds are converted to complex mixtures consisting of numerous and diverse molecular structures. The use of these mixtures as magnetic materials is problematic, because their composition and magnetic characteristics can vary over broad ranges depending, for example, on the amorphous structure and on the preparation and annealing conditions. This is why the integrated problems that come to the forefront include the atomically precise step-by-step assembly of graphene nanostructures with specified magnetic properties, development of systems for quality control and physical manipulation of separate graphene molecules and design of the interfaces between these molecules and the final consumer. 24,25
The present review considers the modern trends in the development of synthetic chemistry of graphene nanostructures. The methods for the preparation of magnetically active graphene materials are integrated, focusing on spin-labelled graphene structures. Such structures are required for the development of prospective technologies in the organic and graphene spintronics and magnetronics; therefore, a considerable increase in the number of relevant publications is to be expected in the near future. We believe that this goal would benefit from this review where we attempted to depict the way from spin-labelled fused polycyclic aromatics to magnetically active graphene nanostructures and to define the challenges faced by researchers in this field and possible solutions.
The following abbreviations and designations are used in the review:
| AGNR | — armchair edge graphene nanoribbon; |
| BTBN | — nitronyl nitroxide-substituted dibromotetrathiafulvalene; |
| DCM | — dichloromethane; |
| DEER | — double electron–electron resonance; |
| dppf | — 1,1'-bis(diphenylphosphino)ferrocene; |
| dppp | — 1,3-bis(diphenylphosphino)propane; |
| ESBN | — benzodiselenafulvalene derivative bearing nitronyl nitroxide; |
| FFT | — fast Fourier transform; |
| GNR | — graphene nanoribbon; |
| HBC | — hexabenzocoronene; |
| hfac | — hexafluoroacetylacetonate; |
| J | — exchange interaction parameter; |
| I | — intensity; |
| kB | — Boltzmann constant; |
| K region | — convex armchair edge in the graphene nomenclature; |
| NIT | — nitronyl nitroxide; |
| p-TSA | — p-toluenesulfonic acid; |
| t | — time; |
| TBAF | — tetrabutylammonium fluoride; |
| TFA | — trifluoroacetic acid; |
| TIPS | — triisopropylsilyloxy; |
| TSBN | — spin-labelled derivative of benzotetraselenafulvalene; |
| TTF | — tetrathiafulvalene; |
| V | — voltage; |
| ZGNR | — zigzag-edge graphene nanoribbon; |
| χ | — magnetic susceptibility. |
2. Nanoscale graphene structures
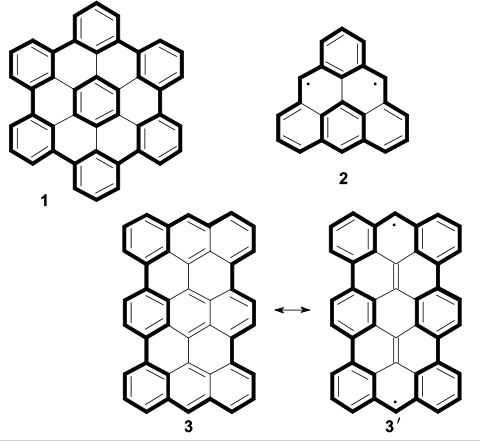
The design of magnetically active graphene nanostructures is a nascent area of research; therefore, it is reasonable to consider the trends in which (and at the interfaces between which) considerable advances were made. First of all, this refers to the studies of graphene, a two-dimensional carbon allotrope, which attracts attention of researchers and engineers. 26 The results of studying graphene gave rise to the general concept of the development of a broad class of sp2-materials, including polycyclic aromatic molecules, fullerenes, carbon nanotubes, graphene quantum dots, graphene nanoribbons and their derivatives obtained by structuring, chemical treatment, insertion of non-carbon atoms or defects. Perfect graphene is non-magnetic by itself, but its derivatives show various magnetic properties depending on the structure. 20 Particular interest is aroused by graphene structures with zigzag edges such as quantum dots and nanoribbons. 27 – 30 Below are given examples of such structures: hexabenzocoronene (HBC, 1) and fused polycyclic aromatic hydrocarbons with armchair and zigzag edges (2, 3). The edges and selected benzene rings are indicated in bold; for the Kekule polyaromatic hydrocarbon 3, one of the biradical resonance structures is shown (3').
Zigzag-edge nanoribbons are antiferromagnetic, their spin density is concentrated at the edges and their band gap is <0.1 eV. Despite the reasoned skepticism, 31 these nanostructures are considered as working devices for spintronics in which spin current rather than electrical current (as in electronics) serves as the information carrier. This opens up prospects for information processing and transfer at higher rates and with lower energy consumption.
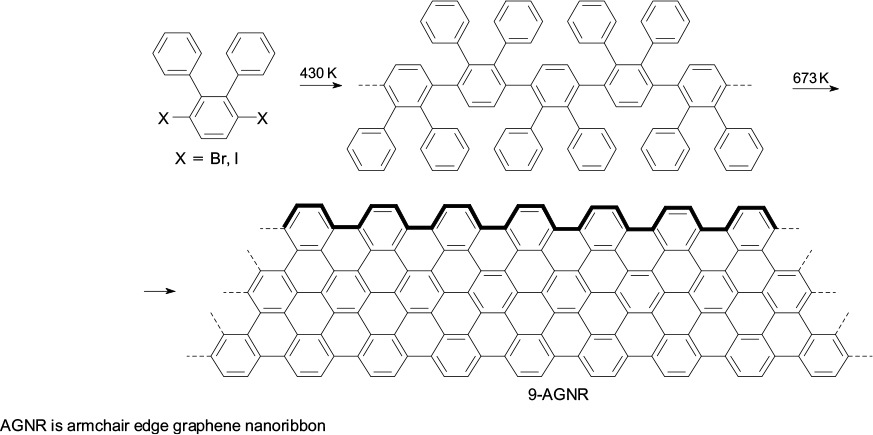
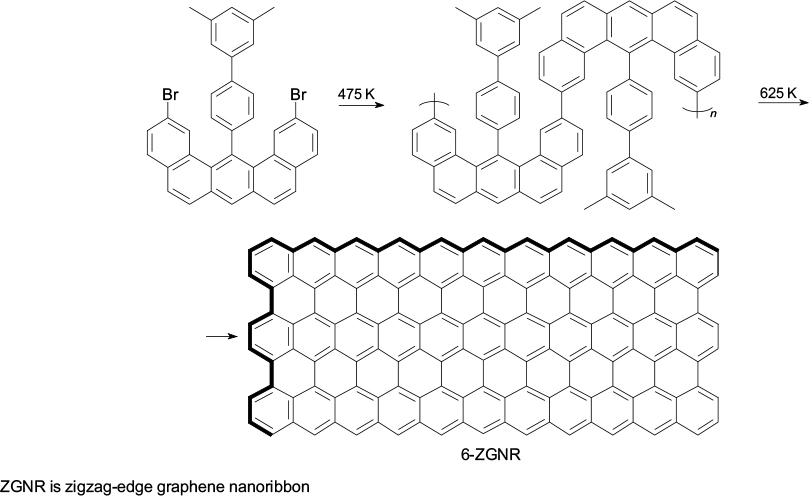
It is possible to distinguish two approaches for targeted synthesis of graphene nanostructures (graphene molecules, quantum dots and nanoribbons). According to one approach, molecules of organic monomers are supposed to be deposited on a hot metal surface, and nanoribbons with required geometry and edge shape are formed between them via carbon – carbon bond formation. In the first stage, monomers are sublimed on the metal surface (silver, gold), which is accompanied by removal of halogen substituents, while the subsequent heat treatment leads to diffusion of the generated radicals over the surface and their addition to one another to give a linear polymer chain (Scheme 1). The second stage is heat treatment at a higher temperature, which induces polymer cyclodehydrogenation and transformation into a narrow graphene ribbon with perfect armchair edge structure (9-AGNR). The illustrated bottom-up approach is widely used; depending on the type of the starting molecules, graphene nanoribbons with diverse structures and shapes were obtained. 32 – 49 Outstanding achievements are the fabrication of the 6-ZGNR graphene nanoribbon with zigzag edges on a gold(III) surface, its imaging by non-contact atomic force microscopy and experimental observation of a localized spin state at the edge of the graphene fragment (Scheme 2). 50 – 52
Structures 1–3
Download figure:
Scheme 1
Download figure:
Scheme 2
Download figure:
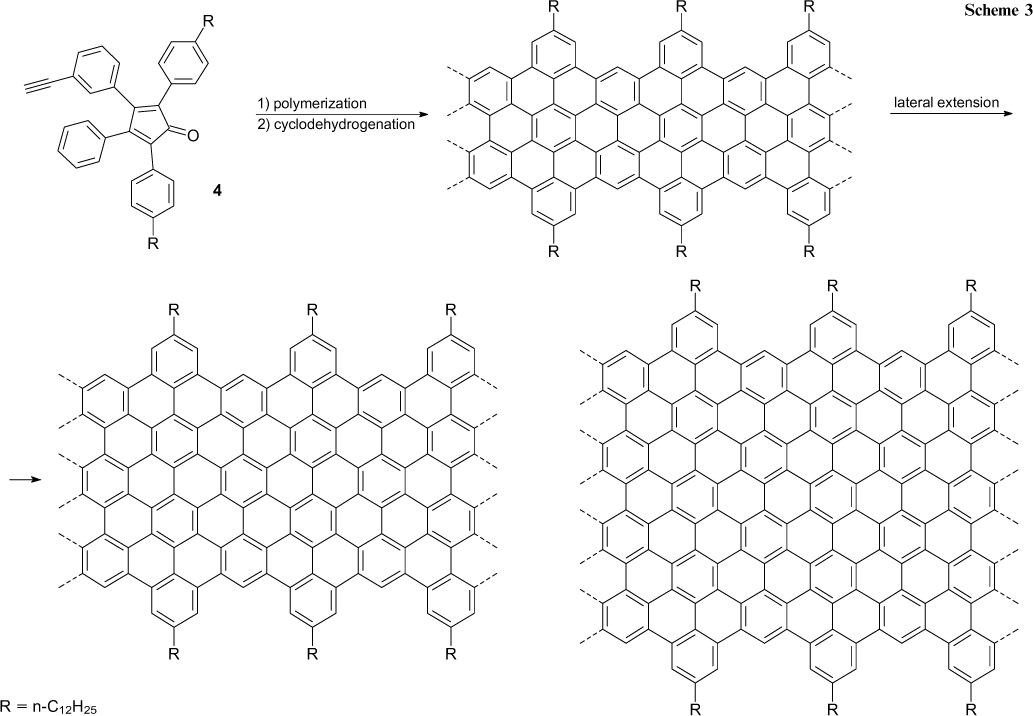
The second approach consists in the step-by-step assembly of graphene structures via reactions conducted in solution. 53 The most vivid example illustrating this approach is the sequence of chemical reactions the key step of which is the Diels – Alder polymerization of monomer 4, giving rise to substituted polyphenylenes with an average molecular mass of ⩾ 6 × 105 Da (Scheme 3). The oxidative cyclodehydrogenation of the polymer gives graphene nanoribbons with an average length exceeding 600 nm and the narrowest width corresponding to the size of the biphenyl group. It is also impressive that the rational design of the monomer structure in terms of the proposed concept resulted in the synthesis of several extended (> 100 nm) nanoribbons with ter- and tetraphenyl moieties in the narrowest part and nanoribbons bearing functional groups, apart from alkyl groups, at the edges (see Scheme 3). 54 – 58
Scheme 3
Download figure:
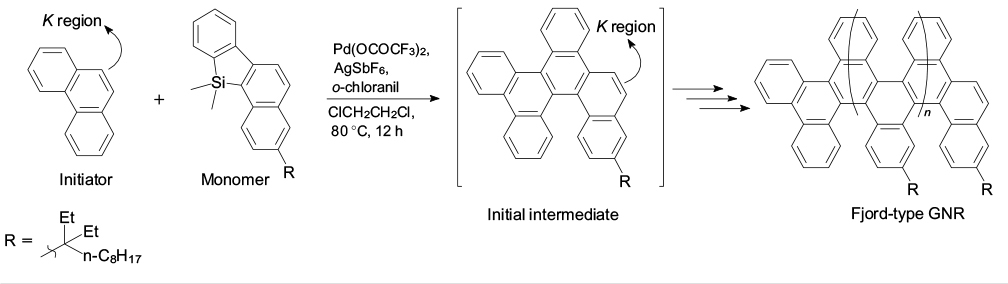
A milestone in the development of this concept is the appearance of the method for fabrication of graphene nanoribbons of not only specified width, but also specified length. The essence of the method is successive Pd-catalyzed annulation of the polyaromatic system under the action of benzonaphthosilole, which ensures chain growth (Scheme 4). The point is in the use, in the first step, of phenanthrene, in which the double bond reactivity (K region is the convex armchair edge in the graphene nomenclature) is much higher than in the annulation products due to steric factors. 59 Generally, this ensures a lower rate of chain growth compared with the initiation rate. With specially selected conditions, polymerization becomes 'living' and, as a consequence, it is possible to prepare narrow-dispersion polymers with substantially different number-average molecular masses, varying from 2.9 × 103 to 1.5 × 105 Da as the monomer to initiator ratio changes from 10 to 500, respectively. 60
Scheme 4
Download figure:
Thus, the structure-oriented synthesis of graphene nanoribbons, which seemed impossible only recently, became a reality. Obviously, this area will be actively developed, in particular, towards the synthesis of magnetically active graphene materials and then towards targeted design of spin devices and required interfaces based on them. Currently, it is necessary to specify the guidelines for further progress in this area considering the accumulated experience. The final goal is to develop methods for the atomically precise synthesis of graphene conductors possessing magnetic properties. It was shown above that under high vacuum, it is possible to prepare graphene nanoribbons wih zigzag edges, on which the spin density is relatively high. Rough quantum chemical calculations showed that the edge carbon atoms of these ribbons in the antiferromagnetic state should have the magnetic moment of ∼0.14 μB. 61 Hence, these atoms are highly reactive. 62 This highly complicates investigation and application of purely graphene magnets and interferes with their preparation by reactions in solutions.
These problems could be solved by using a stable graphene nanoribbon with semiconductor properties bearing stable spin carriers on the edges, which endow the nanomolecule with desired magnetic characteristics. 63,64 Thus, for attaining the above goal, it is necessary to solve a number of key problems, which include:
- —development of the synthesis of graphene nanoribbons suitable for subsequent functionalization with paramagnetic groups in solution;
- —systematic investigation of graphene nanostructures with various spin carriers;
- —implementation and detection of spin-dependent states in magnetically active graphenes.
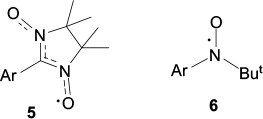
In the synthetic aspect, it appears rational to use stable paramagnetic groups as spin carriers; such groups have been thoroughly studied for the preparation of polyfunctional derivatives, they are convenient for grafting and can efficiently inject spin density into the graphene nanoribbon. 65 Currently, stable nitronyl nitroxides (structure 5) and tert-butyl nitroxides (structure 6) are without a rival in this respect.
Structures 5, 6
Download figure:
For this reason, for methodological assistance to the recently started investigations into spin-labelled graphene nanostructures, we deemed it reasonable, first, to consider the synthesis and properties of their immediate precursors — fused polycyclic aromatic hydrocarbons and heteroaromatic analogues bearing nitroxide groups — and then to formulate problems related to the design of magnetically active graphene materials.
3. Nitronyl nitroxide-substituted fused polyarenes and -hetarenes
Fused polycyclic aromatic hydrocarbons have always served as a testing area for sharpening the skills of targeted organic synthesis and for verification of the theory of structure and reactivity of organic compounds. 66 – 72 It is necessary to give proper respect to chemists whose efforts gave rise to elegant methods for construction of fused polyaromatic structures with different modes of coupling of aromatic rings and provided the synthesis of heteroatomic polyaromatic structures and diverse functional derivatives, which found use as dyes, organic semiconductors and components of optoelectronic devices. 73,74 The research in the field of fused polyarenes greatly benefited from the active use of scanning probe and electron microscopy, which allowed not only visualization of polycyclic aromatic compounds, 28,75 but also recording of the current – voltage characteristics for single molecules. 76 – 80 The results of these unique experiments demonstrated that fused aromatic molecules can act as single-molecule diodes and field effect transistors, i.e., they can provide the charge control of electrical conductivity.
Researchers specializing in organic electronics and in spintronics actively exchange ideas; this gave rise to publications aimed at the development of polyaromatic compounds with spin-dependent conductivity. In this area, two concepts have been proposed, crystal chemical and supramolecular ones. The former concept implies studying the effect of the magnetic field on the conductivity of crystals of spin-labelled polyarenes with a polymer-stacked packing. According to the latter concept, the studies are focused on the spin-dependent conductivity through a single paramagnetic molecule, which is obviously an exceptionally complicated task regarding the arrangement of the required interfaces. A Japanese research team 81,82 succeeded in finding an elegant solution, according to which two closely spaced gold electrodes are treated first with a solution of a paramagnetic organic conductor and then with a solution of gold nanoparticles. This treatment induces the spontaneous formation of a conducting bridge between electrodes, which represents, depending on the conditions, either a network of nanoparticles connected by organic conductors or a network composed of bound nanoparticles and granules (Fig. 1). It was shown that the electrical properties of composite conductors based on the pyrrole-thiophene oligomer, which incorporates paramagnetic compound 7 and that obtained using diamagnetic analogue 8, are considerably different. The former have a markedly lower conductivity at low temperature and a negative magnetoresistance, which is caused by interaction of the spin of electron that tunnels from one nanoparticle to another with the localized spin of the organic conductor.
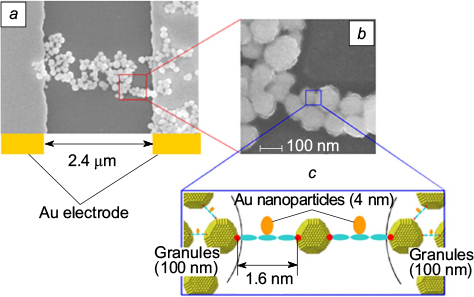
Figure 1. Composite conductor consisting of spin-labelled organic molecules 7 and gold nanoparticles. 81 (a) The chain connecting the electrodes consists of ∼100 nm granules; (b) granules consisting each of ∼5 800 gold nanoparticles connected by paramagnetic organic conductors 7; (c) structure of the contact between the granules ensuring the highest electrical resistance.
Download figure:
Standard imageStructures 7, 8
Download figure:

According to the crystal chemical concept (unlike the supramolecular one), the role of the conductor is played by the single crystal, the packing of which is formed by stacks of organic molecules, rather than by the heteroaromatic oligomer. The stacks can contain identical molecules or ions, alternating neutral and oxidized organic molecules of the same type (D⋯D+·⋯D⋯D+·), or neutral (D⋯A⋯D⋯A) and charged donor (D) and acceptor (A) molecules (D−⋯A+⋯D−⋯A+). The obligatory conditions for the design of magnetically active conductors are, first, high electrical conductivity of the stacks and, second, the presence of spin-labelled molecules in them, the structure of which is favourable for high-energy exchange interaction between the localized unpaired electrons of paramagnetic substituents and conduction electrons. Being guided by these considerations, Japanese researchers 83 focused attention on the development of structures using tetrathiafulvalene derivatives (TTF). The search for appropriate derivatives led them to the design of several paramagnets — ESBN (9, Scheme 5), TSBN (10) and BTBN (11), which are TTF nitronyl nitroxide derivatives.
Structures 10, 11
Download figure:
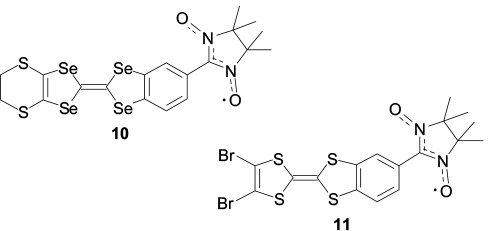
A specific feature of the obtained paramagnetic compounds is that the tetraselenafulvalene moiety possesses a lower single-electron oxidation potential compared to nitronyl nitroxide; therefore, for example, diselena derivative ESBN is converted to the radical ion salt (ESBN)2ClO4 during electro-crystallization. 83 The salt packing is formed by stacks composed of alternating paramagnetic molecules and their biradical cations, thus giving electrical conduction channels. The electron mobility in these channels depends, in a complicated manner, on the applied voltage (V ) and temperature. For example, at 2 K, the current – voltage dependence

typical of the salt (ESBN)2ClO4 sharply changes at V > 4.5 V, in particular, the exponent n increases to 12. This pronounced conductivity dependence is typical of conditions under which the current is determined by the spatial charge arising under the action of strong electric field. 84,85 Under these conditions, which promote the formation of holes, a considerable effect of the magnetic field on the conductivity of (ESBN)2ClO4 was detected. It was shown that the electrical resistance of (ESBN)2ClO4 decreases in magnetic fields (in a ±9 T field, the decrease can be as high as 70%). 83,86 The monotonic decrease in the conductivity with increasing temperature completely stops at ∼20 K, which actually determines the exchange interaction energy (characterized by the parameter J ) between the localized and conduction electrons. Pay attention to the fact that this value is an order of magnitude lower than the exchange interaction energy in a single radical cation ESBN+· (2 J ≈ 200 K), which is formally incorporated in (ESBN)2ClO4 (Scheme 5).
Scheme 5
Download figure:

In reality, according to magnetic measurements, the formula fragment of ESBN has one spin, in particular the spin of the nitroxide group, while the spins of the π-donor moieties are coupled in the conduction band and are perturbed upon application of strong electric fields to give holes, which are delocalized over several ESBN donor groups. Nevertheless, the Japanese researchers succeeded for the first time in designing a purely organic conducting and magnetically active system, for which they demonstrated the giant magnetoresistance effect. This effect is caused by the alignment of spins of the nitronyl nitroxide groups in a magnetic field, which suppresses scattering of conduction electrons generated by the applied electric potential on these spins, and thus reduces the resistance to current. The mechanism of influence of the localized unpaired electron spin on the conductivity of the salt (ESBN)2ClO4 is illustrated in Scheme 6 (the differently coloured arrows designate different orientations of the spins of unpaired electrons and electrons of the highest occupied molecular orbital).
Nearly the same magnitude of negative magnetoresistance effect (–76%) was detected in neutral crystals of the dibromo derivative BTBN at 2 K in a ±5 T magnetic field. 87 Apparently, the crucial role belongs to the packing of BTBN molecules giving conducting stacks, which are, in turn, connected by short intermolecular Br⋯O and Br⋯S contacts to give well-like tetramer structures. The major exchange channels in the solid phase of BTBN are contacts between the nitronyl nitroxide groups within the stacks, which form exchange-coupled chains with J = 6.5 K. In the author's opinion, 87 this fact, more precisely, the fact that the conduction electrons in BTBN interact with the whole cluster of ferromagnetically coupled spins is responsible for a considerably stronger magnetic field dependence of the BTBN conductivity compared with that of the salt (ESBN)2ClO4. These conclusions, however, should be treated with caution, because of scarcity of the data on the relationship between the structures of paramagnetic systems and the electrical conductivity response to the magnetic field. Meanwhile, this relationship can be quite substantial. Indeed, in the crystal of TSBN · THF, the molecules do not form stacks, but are connected into layers via van der Waals interactions between chalcogen atoms. The absence of stacks in the structure of TSBN · THF crucially changes the influence of the magnetic field on the electrical conductivity; at a temperature of 2 K, the resistance decreases by only 5% in a 5 T magnetic field, while at 4 K, it increases by 1% (Ref. 88).
Scheme 6
Download figure:

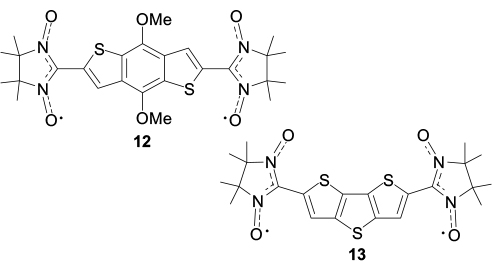
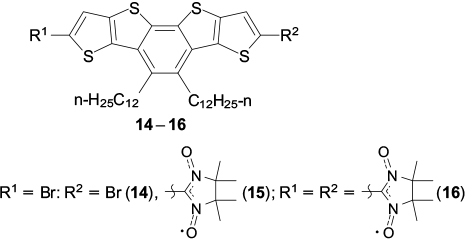
It can be assumed that a spin-dependent type of conduction can arise only if stacks containing spin-labelled polyaromatic molecules are formed in the crystal. In order to conceive how much spin-labelled polyarenes tend to form this type of packing, it appears reasonable to summarize the available structural data about nitronyl nitroxide-substituted poly- and hetarenes in which the aromatic rings are parallel to one another. The first example is biradical 12 with the benzo[1,2-b:4,5-b']dithiophenediyl bridge, the crystals of which contain dimers with a distance between the polyaromatic planes of 3.423 Å. 89 This distance can occur if the biradical molecules are slightly rotated relative to each other, which reduces the electronic and steric repulsion of four nitronyl nitroxide groups (Fig. 2). In the solid phase of biradical 13 with the dithieno[3,2-b:2',3'-d]thiophene bridge, the molecules form stacks with a distance of 3.303 Å between the planes of the aromatic rings. The biradicals are arranged at an angle of 90° relative to one another, and the sulfur atoms of the central thiophene ring are located virtually on one straight line, parallel to the [001] crystallographic axis (Fig. 3). 90 It is also worth noting how the successive replacement of bromine atoms in di(5-bromothieno)[2,3-d;2',3'-d']-4,5-didodecylbenzo[2,1-b:3,4-b' ]dithiophene 14 affects the crystal structures of compounds 15 and 16. Whereas dibromide 14 tends to be packed in pairs with a 3.42 Å distance between the ring planes and a slight (like in structure 12) rotation of molecules relative to each other (Fig. 4 a), 91 monoradicals 15 form centrosymmetric dimers in the solid phase, which are arranged in stacks with a distance of 3.58 Å between the sulfur atoms (Fig. 4 b). 92 In the crystals of biradical 16, two independent molecules form pairs of centrosymmetric dimers A⋯A and B⋯B, respectively, which alternate in inclined stacks with the shortest distance between the sulfur atoms of the biradicals of neighbouring dimers being 3.56 Å (Fig. 4 c).
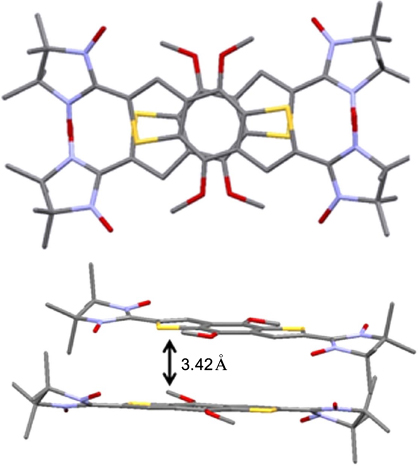
Figure 2. Structure of the dimer in the crystals of nitronyl nitroxide biradical 12. 89
Download figure:
Standard image
Figure 3. Packing of molecules of biradical 13 in stacks. 90
Download figure:
Standard image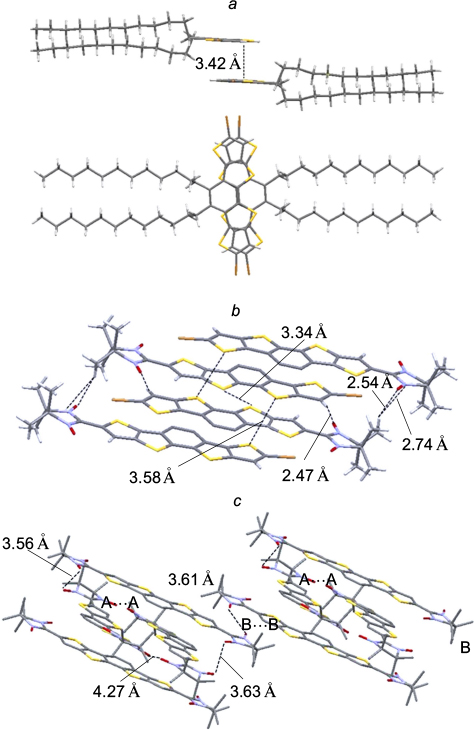
Figure 4. Fragments of the packing of dibromide 14 (a), 91 monoradical 15 (b) 92 and biradical 16 (c). 92
Download figure:
Standard imageStructures 12, 13
Download figure:
Structures 14–16
Download figure:
The above examples demonstrate that the introduction of the nitronyl nitroxide group into a polyaromatic structure causes a considerable perturbation to the packing of the initial diamagnetic molecules; interactions involving the paramagnetic group (exchange interactions, C–H⋯O no hydrogen bonds) may come to the forefront, while stacking interactions become less significant. A typical example is pyrene and some of its functional derivatives, which form diverse supramolecular structures via π–π stacking. 73,93 However if a nitronyl nitroxide group is introduced into the pyrene molecule, 94 the proneness of the molecules to form stacks is surpassed by stronger interactions. There is only one example pointing, to some extent, to the opposite, namely, the nitronyl nitroxide biradical 17 with a tetramethoxypyrene bridge. In the crystals of this compound, the molecules are arranged in layers in the herringbone pattern in such a way that the distances between the pyrene planes of the biradicals located in neighbouring layers are 3.730 Å (Fig. 5). Curiously, the replacement of one nitronyl nitroxide group by an imino nitroxide group (biradical 18) does not affect the general packing motif, but induces a considerable increase in the interplanar distances (up to 4.258 Å). 95
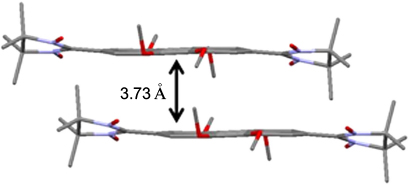
Figure 5. Dimeric moiety in structure 17. 95
Download figure:
Standard imageStructures 17, 18
Download figure:
As a whole, it can be stated that nitronyl nitroxides with fused polyaromatic substituents do not tend to form uniform stacks in the crystals. This is due to relatively low energy of stacking interactions between neutral aromatic rings, which is lower than the energy of van der Waals interactions involving the nitronyl nitroxide group. The stacking interactions can be enhanced by using charge transfer complexes, radical ion salts or cation – anion systems in which functionally substituted nitronyl nitroxides serve as active components. For example, crystallization of pyren-1-yl-substituted nitronyl nitroxide 19 from perfluorobenzene or co-crystallization of compound 19 with perfluoronaphthalene gives solid phases composed of packed triads 19 · Cn Fm · 19 (Fig. 6). 94,96 In triads 19 · C10F8 · 19 and 19 · C6F6 · 19, the angles between the planes of parallel pyrene rings and perfluoroarene are 2.58 and 7.15°, while the distances between the centroids of the pyrene moieties and perfluoroarenes are 3.407 and 3.524 Å, respectively. These triads are packed into stacks with relatively short distances between the pyrene planes (3.328 and 3.381 Å). Thus, the use of perfluoroarenes enforces the stacking interactions between pyren-1-yl-substituted nitronyl nitroxides, and this can be employed for the design of electrically conducting crystals of spin-labelled fused benzoid structures. In addition, it is of interest to prepare adducts of graphene-like quantum dots or nanoribbons with perfluoroaromatic compounds. The formation of these adducts may induce disaggregation of graphene nanostructures, increase their solubility and provide control over the electronic and magnetic properties. The development of this method requires separate comprehensive investigations.

Figure 6. Structures of triads in the co-crystals of pyren-1-yl-substituted nitronyl nitroxide 19 with perfluorobenzene (a) and perfluoronaphthalene (b). 94,96
Download figure:
Standard imageStructure 19
Download figure:
4. tert-Butyl nitroxide-substituted fused polycyclic arenes
In the nitronyl nitroxides considered in the previous Section, the spin density on the carbon atom of the paramagnetic ONCNO moiety is negative and, according to polarized neutron diffraction data, it is 2 – 3 times lower than the spin density on the nitrogen and oxygen atoms. 97,98 Furthermore, the nitronyl nitroxide group is often rotated through a considerable angle relative to the plane of the polyaromatic substituent. All this results in a low proportion of unpaired electron density in polyarene, which has an adverse effect on the performance of spintronic devices with working elements based on nitronyl nitroxide radicals. For this reason, in the design of magnetically active organic conductors and magnets, it is better to use kinetically stable radical groups (for example, tert-butyl nitroxide) able to efficiently inject spin density into the fused aromatic system. 99 The active use of the tert-butyl nitroxide group allowed researchers of the outstanding Hiizu Iwamura's school to design a number of high-spin organic compounds with high energy of exchange interactions between the paramagnetic centres (J/kB > 50 K) and to produce magnets based on manganese complexes with tert-butyl aryl nitroxides, with magnetic ordering temperatures reaching 46 K. 100,101
In recent years, research in the chemistry of tert-butyl aryl nitroxides has been focused on the development of dynamic spin systems containing a paramagnetic group (or several such groups) and a fused polyaromatic moiety. 102 For example, a family of spin-labelled naphthalene-1,8:4,5-bis(dicarboximide) and perylene-3,4:9,10-bis(dicarboximide) derivatives (20 – 23), parent compounds for a group of photo- and thermally stable dyes with strong visible absorption, were prepared and found wide use as photoactive materials in electronics. 103 These compounds containing a tert-butyl nitroxide group were used to detect photoexcited spin states, the initial polarization and evolution of which are determined by the structure, more precisely, by the π-topology of the group † binding the spin systems. 104,105 In view of the high proneness of naphthalene-1,8:4,5-bis(dicarboximide) (20) and perylene-3,4:9,10-bis(dicarboximide) derivatives (21 – 23) to self-assembly of organized structures, the results indicate the possibility of formation of supramolecular photoinduced polarized spin ensembles, in which coherent transfer of spin information takes place during relaxation.
Considerable interest is also attracted by the properties of spin-labelled pyrene compounds. Targeted synthesis of tert-butyl nitroxide mono- (24, 25) and biradicals (26 – 29) with regularly varying structure was implemented. A tetramethoxy-substituted pyrene serves as the parent structure. 106 – 108 According to X-ray diffraction data, spin-labelled pyrenes 24, 25 and 28 form dimers and stacks with a pronounced shift of aromatic systems relative to one another in such a way that the methoxy groups of neighbouring molecules are actually located above and below each aromatic system (Fig. 7). Quantum chemical model calculations [UBLYP/6-31g(d)] indicate that the singlet – triplet splitting energies (ΔEst = 2 J/kB) for isomeric biradicals 26 – 28 differ by an order of magnitude, being –216, –243 and –2010 K, respectively, with their absolute values being markedly higher than the absolute value of this energy for biradical 29, which amounts to –60 K.
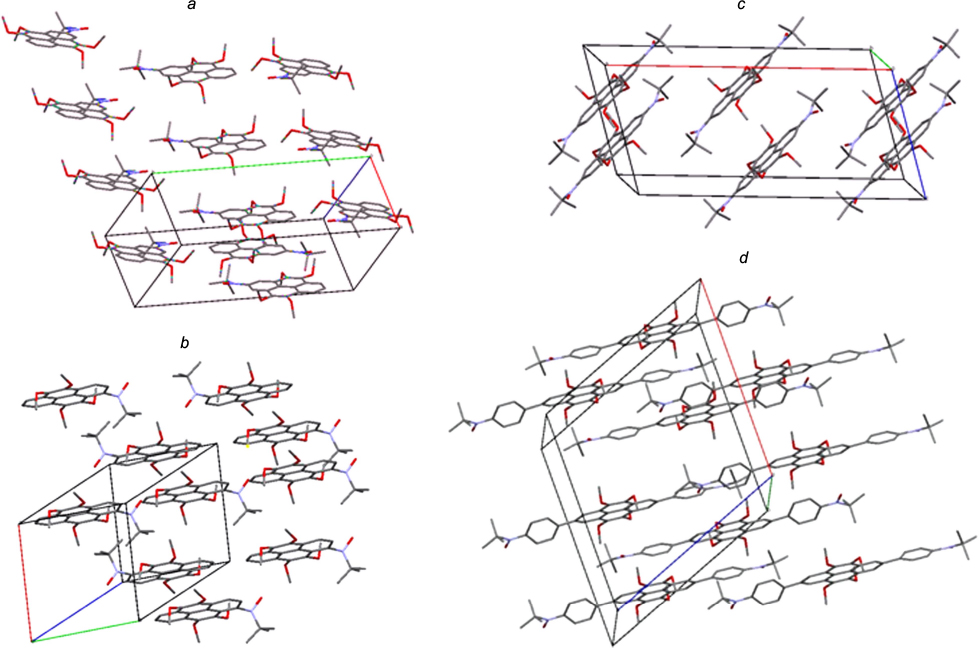
Figure 7. Fragments of the crystal packing of spin-labelled pyrenes 24 (a), 25 (b), 28 (c) and 29 (d ). 106 – 108
Download figure:
Standard imageStructures 20–29
Download figure:
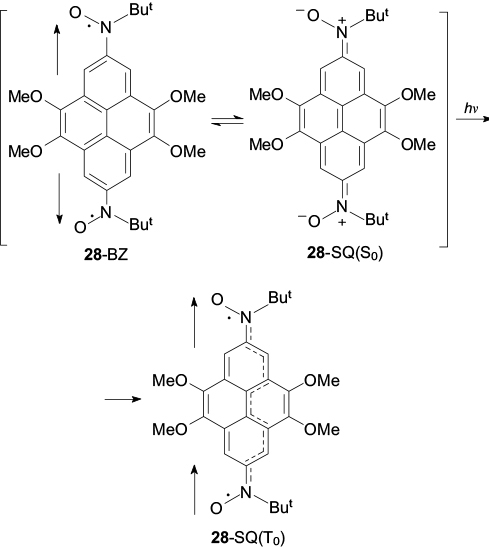
The presence of strong antiferromagnetic exchange interaction in biradical 28 was confirmed by experiments using temperature-dependent EPR spectroscopy. It was shown that raising the temperature of a single-crystalline sample from 273 to 353 K results in a considerable growth of the EPR signal. Theoretical analysis of the obtained dependence in terms of the Bleaney – Bowers equation 109 resulted in the singlet – triplet splitting energy equal to 1185 K. Actually, this means that under ambient conditions, the major contribution to the structure of compound 28 is made by the quinoid resonance structure 28-SQ(S0), located markedly below triplet biradical 28-SQ(T0) in the energy diagram [Scheme 7; the vertical arrows show the triplet ground state 28-SQ(T0) and the singlet state 28-BZ]. As a consequence, the N–O groups in molecule 28 (see Fig. 7 c), unlike those in radicals 24, 25 and 29 (see Figs 6 a,b,d ) lie virtually in the plane of the neighbouring aromatic ring. It is of interest that, according to UV spectroscopy, compound 28-SQ occurs in solution in an equilibrium with benzoid structure 28-BZ, the conversion to which proceeds apparently via triplet state 28-SQ(T0). 106
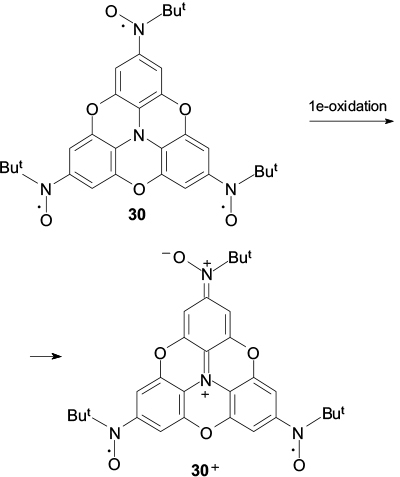
It was noted above (see Section 2) that the targeted formation of stacks for nitronyl nitroxide radicals should reasonably be performed using charged spin-labelled components such as biradical 30+ (Scheme 8) with a low-lying triplet state (J/kB > 103 K). The paramagnetic cation 30+ was obtained by single-electron oxidation of trioxytriphenylamine substituted with three tert-butyl nitroxide groups and isolated as stable salt 30+SbCl6 − (Ref. 110). According to X-ray diffraction analysis, the core of cation 30+ was planar; therefore, this triplet biradical can be used for the formation of conducting magnetically active stacks held together by Coulomb forces.
Scheme 7
Download figure:
Scheme 8
Download figure:
Thus, a skilled approach to the design of high-spin systems can furnish neutral and charged biradicals with a planar core and a triplet or singlet ground state, which is specified by the π-topology of the group linking the spin systems. This experience can be applied to design spin-labelled graphene materials.
5. Magnetically active graphene nanostructures
The magnetism of carbon structures is quite a broad notion covering the main carbon allotropes: diamond, graphite, nanographite, nanotubes, fullerenes, graphene and their reduced and oxidized forms. The structures of these allotropes can be either perfect or defective, they can contain heteroatoms and various (including magnetic) impurities and form hybrid structures. The latter can represent, for example, complexes of carbon materials with molecules or ions serving as paramagnetic centres or exchange-coupled clusters of these centres.
The present review does not consider the major part of magnetic carbon structures to which the notion molecular magnetism is, in principle, inapplicable, because their magnetism is random and the structural unit responsible for the magnetism has not been defined. The attention is concentrated on the advances in the targeted design of carbon materials containing paramagnetic centres, which is done using three approaches
- —preparation of various host – guest complexes in which a carbon nanostructure (fullerene, nanotube, graphene) serves as the host, while a carrier of one or several paramagnetic centres is the guest; despite the fact that the magnetic properties of such complexes are borne by the paramagnetic host molecule, its quantum characteristics and spin system dynamics substantially depend on the structure and state of the nanocarbon environment (Fig. 8); 111
- —targeted synthesis of carbon nanostructures with zigzag edges implemented by deposition of specially designed organic molecules on a hot surface; zigzag-edge ribbons are expected to have antiferromagnetic properties and a band gap of <0.1 eV, which monotonically decreases with increasing width of the ribbon; 112 these magnets possess high reactivity; therefore, they can be handled only in high vacuum; 50,52,113,114
- —synthesis of spin-labelled kinetically stable graphene nanostructures incorporating a chemically inert graphene nanostructure and a stable carrier of an unpaired electron, for example, a nitronyl nitroxide or tert-butyl nitroxide group; comprehensive investigation of magnetic structural correlations inherent in these systems would make it possible to evaluate their applicability to spintronics.
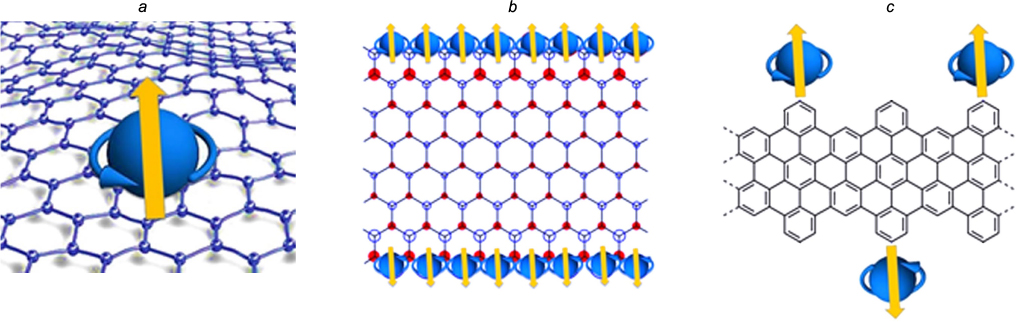
Figure 8. Approaches to the molecular design of magnetically active carbon nanostructures.(a) Graphene – paramagnetic guest complexes; (b) paramagnetic carbon nanostructures; (c) spin-labelled carbon nanomolecules.
Download figure:
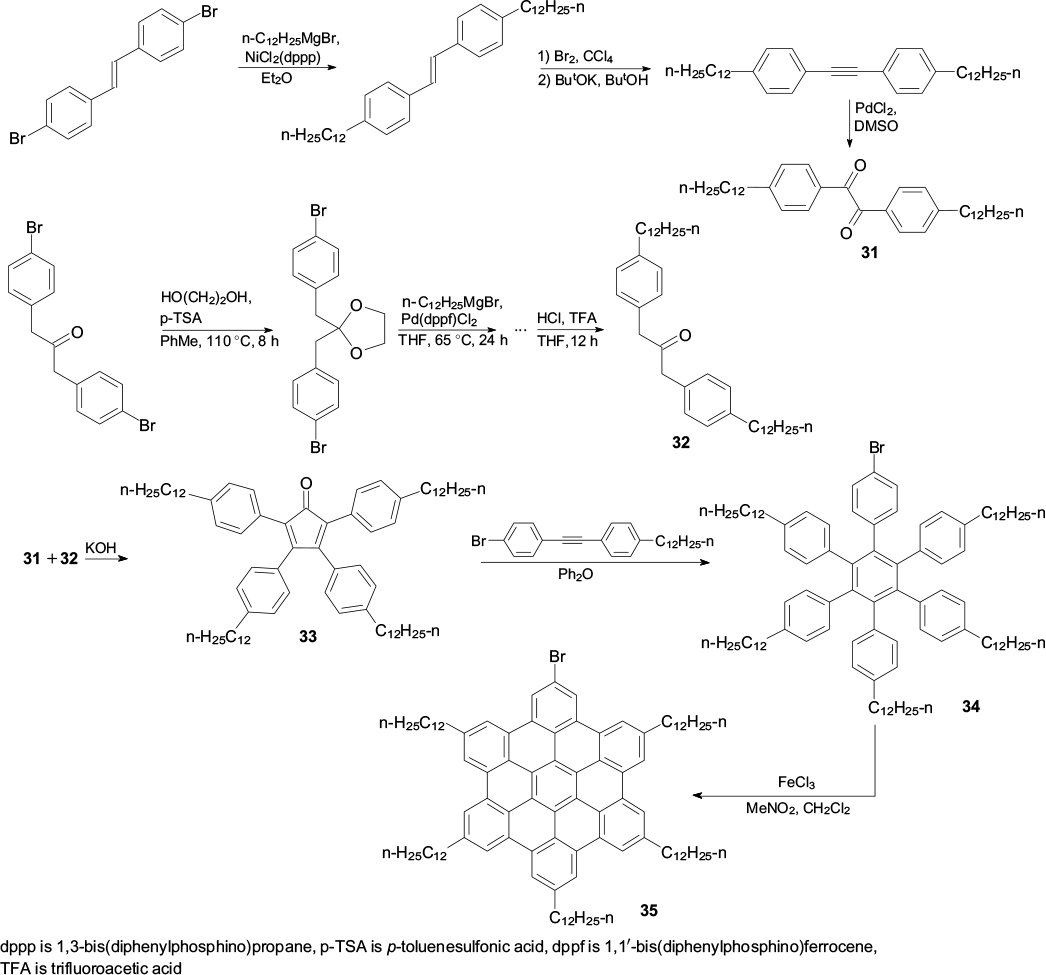
Standard imageA road towards spin-labelled graphene materials was paved by the previously mentioned pioneering works, which developed approaches to step-by-step assembly of graphene nanostructures from simple molecular precursors. Using these approaches, it was possible to obtain molecules comprising 48, 60 and even 96 carbon atoms in the fused aromatic part. 54,60,69,115 Especially significant in this respect is a series of works, which resulted in a universal method for the construction of halogen-substituted graphene quantum dots and nanoribbons, because the presence of a halogen (in particular bromine) atom enables functionalization of giant molecules. The method is based on the synthesis of two key compounds: dodecyl-substituted α-diketone (31) and 1,3-diphenylacetone (32), 63,116 which are condensed with each other to give tetraphenyl-substituted cyclopentadienone 33. The Diels – Alder reaction of compound 33 with diphenylacetylene in refluxing diphenyl ether affords hexaphenylbenzene 34. Graphitization of this product is induced by FeCl3 in nitromethane; the yield of the resulting hexabenzocoronene 35 is up to 90% (Scheme 9). 44 – 48
Compound 35 obtained in this way was subjected to cross-coupling with boronic acid bearing a protected tert-butyl(hydroxy)amino group to give compound 36, the subsequent hydrolysis and oxidation of which furnished the first conjugated spin-labelled derivative 37 (Scheme 10). 117 Using differential scanning calorimetry and wide-angle X-ray scattering, it was shown that nitroxide 37 transforms from the hexagonal crystalline to columnar liquid-crystalline phase at a temperature of >368 K. The reverse transformation takes place on cooling in the range of 320 – 310 K and leads to enhancement of magnetic interactions in the resulting crystalline phase within the stacks (Fig. 9), which is manifested, in particular, as a half-field signal in the EPR spectra at 140 K.
Scheme 9
Download figure:
Scheme 10
Download figure:
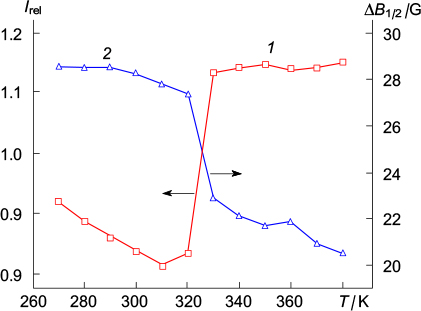
Figure 9. Variation of the relative signal intensity (1) and EPR line width (2) on cooling from 380 to 270 K. 117 ΔB1/2 is the full width at half maximum.
Download figure:
Standard imageFor a skilled organic chemist, it is sufficient to have a look on Scheme 9 to assess its synthetic potential. It is evident that by varying substituents in the substrates subjected to the Diels – Alder reaction, it is possible to prepare differently substituted hexaphenylbenzenes and their derivatives (see structures 34 and 35, respectively). Despite the drastic conditions of the Diels – Alder reaction, this scheme proved to be applicable for the preparation of a number of bromo- and dibromo-substituted polyalkylated HBC analogues. It was shown that they possess sufficient solubility and can participate in many reactions giving carbon – carbon or carbon – heteroatom bonds, which made it possible to synthesize a broad range of functionalized HBC derivatives. 118 – 125 This means that the preparation of a highly interesting family of magnetically active systems with a hexabenzocoronene core and similar graphene nanostructures can be expected in the near future. There are two probable routes of further development in the field of spin-labelled hexabenzocoronenes and their analogues:
- —the first route is specified by the above-considered compound 37 in which the paramagnetic substituent is conjugated with the polyaromatic core and injects spin density into it;
- —the second route is associated with yet the only HBC 38 bearing the nitronyl nitroxide group at the end of a long chain. 126
The reaction of Co(hfac)2 (hfac is hexafluoroacetylacetonate) with HBC derivative 38 followed by product precipitation affords the heterospin complex [Co(hfac)2 · 38]n (Fig. 10, method 1). Its magnetic properties crucially differ from those of the complex [Co(hfac)2]m . {38}n , which was prepared by the reaction of Co(hfac)2 with nanotubular {38}n precipitated in advance (Fig. 10, method 2). On cooling of the complex [Co(hfac)2 · 38]n down to a temperature of >10 K, the product of magnetic susceptibility and temperature (χ T ), inherent in this compound, sharply increases, whereas for analogue [Co(hfac)2]m · {38}n , this value continues to monotonically decrease (Fig. 11). The observed difference between the magnetic properties of the complexes [Co(hfac)2 · 38]n and [Co(hfac)2]m · {38}n is probably attributable to different structures of exchange clusters they contain. When Co(hfac)2 reacts with compound 38, polymer chains –(Co–38)n – are initially formed via binding of nitronyl nitroxide groups (ONCNO) to cobalt ions. Subsequently, during precipitation of [Co(hfac)2 · 38]n and formation of the nanotubular structure, the polymer chains –(Co–ONCNO)n – are laid down on the surface of nanotubes in a somewhat ordered fashion, 126 which induces weak magnetic ordering effects (see Fig. 10). Meanwhile, during the reaction of Co(hfac)2 with polymer {38}n precipitated in advance, cobalt ions randomly bind paramagnetic groups of molecules 38 packed in nanotubes; the polymer chains –(Co–ONCNO)n are formed in an irregular manner, if at all; therefore, the cooperative magnetic effects do not appear. The magnetic structure correlations revealed in the studies of complex formation of Co(hfac)2 with compound 38 are unique, and they should be taken into account in the design of heterospin systems based on complexes of paramagnetic metal ions with spin-labelled graphene nanostructures.
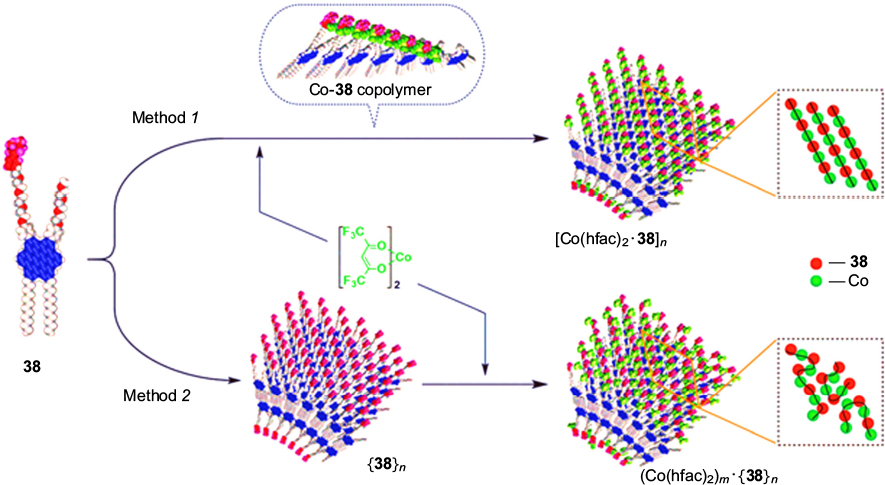
Figure 10. Illustration of formation of different heterospin structures upon isolation of the complex [Co(hfac)2 · 38]n into the solid phase (method 1) and upon the reaction of Co(hfac)2 with the polymer {38}n precipitated in advance (method 2 ). 126
Download figure:
Standard image
Figure 11. Plots χ T – T for [Co(hfac)2 · 38]n (a) and (Co(hfac)2)m · {38}n (b). 126
Download figure:
Standard imageStructure 38
Download figure:

It is noteworthy that, being guided by the synthetic paradigm of the general approach to the preparation of nanographene structures (see Schemes 9, 10), one can prepare not only substituted HBC, but also graphene nanoribbons, and, what is especially valuable from the synthetic standpoint, unique polybrominated nanoribbon derivatives. The synthetic route to bromine-substituted nanoribbons includes the production of monoalkylated diketone 39, bearing a protected acetylenic group, production of 1,3-diphenylacetone 40 and the subsequent Knoevenagel condensation of these ketones to give functionalized cyclopentadienone 41. The deprotection of the acetylenic moiety of compound 41 yields desired monomer 42 (Scheme 11). 63,116
Cyclopentadienone 42 polymerizes according to the Diels – Alder reaction on heating in Ph2O to give polyphenylene polymer 43 with the average molecular mass of ∼1.2 × 105 g mol−1. The subsequent graphitization of this polymer on treatment with FeCl3 in nitromethane affords black-coloured poorly soluble compound 44 as a brominated graphene nanoribbon. The average length of the nanoribbon was estimated as ∼100 nm. The palladium-catalyzed cross-coupling of polymer 43 or graphene nanoribbon 44 with excess gold(I) complex 45 furnishes spin-labelled polymer 46 as a reference sample and nanoribbon 47 (Scheme 12). 63
The introduction of radical groups into the phenylene polymer and graphene nanoribbon was confirmed by thorough (in particular, quantitative) EPR experiments (see Fig. 12 a); the degrees of substitution of nitronyl nitroxide groups for bromine atoms in the spin-labelled samples 46 and 47 were found to be ∼0.8% and 1.3%, respectively. 127 Actually, the distribution of radical groups in these molecules is random, but not equiprobable, if for no other reason, because of the fact that more lightweight components of a mixture of polybrominated substrates 46 or 47 are more soluble and, hence, they are more prone to undergo the cross-coupling reaction with organogold derivative 45.
Scheme 11
Download figure:

Scheme 12
Download figure:
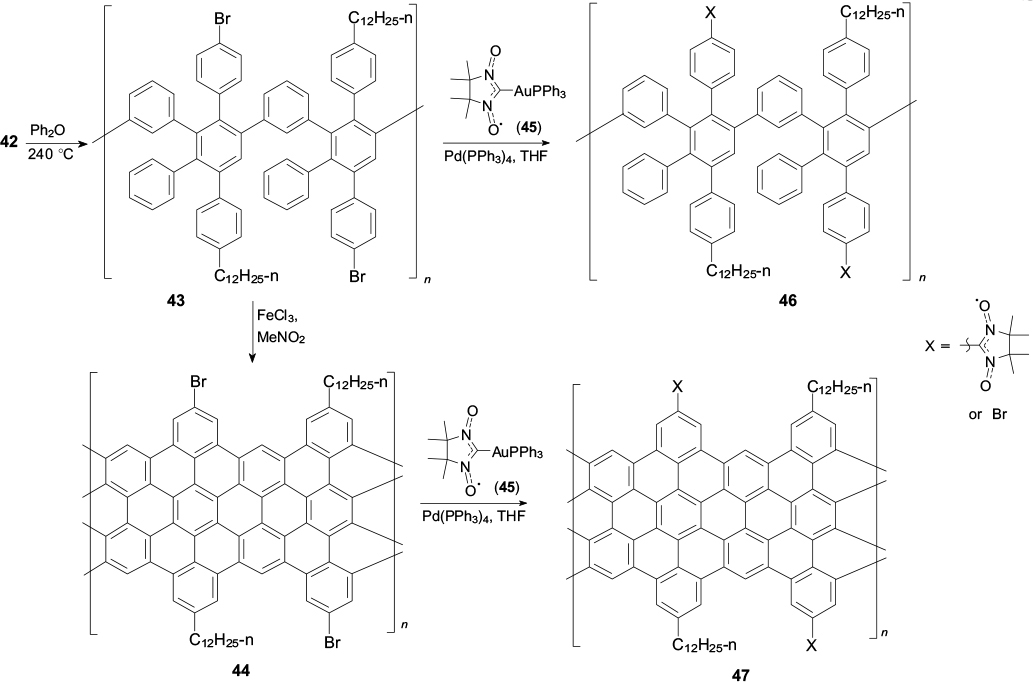

Figure 12. Multifrequency EPR spectra of polymer 46 (1 ) and nanoribbon 47 (2) and simulations (1', 2') plotted against the magnetic field from the edge state resonance (the DEER pump and probe windows are designated by β1 and β2, respectively) (a) and interaction pathways for radical spins with indicated exchange (J1 and J2, blue dashed lines) and dipolar (D1 and D2, orange lines) interactions (b). 63 In Fig. a, wavelength ranges are indicated on the right; NIT is the nitronyl nitroxide; ΔB is the change in the magnetic induction.
Download figure:
Standard imageAccording to EPR spectroscopy data, in nanoribbon 47, parameters of exchange interactions between the unpaired electrons of nitronyl nitroxide groups located on opposite sides (J1) or on the same side (J2) of the nanoribbon differ in the sign and amount to –25 ± 5 MHz (–8.3 × 10−4 cm−1) and 12 ± 3 MHz (4.0 × 10−4 cm−1), respectively (see Fig. 12 b). The signs of exchange parameters correspond to those found in theoretical studies, 128 but the absolute values are markedly lower than those found by quantum chemical calculations for model nitronyl nitroxide-substituted graphene nanoribbons. 129 Indeed, the calculated parameters J1 between the most proximate nitroxide groups are in the range from –0.4 to –0.2 cm−1, while the J2 values are close to 0.06 cm−1. The cause behind the difference of oders of magnitude between the experimental and calculated Jn values is the low content of nitronyl nitroxide groups in nanoribbon 47. Nevertheless, this content proved to be sufficient for conducting comparative EPR experiments, which revealed fundamental differences between the magnetic properties of spin-labelled nanoribbon 47 and polymer 46.
To begin with, the EPR spectra of nanoribbon 47 exhibit, apart from the resonance signal of the nitronyl nitroxide groups, an additional signal with g∥ = 2.0024(3) and g⊥ = 2.0041(2) associated with the edge π-electron state of the graphene nanostructure (see Fig. 12). 63,130 This indicates that the magnetic behaviour of compound 47 is unique, as it contains two unpaired electron ensembles associated with nitronyl nitroxide groups and graphene nanoribbon edges. For studying the transitions between the Zeeman levels of nanoribbon 47, virtually the whole range of available EPR spectroscopy techniques was used. 63 First of all, paramagnetic relaxation times were measured, in particular the spin – spin relaxation time T2, which defines the phase memory time of the system, and the spin – lattice relaxation time T1, which determines the return time of the excited spin to the ground state. According to the data of time-resolved EPR for paramagnetic graphene 47 at a near-room temperature, T1 is ∼10 s, which corresponds to theoretical predictions. 131 As temperature decreases, T1 increases and reaches 103 – 104 s at temperature <25 K, i.e., when the spin–phonon mechanism of spin relaxation becomes predominant.
Analysis of the results of experimental determination of the spin echo dephasing time (Tm) for compound 47 demonstrated that the major contribution to the loss of spin coherence is made by the spin – spin relaxation, which means that the times Tm and T2 are similar. The Tm values proved to be 0.5 and 1.1 s at 300 and 85 K, respectively, which is orders of magnitude higher than this value for previously described graphene-based spintronic devices. 132 However, the key distinctive feature of nanoribbon 47 is the above-noted presence of two paramagnetic subsystems — the proper nitroxide and the edge ones. The electron – electron interaction between these subsystems was studied using four-pulse double electron – electron resonance (DEER). In the experiment, system excitation and detection of the signal were performed at the resonant frequency of nitroxide radicals, and perturbation was at the edge state resonant frequency. The obtained DEER spectrum of nanoribbon 47 represents a superposition of fast and slow oscillations. The former are due to dipole – dipole interactions D1 and D2 (see Fig. 12 b), while the latter are attributable to the interaction of spins of the nitroxide group and spins of the edge states (Fig. 13). Fourier transformation of the DEER signal gives the electron – electron interaction energy of 1.5 MHz, which corresponds to the characteristic inversion time of spin subsystems (330 ns). This time is markedly shorter than Tm, which enables coherent inversion operations using graphene edge states and nitroxide spins.

Figure 13. Decay of the substrate-corrected DEER signals of polyphenylene 46 (1) and graphene nanoribbon 47 (2); low-frequency component of the DEER spectrum (3) (a) and fast Fourier transform (FFT) of the decay of the DEER signal for nanoribbon 47, demonstrating the interaction energy spectrum characteristic of two-spin operations (b). 63 In Fig. b, the black-coloured line indicates the contribution of edge states interacting with localized spins.
Download figure:
Standard imageCurrently, there are conditions for initiation of a new research area pertaining to molecular magnetism at the junction of the synthetic chemistry of stable radicals and studying of graphene carbon nanostructures, namely, synthesis and studies of stable spin-labelled graphene structures. Certainly, this area would be of interest for specialists and would become a part of the synthetic chemistry of graphene directed towards the manufacture of operating spintronic devices.
6. Conclusion
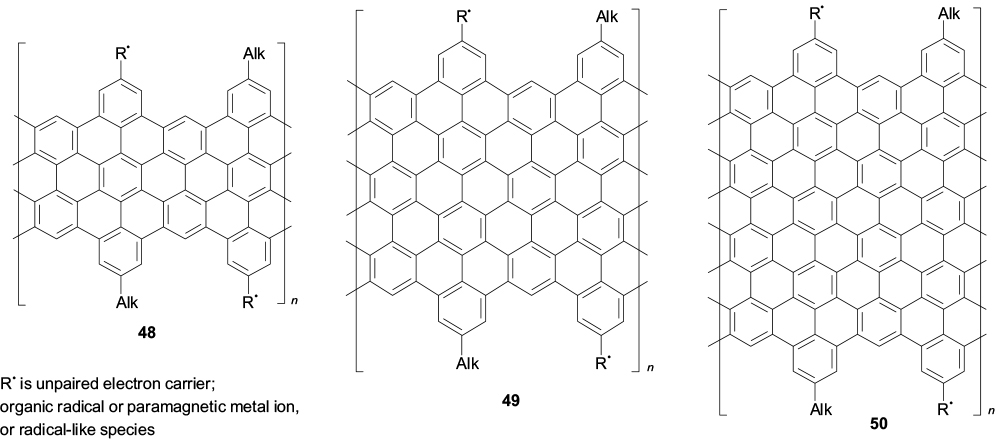
The review summarizes the data on the synthesis and study of spin-labelled fused polyaromatic compounds, including graphene nanostructures such as hexabenzocoronene and graphene nanoribbons. Advantages of the latter include stability and the conceptual possibility for them to be prepared with atomic precision, which determines the whole set of their physical properties. For efficient use of these advantages, in particular for approaching the design of valuable materials for molecular spintronics, it is necessary to define the immediate tasks and prospective challenges. First of all, it is impossible to do without elucidation of magnetic structure correlations of spin-labelled graphene nanostructures. For example, the synthesis of regularly spin-labelled graphene nanoribbons of different width is to be implemented and the effect of the structure of the aromatic core and the nature of paramagnetic substituents on the interaction of unpaired electrons is to be elucidated (Structures 48 – 50).
In addition, with the goal of manufacturing materials for spintronics, it would be highly valuable to synthesize graphene nanoribbons with various spin carriers, especially systems with a regular structure, in which the geometry of exchange channels is strictly specified (structure 51).
Structures 48–50
Download figure:
Structures 51
Download figure:
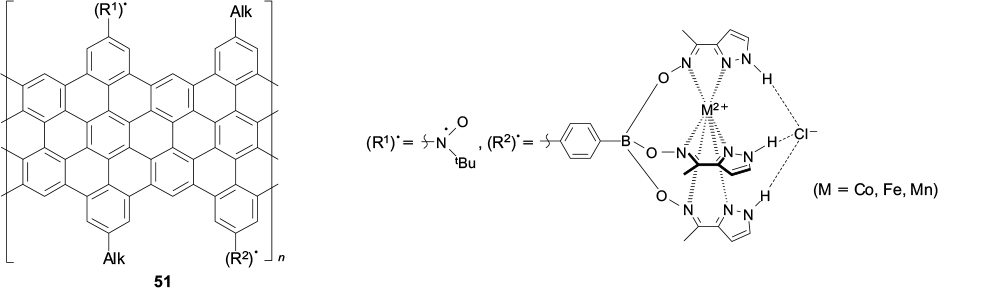
Spin carriers can be represented by stable organic radicals, chelated or clathrochelated paramagnetic metal ions, including those undergoing spin transitions, and various systems with a thermally accessible high-spin state. It is of particular interest to obtain graphene nanoribbons incorporating single-molecule magnets capable of magnetic bistability and, hence, acting as single qubits.
A special challenge is to characterize spin-labelled graphene nanoribbons. This can be done relying mainly on the advanced scanning electron and atomic force microscopy methods and, for elucidation of magnetic properties, resorting to micro-SQUID magnetometry; 133,134 this requires the development of methods for mechanical manipulation of single molecules. The latter is by the way necessary to design experiments on measuring the conductivity of single molecules of spin-labelled graphene nanostructures, in particular in magnetic fields. In this respect, a number of nanotechnological problems are to be solved, in particular creating contacts between gold electrodes and nanoribbons. One of the ideas how to do this is as follows. The –S–SC6H13-n groups should be introduced at the end faces of spin-labelled graphene nanoribbons; this would enable grafting of nanoribbons either directly to gold electrodes or to gold electrodes with the participation of gold nanoparticles (see Fig. 1). It can be expected that self-assembly of these conductors would be accompanied by selection of graphene nanoribbons in terms of length corresponding to the distance between the electrodes.
Considerable attention will be attracted by the preparation of charge transfer complexes, radical ion salts or cation–anion systems with spin-labelled hexabenzocoronenes (or their analogues) as active components. In particular, stacked structures, resulting from co-crystallization of spin-labelled hexabenzocoronenes with perfluoroaromatic (especially fused) systems, may prove to be promising. These structures are expected to show an interplay between the structure, conductivity, magnetic properties and responses to external stimuli.
Obviously, the design of spin-labelled graphene nanostructures will open up prospects for unique experiments, for example, related to detection of the spin state of paramagnetic substituents (including spin-polarized ones) by measuring the current of electrons; control of the spin currents by variation of electric fields; design of spin diodes, transistors and capacitors; transfer of quantum information by conduction electrons along the spin ensembles of paramagnetic substituents and edge states interacting with each other. The project of elaboration of magnetic and spin logic elements using spin-labelled graphene nanoribbons connected or superimposed onto each other seems quite practicable.
All these challenges in the field of chemistry and physics of spin-labelled graphene nanostructures are highly sophisticated, their list would be expanded, but their solution would create prerequisites for the onset of the graphene era in spintronics and magnetronics.
This review was written with the financial support of the Russian Science Foundation (Project No. 18-13-00173).
Figure 1 is courtesy of American Physical Society publishers; Figure 9 is courtesy of American Chemical Society publishers; Figures 10 and 11 are courtesy of the Royal Society of Chemistry; Figures 12 and 13 are courtesy of Macmillan Publishers Limited, part of Springer Nature.
Footnotes
- †
By π-topology of a group is meant the relation of this group to a definite canonical hydrocarbon biradical: trimethylenemethane, trimethylenepropane, m- or p-phenylene, etc.