


Abstract

Aspartoacylase is a key enzyme in the human central nervous system. Catalytic deficiency of aspartoacylase is associated with several neurodegenerative disorders, which accounts for the enhanced interest in description of the catalytic action and regulatory mechanisms of this enzyme. This review focuses on molecular modelling of the catalytic function of aspartoacylase and the ways of its allosteric regulation in terms of modern theoretical chemistry approaches. The conclusions based on experimental data on the structure and functions of aspartoacylase accumulated during the last 20 years and conclusions drawn from computer modelling results of the last 5 years are analyzed in detail.
The bibliography includes 109 references.
Export citation and abstract BibTeX RIS
| E.D.Kots. PhD in Physics and Mathematics, Researcher at the Division of Physical Chemistry of the Chemistry Department, MSU; Junior Researcher at the IBCP RAS. |
| Telephone: +8(495)939 - 4840, e-mail: kots.katya@gmail.com |
| M.G.Khrenova. Doctor of Physical and Mathematical Sciences, Leading Researcher at the Division of Physical Chemistry of the Chemistry Department, MSU and at the FRC 'Fundamentals of Biotechnology', RAS. |
| Telephone: +7(495)939 - 2035, e-mail: khrenova.maria@gmail.com |
| Current research interests of the authors: quantum chemistry, mathemat-ical methods in chemistry, biochemistry, enzymology. |
| A.V.Nemukhin. Doctor of Chemical Sciences, Professor, Head of the Laboratory of Chemical Cybernetics of the Division of Physical Chemistry of the Chemistry Department, MSU and the Laboratory of Computational Modelling of Biomolecular Systems and Nanomaterials, IBCP RAS. |
| Telephone: +7(495)939 – 1096, e-mail: anem@lcc.chem.msu.ru |
| Current research interests: quantum chemistry, mathematical methods in chemistry. |
| S.D.Varfolomeev. Corresponding Member of the RAS, Doctor of Che-mical Sciences, Professor, Head of the Division of Chemical Enzymology of the Chemistry Department, MSU; Scientific Adviser at the IBCP RAS. |
| Telephone: +7(499)137 - 6420, e-mail: sdvarf@sky.chph.ras.ru |
| Current research interests: biochemistry, enzymology. |
1. Introduction
Human brain is one of the most sophisticated and attractive investigation objects of modern science. Central nervous system enzymes catalyze brain energy supply, regulation of neurotransmitter synthesis and dissociation, synaptic transmission and many other significant processes.
One of the most mysterious brain systems is the system of N-acetylaspartic acid (NAA) metabolism. The NAA synthesizing and hydrolyzing enzymes function in such a way that the concentration of this metabolite can be as high as 5 mmol litre−1. High NAA levels are dictated by the relatively low activity of the hydrolyzing enzyme, N-acetyl-aspartate hydrolase (aspartoacylase).
It is evident that this enzyme largely determines the behaviour of the whole system of NAA metabolism. The interest in aspartoacylase functioning and activity regulation mechanisms is very high. Publications devoted to the structure, properties and polymorphs of aspartoacylase and its role in metabolic processes appear every year; however, no reviews on this subject have been published to date.
In recent years, a new tool appeared for studying protein molecules — molecular modelling that includes investigation of system properties from the standpoints of molecular mechanics, molecular dynamics and combined quantum mechanics/molecular mechanics approaches. The increase in the potential of this modelling technique is largely determined by the progress of computing technologies and development of supercomputing clusters. Detailed experimental investigations of the structures of biomacromolecules in combination with application of the fundamental laws of quantum and molecular mechanics lead to unique results including analysis of the kinetic behaviour of the systems.
This review presents integration and analysis of the achievements made by this approach as applied to human brain aspartoacylase.
The following abbreviations are used:
- C.N.— coordination number,
- CNS— central nervous system,
- CPA— carboxypeptidase A,
- EDTA— ethylenediaminetetraacetic acid,
- EP1P2— enzyme − reaction product (complex),
- ES— enzyme − substrate (complex),
- hAsp— human aspartoacylase,
- HREUS— Hamiltonian replica exchange umbrella sampling,
- MD— classical molecular dynamics,
- MSM— Markov state model,
- NAA— N-acetylaspartic acid and N-acetylaspartate,
- NAAG— N-acetylaspartyl-glutamic acid and N-acetylaspartyl-glutamate,
- NPA— N-phosphonomethyl-L-aspartic acid and N-phosphonomethyl-L-aspartate,
- OP— o-phenanthroline,
- PDB— Protein Data Bank,
- RMSD— root-mean-square deviation,
- SCC-DFTB— self-consistent charge density functional tight binding,
- SGD— succinylglutamate desuccinylase,
- tICA— time independent component analysis,
- QM/MM— quantum mechanics/molecular mechanics.
2. Structure of human aspartoacylase
Human aspartoacylase (hAsp) (EC 3.15.1.15, UniProt P45381) is a hydrolase like enzyme involved in the metabolism of N-acetyl-L-aspartic acid. This acid is synthesized in neuronal mitochondria from L-aspartate and acetyl-co-enzyme A. 1,2 Aspartoacylase catalyzes cleavage of the NAA peptide bond to give L-aspartate and acetate anions (Scheme 1).
N-Acetyl-L-aspartate is the second most abundant brain metabolite after glutamic acid; 3,4 it is present in all tissues of the central nervous system with the highest concentration occurring in grey matter. 5 During the postnatal development, NAA is found both in neurons and in oligodendrocytes, while in the middle age, it is localized in oligodendrocytes at concentrations of up to 10 – 14 mmol litre−1 (Ref. 6). The exact role of NAA in brain remains the subject of biological and medical research; 7,8 however, the key NAA metabolism pathways have been reported in the literature . For example, NAA is a precursor for the synthesis of the dipeptide N-acetylaspartyl-glutamate (NAAG), which participates in the neuromodulation of metabotropic and NMDA receptors (NMDA is N-methyl-D-aspartate), 9 regulates the intracellular pressure in neurons 10 and is involved in the energy generation from glutamate anions in neuronal mitochondria. 11 It was also found that NAA is a source of acetyl groups for the construction of myelin sheath in the brain; therefore, maintenance of the NAA level ensures proper development and functions of white matter. 12
Scheme 1
Download figure:

Owing to the characteristic 1H NMR chemical shift of the three equivalent protons of the acetate group, NAA is used in medical practice for non-invasive identification of functionally mature neurons. 6,13,14 However, the characteristic NAA peak can also contain minor contributions of other acetylated compounds occurring in neurons. Nevertheless, identification of abnormal total contents of NAA in particular brain areas is an important diagnostic tool. 15,16
For example, it was found that many neurodegenerative diseases, that is, multiple sclerosis, epilepsy and Alzheimer's disease, are associated with reduced NAA contents in white matter. 17 – 19 A mechanical brain damage (traumatic brain injury) causes a reversible decrease in the NAA concentration, 20,21 which is traditionally attributed to energy exchange disorder and to acute stress response of neurons. 22,23 Calculations of the correlation coefficients revealed a relationship between concentrations of NAA and other metabolites detected in the 1H NMR spectra, namely, choline and creatine. 20 These data suggest that activation of the energy-dependent choline and creatine synthesis affects the NAA concentration in the cerebral cortex.
Furthermore, an increased concentration of NAA is a consequence of lethal autosomal recessive leukodystrophy, the Canavan disease, characterized by hAsp dysfunction. 24 Mutation of the gene encoding hAsp gives rise to the enzyme with reduced catalytic activity. The incomplete NAA catabolism generates a deficiency of acetate ions and, as a consequence, leads to low myelinization of neurons and spongy degeneration of white matter. 25,26 The progression rate and degree of the disease may vary depending on the particular mutation; 27 however, all forms are characterized by a pronounced increase in NAA concentration. A 100-fold excess of NAA over the reference is a sufficient diagnostic criterion for the Canavan disease. 28
The Protein Data Bank (PDB) 29 contains eight crystal structures of the human aspartoacylase homodimer, four of which belong to the native form: three structures describe the geometry of apo-hAsp [PDB ID 2O53, 30 2I3C (Ref. 31) and 2Q51 (Ref. 32), an assembly of 16 models], while the fourth one belongs to hAsp with a tetrahedral intermediate analogue in the active site (PDB ID 2O4H). 30 However, the free enzyme structures do not formally correspond to the apo-form of hAsp, since their active sites contain phosphate anions, which occupy the substrate position in the zinc coordination sphere.
According to gel permeation chromatography and mass spectrometry data, the protein hAsp occurs in solution as a homodimer (36 kDa),
33,34
which has symmetry  in the crystalline state.
31
The hAsp interface between two subunits (ASPA and ASPB) has a solvent accessible surface area of 1200 Å2. The quaternary structure is maintained by 12 hydrogen bonds and two salt bridges located symmetrically relative to the cavity centre. The access to the transport channels of hAsp subunits occurs from the interface, which may generate additional steric hindrance to substrate binding.
in the crystalline state.
31
The hAsp interface between two subunits (ASPA and ASPB) has a solvent accessible surface area of 1200 Å2. The quaternary structure is maintained by 12 hydrogen bonds and two salt bridges located symmetrically relative to the cavity centre. The access to the transport channels of hAsp subunits occurs from the interface, which may generate additional steric hindrance to substrate binding.
2.1. Protein structure
Aspartoacylase belongs to the family of zinc-dependent carboxypeptidases. 33 Each polypeptide chain consists of two domains, in particular, the N-domain comprising amino acid residues 1 – 212 and the C-domain consisting of residues 213 – 313 (Fig. 1). The secondary structure of the hAsp N-domain belongs to the αβα-sandwich type (according to the CATH classification) 35 and corresponds to zinc-dependent hydrolases of the carboxypeptidase A (CPA) family. The non-covalent interactions generating the hAsp tertiary structure occur, most of all, between the same secondary structure elements as in carboxypeptidase A. 36

Figure 1. Two aspartoacylase subunits: ASPA and ASPB. 30 Positions of the active sites and of the C- and N-domains. †
Download figure:
Standard imageThe difference between the tertiary structures of hAsp and carboxypeptidase is that the latter is devoid of the C-domain: the carboxypeptidase active site is located on the protein surface; therefore, it becomes accessible to large peptide substrates. The C-domain of hAsp consisting of two β-sheets and non-structured regions sterically shields the access to the catalytic site. The C- and N-domains form deep and narrow transport channels towards the active site in each of the two subunits. The chain segments that form the gate to the transport channels in both ASPA and ASPB consist mainly of positively charged amino acid residues: Arg71, Lys228, Lys291 and Lys292, which may promote binding of negatively charged organic molecules.
The gate to the hAsp transport channel is formed by loops consisting of residues 62 – 74 on one side and residues 282 – 294 on the other side. In the crystal structures of apo-hAsp, the distance between the centres of mass of the backbones of these loops varies between 15.7 and 16.6 Å. Comparison of the positions of the amino acid residues that form the active site gate of the ASPA monomer suggests high mobility of this chain segment (Fig. 2). In the 16 models of the PDB ID 2Q51 structure, the mobilities of loops 62 – 74 and 282 – 294 are virtually identical for the two hAsp subunits (ASPA and ASPB, respectively). The most labile amino acid residues, Arg71 and Glu293, support both the open and closed conformations. The reduced electron density around the Arg71 side chain of one PDB ID 2O53 monomer is also indicative of a high degree of mobility of this apo-hAsp segment and of the possible orientation of this segment both inwards into the transport channel and outwards into the solution.

Figure 2. Positions of amino acids forming the gate to the hAsp transport channel in the PDB ID 2Q51 crystal structure.(1) ASPA, (2) ASPB. 30 – 32
Download figure:
Standard imageOn binding of the tetrahedral intermediate analogue, N-phosphonomethyl-L-aspartate (NPA), in the hAsp active site, the side chains of some residues of transport channel gates change their positions. The Arg71 carboxamide group shifts by 4 – 5 Å relative to the position in the apo-form and thus forms stable electrostatic contacts with one carboxyl group of the substrate and participates in the substrate binding in the active site. The aromatic group of Tyr164 shifts by 45° relative to the Cα–Cβ bond to form a hydrogen bond between the tyrosine hydroxy group and the NAA carboxyl oxygen atom.
The superposition of the crystal structures of hAsp 2I3C and 2O53 with the phosphate anion in the zinc coordination sphere demonstrated that the greatest contribution to the root-mean-square deviation (RMSD) between them is made by the 158 – 164 segment (RMSDmax = 7.1 Å). 30 The authors attributed this discrepancy to different concentrations of phosphate anions during crystallization. However, it should be noted that loop 158 – 164 is arranged above the gate to the hAsp catalytic site and, therefore, it can participate in the regulation of substrate binding. 31
2.2. Active site structure
The active site of aspartoacylase is located on the bottom of the narrow transport channel at a ∼12 Å distance from the enzyme surface and consists of three regions, differing in their function:
— Zn2+ coordination region: His21, Glu24 and His116;
— NAA binding region: Arg63, Asn70, Arg71, Tyr164, Arg168 and Tyr288;
— catalytic base Glu178 acting as a proton acceptor.
All of the crystal structures of hAsp present in the Protein Data Bank contain one zinc cation in the active site. The Zn2+ coordination sphere is composed of three conserved amino acid residues: His21, Glu24 and His116 (Fig. 3). 37

Figure 3. Schematic view of the aspartoacylase active site. 37 Blue colour indicates permanent ligands of the zinc cation, while green designates Glu178, NAA and water molecule, which are directly involved in the catalytic hydrolysis.
Download figure:
Standard imageThe composition of zinc coordination sphere was initially determined by comparison of the hAsp amino acid sequence with those of CPA and succinylglutamate desuccinylase (SGD). 38 In zinc-dependent carboxypeptidases, the zinc-binding domain is specified by the short conserved HisXXGlu motif and the long LeuHisGlyGlyX motif located in amino acids 95 – 135 at the C-terminus. Three zinc ligands, His69, Glu72, His196 in CPA and His56, Glu59, His149 in SGD, are equivalent to the His21, Glu24, His116 site in hAsp. The 91-residue range in the amino acid sequence between Glu24 and His116 in hAsp is also consistent with the CPA and SGD structures (123 and 89, respectively). The comparison also revealed that the hAsp catalytic base is Glu178, which corresponds to Glu270 and Glu214 in CPA and SGD, respectively.
The successive replacement of each of these amino acids by alanine results in complete loss of the activity, 39 the same is true for the pair-to-pair replacements, His21Glu/Glu24His and Glu24His/His116Glu. 40 The nature of the fourth ligand of the zinc ion depends, apparently, on the active site structure: in the pseudo-apo-hAsp, one oxygen atom of the phosphate group is incorporated in the zinc coordination sphere, while in the system with the tetrahedral intermediate analogue, this site is occupied by phosphonic group oxygen. In the absence of ligands, the zinc coordination sphere contains a water molecule.
The hAsp crystal structure with the tetrahedral intermediate analogue (PDB ID 2O4H) can be used to describe the interactions in the active site responsible for substrate binding and stabilization of intermediates of NAA hydrolysis (Fig. 4). The NPA carboxyl groups are positioned by the electrostatic interactions with Arg71 and Arg168 and by hydrogen bonds with Asn70, Tyr164 and Tyr288.

Figure 4. Geometry of the aspartoacylase active site (PDB ID 2O4H). 30 Here and below, the distances are in Å.
Download figure:
Standard imageThe tetrahedral phosphonyl group is coordinated by the zinc cation and by the Arg63 side chain. The distance between the nitrogen atoms of the guanidine group of arginine and the phosphorus atom exceeds 4.5 Å; therefore, there is a probability that Arg63 participates only in the stabilization of intermediates, but does not make a significant contribution to the formation of the protein – substrate complex.
The contribution of Arg71 to stabilization of the subsequent hydrolysis intermediates can be illustrated by the results of directed mutagenesis. The conservative replacement of Arg71Lys results in a 20-fold decrease in the rate constant of hydrolysis (kcat), while the Michaelis constant (Km) remains virtually invariable. 30 The Arg71Asn replacement completely eliminates the catalytic activity. 40
The replacement of Tyr164Phe reduces kcat by 2.5 orders of magnitude, while the Km value increases 6-fold in comparison with wild-type hAsp, indicating, in particular, a decrease in the substrate binding affinity to the active site. 30
Single conservative replacements, Arg168Lys and Tyr288Phe, decrease the kcat value by an order of magnitude. 30 The Tyr288 hydroxyl oxygen atom is located at 2.7 Å distance from the carboxyl oxygen atom of the substrate and at 2.8 Å distance from the amino group nitrogen atom. As a result, two hydrogen bonds are formed, which stabilize the intermediate during peptide bond cleavage.
The residue Glu178 acts as a base by accepting proton from the catalytic water molecule in the first step of hydrolysis. 40 – 42 In all crystal structures of the pseudo-apo-form of hAsp, the residue Glu178 is located at a hydrogen bond distance from the phosphate oxygen in the zinc coordination sphere; the location in the complex with NPA is similar. In the enzyme – substrate complex, Glu178 may promote polarization of the catalytic water molecule. The activated hydroxy group attacks the carbon atom of the carbonyl group polarized by the zinc ion and by the residue Arg63, thus giving the tetrahedral intermediate of the reaction. The subsequent transfer of two protons to the NAA nitrogen atom can be mediated by both Glu178 and Tyr288. 38 The site-directed mutagenesis Glu178Gln leads to the complete loss of the hAsp catalytic activity, while the replacement of Glu by aspartic acid decreases kcat by an order of magnitude, with the Km value being virtually invariable. This indicates a higher contribution of Glu178 to stabilization of transition states than to substrate binding in the hAsp active site. 38
The geometric configuration of the first intermediate (I1) derived from the results of QM(DFT/PBE0/6 – 31G**)/MM(AMBER99) calculations is consistent with the positions of heavy atoms in the PDB ID 2O4H crystal structure (Table 1). 37 The fourth ligand site in the optimized structure of I1 is occupied by the Os oxygen atom of the NAA carbonyl group. In the PDB ID 2O4H structure, the zinc coordination number is increased to 5 and the geometry of the complex is close to a trigonal bipyramid (see Table 1). This discrepancy can be attributed to the fact that the oxygen atoms at phosphorus atoms in NPA have more similar electron densities than the Os and Ow oxygen atoms in I1.
Table 1. Key distances (Å) for the calculated geometry of the first intermediate (I1) and crystal structure of 2O4H. 37
| PDB ID 2O4H | Optimized I1 structure | |
|---|---|---|
| Zn−Ow | 2.43 | 2.61 |
| Zn−Os | 2.55 | 1.92 |
| Zn−Nδ1 (His21) | 2.23 | 2.06 |
| Zn−Nδ1 (His116) | 2.29 | 2.02 |
| Zn−Oε1 (Glu24) | 2.15 | 2.05 |
| Zn−Oε2 (Glu24) | 2.62 | 2.58 |
| Ow−Oε2 (Glu178) | 2.93 | 2.66 |
| Ns−Oε1 (Glu178) | 3.74 | 3.40 |
| Oδ1 (NAA)−Nη2 (Arg168) | 2.94 | 2.67 |
| Oδ2 (NAA)−Nη1 (Arg168) | 3.26 | 2.70 |
| Os−Nη1 (Arg63) | 2.84 | 2.73 |
| Os−Nη2 (Arg63) | 3.00 | 3.55 |
2.3. Post-translational modification
After completion of the translation, some side groups of the polypeptide chain of the protein can be involved in additional processes changing its chemical composition. Often, post-translational modifications represent a necessary stage of prote in maturation and determine the functional activity of the protein.
The amino acid sequence of hAsp includes a conserved AsnXThr/Ser N-glycosylation site, Asn117 – Thr118 – Thr119, 43 located in the close vicinity of the active site. Hence, it may happen that the hAsp system contains a covalently bonded oligosaccharide.
The results of early studies of the catalytic activity of hAsp expressed in various systems suggest that binding of oligosaccharide to Asn117 is necessary to attain the catalytic activity observed in the experiment. 33 The hAsp expression in the prokaryotic Escherichia coli system, in which the post-translational modification is impossible, gives the protein with reduced catalytic activity and stability, 34 which may indicate the necessity of this maturation stage for the formation of the functionally active enzyme. On going to the eukaryotic Pichia pastoris systems, in which glycosylation is, in principle, possible, more stable enzyme with a 100-fold higher catalytic activity was obtained. 33
The action of glycosidase on hAsp (P. pastoris) results in a change in the catalytic function. The incubation of the obtained hAsp with PNGase F, an N-glycosidic bond cleaving enzyme, substantially decreases the activity (by 80%). An equal decrease in the activity and stability takes place upon the Asn117Gln single replacement, which prevents glycosylation. 33 Finally, upon the action of glycosidase on hAsp, a matrix assisted desorption – ionization mass spectrometry (MALDI MS) analysis detected a compound with m/z 2544.871, which corresponds to a polysaccharide consisting of monosaccharide residues of six hexoses, six N-acetylglucosamines and one N-acetylneuraminic acid residue (Fig. 5). 33

Figure 5. Structure of the polysaccharide isolated upon treatment of aspartoacylase with glycosidase. 33
Download figure:
Standard imageHowever, electron density map calculation for the structure of hAsp expressed in the P. pastoris system did not reveal additional electron density in the putative glycosylation site. Furthermore, the residue Asn117 is located in the enzyme active site — the neighbouring residue His116 occupies position of one of coordinated zinc ligands and the Asn117 side chain points to the interior of the protein. 30 The discordance of experimental results from different sources initiated the subsequent investigations of the possibility of post-translational glycosylation of hAsp and its role in the regulation of the enzymatic activity.
According to polyacrylamide gel electrophoresis data, the reference and PNGase-treated hAsp samples have identical masses of 34 kDa. In a similar experiment with ovalbumin, a thoroughly characterized glycoprotein, the sample masses differ by 2 kDa. 44 The absence of a shift between the marker positions for deglycosylated and control hAsp samples attests to the absence of covalently bound polysaccharide. The mass spectrum of native hAsp also does not exhibit any peaks that could be assigned to glycosylated hAsp. 44 Solid phase extraction of glycoprotein (SPEG) using hydrazine derivatives, which is a widely used method for efficient and selective identification of N-glycosylation sites, 45,46 also gives a negative result when applied to hAsp. Analysis of the ionic strength of PNGase-treated hAsp by isoelectric focusing method did not reveal distinctions from the reference sample either, which is untypical of a glycosylated protein because of the presence of terminal sialic acid residues.
The obtained results can be summarized as follows.
- 1.The reduced activity of hAsp samples expressed in E. coli 34 is apparently related to drawbacks of the applied procedure and inadequacy of the measures taken to exclude oxidation of the samples. 44 In the repeated, more properly designed expression of hAsp in E. coli, the calculated specific activity was 5 – 7 AU mg−1, which is comparable with the value obtained for hAsp expressed in P. pastoris (5 – 10 AU mg−1). 44
- 2.The treatment of hAsp with the deglycosylating enzyme PNGase was carried out in the presence of EDTA as a chelating agent at pH 7.5. 33 Thus, the decrease in the hAsp activity may be caused by removal of zinc from the active site. In the absence of EDTA or when pH is below the value optimal for zinc binding, the catalytic activity of hAsp is not lost after the incubation with PNGase. 44
- 3.The decrease in the hAsp activity upon Asn117Gln replacement is attributable to the proximate position of this amino acid to His116, which is incorporated in the zinc coordination sphere. According to the modelling of the mutant form using SWISS-MODEL server, 47 an elongation of the side chain introduces considerable distortions into the enzyme active site, which may be a reason for activity decline (Fig. 6).
- 4.Most difficulties arise when one attempts to interpret the discrepancy in the results of mass spectroscopy for the polysaccharide eliminated from hAsp. The eliminated glycan was identified by comparing m/z with the values for known polysaccharides. 34 The recorded peak may correspond to a side product.

Figure 6. Possible change in the aspartoacylase structure upon the Asn117Gln point mutation (N117Q) according to molecular modelling data. 44
Download figure:
Standard image2.4. Molecular polymorphism
The molecular polymorphism of hAsp includes, in particular, a set of clinically (or catalytically) significant mutations responsible for the Canavan disease. Currently, 80 replacements resulting in a change in the hAsp catalytic activity are known. 48 Most of them are located outside the enzyme active site.
The Canavan disease occurs most frequently in Ashkenazi Jews, with > 96% of cases being associated with two point mutations of hAsp: Glu285Ala and Tyr231X. The carrier frequency of these mutations among the Ashkenazi population varies from 1 : 37 to 1 : 40 and the highest prevalence rate is one per 6400 – 13 500. 49 The Canavan disease in other ethnic groups is associated with a more diverse range of mutations, the Ala305Glu replacement being the most frequent.
The point mutations of the active site amino acids either preclude zinc cation coordination (His21Pro, Glu24Gly) or reduce the active site capacity for substrate binding (Asn70Ser, Arg71His, Arg168His, Arg168Cys, Tyr288Cys). In most cases, the presence of mutations in the active site results in undetectable catalytic activity. 40,50
Among the clinically significant mutations of amino acids that play a key role in stabilization of the catalytically active conformation of hAsp, noteworthy are three mutations of Cys152 (Cys152Arg, Cys152Tyr, Cys152Trp). The residue Cys152 forms a disulfide bridge with Cys124; without this bridge, the catalytic activity decreases twofold with respect to the native enzyme, 40 as shown by alanine scanning and measurement of the specific catalytic activity. It is worth noting that the Cys152Trp and Cys152Arg mutations induce a severe form of the Canavan disease and the catalytic activity of the mutants does not exceed 1% of the native form activity (Table 2). 50
Table 2. Biochemical properties of selected clinically significant aspartoacylase mutants. 50
| Relative activity of hAsp(%) | Disease form | Thermal stability / °C | Denaturation stability / mM urea | |
|---|---|---|---|---|
| Native form | 100 | 59.9 | 1150 | |
| Glu285Ala | 0.3 | Variable | 48.5 | 125 |
| Glu285Ala/Pro181Thr | 32 | Mild | 58.3 | 600 |
| Tyr231Cys | 24 | Mild | 49.6 | 1050 |
| Phe295Ser | 10 | Variable | 56.6 | 1050 |
| Arg71His | 11 | Mild | 57.2 | 650 |
| Cys152Trp | 1 | Severe | 58.5 | 1100 |
| Cys152Arg | 0.5 | Severe | 51.2 | 300 |
| Ala305Glu | 10 | Variable | 58.9 | 900 |
The Glu285Ala mutation, which is most often encountered in the Ashkenazi population and is responsible for 85% of Canavan disease cases, 51 is located near the active site (Fig. 7), but does not directly participate in the hydrolysis. The catalytic activity of this mutant protein is not more than 1% of the native form activity. The stabilities against denaturation induced by heating (thermal stability) and by a 1M urea solution (conformational stability) are considerably lower for the mutant than for native forms of the enzyme (see Table 2).

Figure 7. Structural changes in the local environment upon the Glu285Ala replacement. 52
Download figure:
Standard imageThe general appearance of the tertiary and quaternary structures is the same for the mutant and native forms (RMSD = 0.37 Å). 52 However replacement of glutamic acid by alanine leads to considerable disruption of the active site. The shift of active site loops extends the transport channel and, as a consequence, increases the active site accessibility even in the presence of the substrate (Fig. 8). Cleavage of the hydrogen bond between Glu285 and Thr118 induces shifts of helix 285 – 290 and loop 290 – 294. The residue Tyr288 participating in the substrate binding in the native form shifts by 4.5 Å, which precludes hydrogen bonding between its hydroxy group and the substrate amino group. The loop containing Tyr164, which is hydrogen-bonded to the β-carboxy group of the substrate in the native form, is also displaced.

Figure 8. Comparison of the active site geometry for the native and mutant (Glu285Ala) forms of aspartoacylase. 52
Download figure:
Standard imageEven after substrate binding, the active site of the Glu285Ala mutant remains open. The side chains of Arg71 and Lys291 are not involved in the salt bride formation and the Tyr64 – Glu290 hydrogen bond, responsible for stabilization of the closed active site conformation in the native form, is cleaved (see Fig. 8).
The vast majority (∼80%) of known clinically significant mutations of hAsp are located far from the active site. It was hypothesized that the loss of the enzyme function may be due to structure destabilization in the hAsp mutants. Analysis of the kinetic and thermodynamic properties of 16 clinically significant hAsp mutants corresponding to different severity levels of the Canavan disease did not reveal an unambiguous correlation between these values and decrease in the catalytic activity (Fig. 9). 50 Considering the available data, the severity of the disease is not also correlated with the enzymatic activity of clinically significant hAsp mutants. An exception to the expected dependence is the mild form of the disease, which is caused by the Tyr288Cys and Arg71His mutations of active site amino acids, which critically reduce, as expected, the specific activity of hAsp down to 4% and 11% of the native form activity, respectively.

Figure 9. Stability of the mutant forms of aspartoacylase and correlation with the loss of enzyme activity and the form of the Canavan disease. 50 Data for severe (raspberry-red), mild (green) and variable (grey) forms of the disease are given.
Download figure:
Standard imageExploration of the mechanisms of influence of mutations remote from the active site led to calculation of the structures of several mutants — Phe295Ser, Tyr231Cys and Lys213Glu — with the tetrahedral intermediate analogue (NPA) in the active site 52 (PDB ID 4NFR, 4MRI, 4TNU). The positions of these mutations do not imply direct participation in the catalytic hydrolysis; however, each of them can cause Canavan disease of different severity levels. 53,54
The Phe295Ser mutation is responsible for various forms of Canavan disease. The replacement of a bulky nonpolar side chain of phenylalanine by serine induces the displacement of the Met261 side chain through 160° around the S−C bond. The side chain of Lys297 is also displaced from the position in the native enzyme and occupies the vacancy formed as a result of mutation. Judging by the active site structure, substrate binding is similar to that for the native form of hAsp (Fig. 10). It was suggested that a decrease in the activity (down to 10% of the native form activity) is caused by decreasing stability of the enzyme structure. 53 However, investigations of thermal stability and stability against denaturation demonstrated a minor decrease with respect to the native form (see Table 2).

Figure 10. Comparison of the active site geometry for native aspartoacylase and the Phe295Ser mutant. 52
Download figure:
Standard imageMutation of Tyr231 is one of the principal ones in the Ashkenazi population. 51 This residue is located in the loop at the start of the C-domain of hAsp and forms hydrogen bonds with the Tyr289 hydroxy group and the Asn284 carbonyl oxygen. These residues are located in the hydrophobic region (Fig. 11), which adjoins the active site, and the substrate-binding Tyr288 is located in the same loop as Tyr231. The superposition of the crystal structures of hAsp native form and mutant form (Tyr231Cys) (PDB ID 4TNU) was used to describe the deviation between the positions of amino acid residues adjacent to the point of mutation. 53 The Cα-atom of Cys231 is shifted by 0.5 Å with respect to the native structure, while its side chain is not involved in any interactions (see Fig. 11). The shift of Asn284 towards Tyr289 decreases its interaction with the Leu187 backbone of the other subunit (ASPB). The replacement of tyrosine by cysteine causes a local structure perturbation and thus enhances the mobility of the loop (Bfactor > 100 Å2), which is stabilized, in particular, by interactions with the solvent.

Figure 11. Change in the geometry of the local environment of Tyr231 upon its replacement by cysteine. 52
Download figure:
Standard imageThe Tyr231Cys mutation was detected in the mild form of Canavan disease, with the catalytic activity of this mutant being relatively high (24% of the native form activity). 50 Despite the location near the active site, the Tyr231Cys mutation does not have a significant influence on the binding, but the induced structure perturbation may be responsible for a decrease in thermal stability (see Table 2). 50 It should also be noted that Tyr231 is located at the start of the C-domain of hAsp, which can also be crucially important for proper folding of the C-domain polypeptide chain.
The hAsp mutant, Lys213Glu, is described by the PDB ID 4MXU crystal structure. In an organism, the Lys213Glu mutation is, most often, accompanied by Gly274Arg. 53 It was believed for long that in the case of double mutation, Lys213Glu/Gly274Arg, the Lys213Glu replacement enhances the activity of the mutant hAsp. However, in a series of experiments combining point mutagenesis and specific activity measurements, it was found that hAsp(Lys213Glu) has indeed the activity equal to that of native hAsp; however, the catalytic properties of hAsp(Gly274Arg) and hAsp(Gly274Arg/Lys213Glu) do not differ from each other.
3. Mechanism of catalysis and effect of substrate activation and inhibition
Apart from the important biological function performed by hAsp, the interest in the description of the catalytic properties of this enzyme is also related to uncommon kinetic parameters — a broad range of methods for regulation of the enzymatic activity, including allosteric self-activation and self-inhibition. 33
The kinetic parameters for aspartoacylase were determined in the presence of a 1000-fold excess of the substrate; the results were processed according to the Michaelis – Menten approach. The kcat value for recombinant hAsp depends on the method used to determine the product concentration. The value kcat = 12.7±0.05 s−1 was determined by UV spectrophotometry by measuring the concentration of fumarate synthesized from L-aspartate. 33 For determination of the product concentration by spectrophotometry, L-aspartate was converted to a chromogenic product by treatment with 2,4,6-trinitrobenzenesulfonic acid. The kcat found for recombinant hAsp was 14.22±0.48 s−1 (Ref 33).
A study of the hAsp activity as a function of substrate concentration showed an s-shaped curve at low concentrations (<0.3 mmol litre−1), which may attest to self-activation effect. 33 However, at high concentrations (>1 mmol litre−1 NAA), the enzyme activity decreases, indicating the possible existence of an additional NAA binding site, which regulates the hAsp inhibition.
The dependence of the catalytic function of hAsp on the presence of a zinc ion was confirmed in a kinetic experiment. Treatment with a chelating agent (o-phenanthroline, OP) markedly reduced the catalytic activity of hAsp (14%), while slow addition of a dilute ZnCl2 solution to hAsp dialyzed to remove phenanthroline restored the activity; excess of the metal ions also inhibits the enzymatic hydrolysis. 33 According to inductively coupled plasma mass spectrometry data, the ratio of zinc ions and active sites is 1 : 1. 33
Determination of the kinetic parameters of hAsp for several NAA analogues revealed limited substrate specificity for NAA (Table 3). The kcat values were higher for all substrate analogues than for the native form, which is attributable to increasing polarizability of the bond being cleaved on going to other substituents. However, the Km value varies depending on the size and polarity of the functional group in place of acetate: for larger molecules — 2-[(2,2-dichloroacetyl)amino]butanedioic acid (N-dichloroacetyl-L-aspartate) and 2-[(2,2,2-trifluoroacetyl)amino]butanedioic acid (N-trifluoroacetyl-L-aspartate) — the active site affinity increases 1.5-fold; meanwhile, for less bulky 2-(formylamino)butanedioic acid (N-formyl-L-aspartate) and for similar-size but more polar 2-[(2-chloroacetyl)amino]butanedioic acid (N-chloroacetyl-L-aspartate), the binding affinity is two and three times lower, respectively. 34
Table 3. Kinetic parameters of enzymatic hydrolysis of N-acetylaspartate analogues. 34
| Substrate | kcat / s−1 | Km / mmol litre−1 | kcat / Km / litre mol−1 s−1 |
|---|---|---|---|
| N-Acetyl-L-aspartate | 0.083 ± 0.006 | 0.36 ± 0.06 | 2.3×102 |
| N-Formyl-L-aspartate | 0.25 ± 0.03 | 0.95 ± 0.23 | 2.6×102 |
| N-Chloroacetyl-L-aspartate | 0.62 ± 0.04 | 0.59 ± 0.09 | 1.0×103 |
| N-Dichloroacetyl-L-aspartate | 0.77 ± 0.03 | 0.23 ± 0.03 | 3.4×103 |
| N-Trifluoroacetyl-L-aspartate | 1.2 ± 0.09 | 0.21 ± 0.04 | 5.8×103 |
3.1. Catalytic cycle
The first assumptions about the mechanism of enzymatic hydrolysis of NAA were based on the similarity of the hAsp and carboxypeptidase A active sites. 31 The composition and location of the active site residues of hAsp and carboxypeptidase A (PDB ID 1M4L) are roughly identical (Fig. 12). Their root-mean-square deviation is 0.7 Å, which is typical of different crystal structures of the same protein. Presumably, the mechanism of NAA hydrolysis is similar to that known for CPA: first, nucleophilic attack by a water molecule gives the tetrahedral intermediate and next peptide bond cleavage takes place to give acetic and aspartic acids.

Figure 12. Comparison of the active site geometry for carboxypeptidase A (yellow carbon) and aspartoacylase (light-green carbon). 31
Download figure:
Standard imageThe most comprehensive view of the sequence of elementary chemical reactions of hAsp-catalyzed hydrolysis of NAA can be gained using advanced molecular modelling methods. The progress of computing technologies and availability of supercomputer resources 55 provide practical opportunities for such computations.
Quantum chemical description of the mechanism of enzymatic hydrolysis of NAA was developed using the combined quantum mechanics/molecular mechanics (QM/MM) method. This approach implies splitting of the system into two parts: the quantum subsystem comprising the active site and the MM subsystem comprising the major part of the protein and the shell of water molecules. 56 – 69 Then the total Hamiltonian (Htotal) of the system can be written down as expression (1)
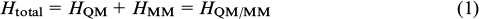
where the terms HQM and HMM include the interaction energy of all atoms of isolated QM and MM subsystems, respectively. Of most difficulty is the calculation of the term HQM/MM including the energy contributions of all pair interactions of atoms of the QM and MM subsystems.
The Hamiltonian for the quantum part was calculated by Kots et al. 37 using density functional theory (DFT/PBE0/6-31G**). The energy of the MM subsystem was described by the AMBER99 force field. The HQM/MM contribution was calculated by the electronic embedding scheme taking into account polarization of the QM subsystem by the protein environment. 70 To confirm the key conclusions of the study, the geometry parameters of the stationary points were also optimized with an extended QM system. 37
The calculated profile in the minimum-energy pathway includes six successive steps (Fig. 13). The enthalpy gain on going from the enzyme — substrate complex (ES) to the enzyme — product complex (EP1P2) was 3.5 kcal mol−1 (taking account of the zero-point energy). Most energy is spent for the transition from the ES complex to the tetrahedral intermediate (12.5 kcal mol−1). The lowest potential energy corresponds to I2 (−2.7 kcal mol−1), which suggests accumulation of this intermediate.

Figure 13. Potential energy profile of the chemical step of the catalytic hydrolysis of N-acetylaspartate. 37 The energies are given with zero-point energy corrections.
Download figure:
Standard image3.1.1. The reaction pathway from the enzyme – substrate complex to peptide bond cleavage
The first step of NAA hydrolysis includes nucleophilic attack by the catalytic water molecule on the carbonyl carbon atom of the substrate to give the tetrahedral intermediate (Fig. 14). The calculated barrier height, 12.5 kcal mol−1, is in line with the results obtained previously for zinc-dependent metalloproteinase-2. 71

Figure 14. Reaction intermediates preceding the peptide bond cleavage. 37 Here and below the energies are in kcal mol−1.
Download figure:
Standard imageAfter the first step, the peptide N−C bond length in NAA increases insignificantly: from 1.33 Å in the ES complex to 1.44 Å in I1. In order to facilitate the bond cleavage, the Hw1 proton transferred to Glu178 in the first step should migrate to the substrate nitrogen atom. The migration distance is 3.07 Å and the barrier for this step does not exceed 1 kcal mol−1. The peptide N−C bond length continues to increase to reach 1.57 Å.
In protein systems, the zinc ion may have a labile coordination sphere in which not only the composition, but also the coordination number (C.N.) can change. 72,73 In the optimized ES complex structure, the oxygen atom of the catalytic water molecule (Ow) forms a coordination bond with the zinc cation, with the Zn−Ow distance being 2.04 Å. The zinc C.N. is retained on going from the ES complex to I1, only the ligand composition being changed. The Ow position in the coordination sphere is occupied by the substrate carbonyl oxygen atom (Os). Furthermore, on going from the ES complex to I1, the distance between the Ow and C atoms decreases from 2.66 to 1.48 Å, which corresponds to the classical position of the nucleophilic attack in zinc-dependent peptidases. 74
3.1.2. N−C bond cleavage
The distance between the N and C atoms was chosen as the reaction coordinate in the step of peptide bond cleavage in the substrate. The peptide bond cleavage is accompanied by spontaneous transfer of the second Hw2 proton onto the oxygen atom of the Glu178 carboxyl group. The distance between Oε1 and Hw2 is 1.1 Å after the third step, while the C−N distance increases to 2.54 Å in the optimized structure of intermediate I3 (Fig. 15).

Figure 15. Intermediates of peptide bond cleavage and Glu178 reorientation steps. 37
Download figure:
Standard imageFor complete regeneration of hAsp, the Hw2 proton should be transferred from Glu178 to the aspartic acid amino group. Therefore, in the next step, the Glu178 carboxyl group is rotated and the Cα−Cβ−Cδ−Cγ dihedral angle changes from 108.1° to 160.5° (see Fig. 15). The calculated rotation barrier is 8.6 kcal mol−1, which is comparable with the barrier for nucleophilic attack. The activation energy for active site rearrangement may be attributed to the necessity to break the strong hydrogen bond between the Ow and Oε1 oxygens.
3.1.3. Formation of the reaction products
The Hw2 proton transfer to the nitrogen atom is expected to give the positively charged ammonium group of L-aspartate. However, the hAsp active site is saturated with three residues, Arg63, Arg71 and Arg168; therefore, the formation of additional positive charge on the product may be hampered. The conformational search for the active site geometry favourable for Hw2 transfer from Glu178 was based on a 15 ps molecular dynamics trajectory with the potentials QM(PBE/DZVP-PW)/MM(CHARMM36) computed for I4 in a canonical ensemble at 300 K and 1 atm. 37
Along the MD trajectory, the system oscillates between two ground states: the protonated residue Glu178 corresponds to the initial position of the acetate anion relative to L-aspartate (Fig. 16 a,b), and the ammonium group is formed as one acetate oxygen atom approaches the nitrogen atom to a distance of hydrogen bonding (see Fig. 16 c,d). The TS5 and TS6 barriers reflect a rather shallow potential energy surface in this segment and are not of particular interest for further analysis.

Figure 16. Conformational search for the route of Hw2 proton transfer to the products. 37 (a, b) Protonated Glu178 and initial position of the acetate anion; (c, d ) formation of the ammonium group and approach of one acetate O atom to the N atom to form a hydrogen bond.
Download figure:
Standard imageComparison of the EP1P2 and ES structures showed that after the composition of zinc ion ligands has changed in the first step, the fourth ligand position is occupied again by the Ow atom in the I4 to EP1P2 transition steps (see Fig. 16).
The computed mechanism of the catalytic hydrolysis of NAA was confirmed by calculation of the key stationary points with the extended QM part. 37 Despite the change in the energy values, the key conclusions of the previous calculation are retained on going to the extended QM part. In view of the fact that the energy of stationary points tends to be underestimated as the QM model is extended, the use of the first, nucleophilic attack, step as the rate-limiting step does not seem substantiated.
The calculation of the potential energy profile for the chemical step of the enzymatic reaction is obviously a key issue in the theoretical description of the mechanism. However, for adequate comparison with the experiment, it is necessary also to take into account the step of formation of the ES complex and release of the reaction products (acetate and aspartate) to the solution. This is even more necessary for comparison of computed parameters with the experimental constants of the Michaelis – Menten equation, because these steps determine the Km and kcat values.
3.1.4. Enzyme – substrate complex formation and product release steps
The non-chemical steps of NAA hydrolysis were described by classical molecular dynamics (MD) methods.
The free energy profiles for NAA binding in the enzyme active site and for release of hydrolysis products to the solution were calculated by the Hamiltonian replica exchange umbrella sampling (HREUS) method. 75 According to this procedure, n replicas of the system travelling along the molecular dynamics trajectories are generated. During the computation, the energies of the adjacent replicas are periodically compared and the current coordinates of the replicas are exchanged.
For each process, 15 ns molecular dynamics trajectories were calculated for each system replica.
Formation of the protein – substrate complex. Binding of NAA is associated with transport through the gate to the active site formed by amino acid pairs facing each other, Tyr64 – Glu290, Asp68 – Lys291 and Arg71 – Glu293. According to the computed free energy profile, the barrier for NAA insertion through the gate was 1.7 kcal mol−1, while ΔG ° = −6.7 kcal mol−1 (Fig. 17). Only the open-gate conformation of the active site was used in the calculations; therefore, the energy barrier may be underestimated.

Figure 17. Free energy profile for N-acetylaspartate entering the aspartoacylase active site (a) and view of the open-gate conformation of hAsp (b). 37
Download figure:
Standard imageRelease of the reaction products to the solution. Hydrolysis of NAA in the hAsp active site gives acetate and L-aspartate anions.
Analysis of the EP1P2 structure revealed the presence of two routes for product release: the first one runs along the main transport channel, while the other one is along the secondary transport channel located on the opposite side of the cavity and gated by the amino acid residues Gln184 and Ala286 (Fig. 18). Considering the size of the N-product molecule, this product can be extracted only via the main transport channel. Meanwhile, the acetate anion is sufficiently small and, hence, it can use also the auxiliary route. Thus, the C-product can either follow the N-product via the main channel or move independently by the second route.

Figure 18. Directions of the release of products of N-acetylaspartate enzymatic hydrolysis. 37
Download figure:
Standard imageThe free activation energy for L-aspartate computed with the distance between the Cα atoms of His116 and the product being used as the coordinate was 3.2 kcal mol−1, while the free energy ΔG ° was 2.6 kcal mol−1. The most pronounced free energy expenditure is associated with overcoming the electrostatic interactions and cleavage of the hydrogen bond between Arg71 and the N-product.
The release of the C-product by the same route as the aspartate was described by varying the distance between the Cα atom of Asn117 and the carboxyl carbon atom of the product. The reaction pathway length was 9.5 Å; the energy ΔG ° was 2.2 kcal mol−1.
The free energy profile for the acetate anion release through the Asn23, Arg63, Gln184 and Glu290 gate was calculated using a pulling vector aligned between the Cα atom of Leu128 and the carboxyl carbon atom of the product. The relatively high activation barrier for this process (7.1 kcal mol−1) can be rationalized by cleavage of the coordination bond between the Os atom and the Zn2+ cation. The ΔG ° energy (−3.9 kcal mol−1) makes this reaction pathway thermodynamically more favourable. Thus, in the free energy profile of the complete catalytic cycle of hAsp, the energy of acetate release step corresponds to the value obtained for the secondary route.
A comparison of the PDB ID 2Q51 structure and the calculated structure of the recovered hAsp enzyme showed a satisfactory agreement of the geometric parameters derived from computations and from crystal structure measurements (Fig. 19).

Figure 19. Superposition of the apo-hAsp structure after modelling (pink carbon) and PDB ID 2Q51 (green carbon). 37
Download figure:
Standard imageThe largest root-mean-square deviation of the calculated geometry from experimental one is found for the Glu178 side chain (RMSD = 3.7 Å), which is attributable to the fact that the carboxyl oxygen of Glu178 in the calculated structure acts as the fourth ligand of the zinc ion, whereas in the crystal structure, the zinc coordination sphere incorporates the phosphate anion.
A curious feature of zinc in enzyme systems is the ability to change the coordination number depending on the environment. In aqueous solutions, zinc cation forms an octahedron with six water molecules, the standard Zn−O bond length being 2.10 Å. 76 In zinc-finger proteins and in the vast majority of other zinc-binding enzymes, the zinc ion coordinates four ligands, but there are also known structures in which the zinc C.N. increases to five or even six. 77,78 The decrease in the zinc coordination number in the protein environment is commonly attributed to the possibility of charge transfer from amino acid ligands to the zinc cation, which decreases the positive charge. This effect is especially pronounced in the systems where zinc coordinates a cysteine residue. 79
Molecular modelling using classical force fields does not explicitly take into account the charge transfer to the cation, as the Zn2+ partial charge is constant (+2e) and does not depend on the ligand number and composition. The polarization effect is not directly taken into account either; however, the use of total atomic charge enables implicit use of some polarization corrections. 80
The zinc coordination sphere in the CHARMM36 force field was described by the Coulomb potential and Lennard-Jones approximation. The van der Waals parameters for the zinc cation were selected in such a way as to describe most closely the experimental radial distribution function and calculate the free energy of solvation of model systems with high accuracy. 81 The long-range non-covalent interactions were taken into account within a 15 Å radius. The carboxypeptidase A structure was one of the model systems used for CHARMM parameterization. The error of calculation of coordination bond lengths in comparison with the crystal structure did not exceed 0.1 Å for the model system. 81 In view of the structural and functional similarity of carboxypeptidase A and hAsp, the description of zinc interactions by means of a simple non-bonded model can be reasonably accurate.
According to QM/MM optimization of stationary points, the zinc coordination sphere is fairly stable throughout all elementary steps of the chemical reaction: three amino acid ligands, His21, Glu24 and His116, retain their bonds with the zinc cation. However, the fourth ligand position is occupied by the Ow atom of the catalytic water molecule in ES and EP1P2 systems, while in the intermediate structures, it is replaced by the Os oxygen atom of the NAA carbonyl group. The geometric configurations obtained by QM/MM optimization were additionally verified by molecular dynamics methods.
The zinc coordination sphere was described using the potentials QM(PBE/DZVP-PW)/MM(CHARMM). The QM/MM-optimized geometry of the ES complex was used for the initial approximation. The QM part included all functional groups of the QM/MM simulation. In the computed trajectory, the zinc coordination sphere remained fairly stable throughout a 15 ps trajectory length. The average bond lengths in the QM/MM-MD model were consistent with the QM/MM results obtained for different QM subsystems (Table 4). The resulting coordination bond length distributions were compared with the results of classical MD calculation of the ES complex for 8 ns length. The QM/MM-optimized ES structure was also chosen as the starting geometry for the MM system. The average bond lengths calculated using classical MD and QM/MM-MD differed by ∼0.1 Å, which was a reasonably good result for description of the geometry of a transition metal complex in the classical approximation (see Table 4). It is noteworthy that the tetrahedral coordination geometry of zinc is retained throughout the classical MD trajectory.
Table 4. Coordination bond lengths (Å) of the zinc ion in the enzyme – substrate complex according to computations in various approximations. 37
| QM/MM | QM/MM (extended QM part) | QM/MM-MD | Classical MD | |
|---|---|---|---|---|
| Zn−Oε1 (Glu24) | 2.18 | 2.18 | 2.09 ± 0.11 | 2.04 ± 0.06 |
| Zn−Nδ (His21) | 2.18 | 2.16 | 2.13 ± 0.11 | 2.26 ± 0.08 |
| Zn−Nδ (His116) | 2.06 | 2.04 | 2.06 ± 0.08 | 2.18 ± 0.06 |
| Zn−H2O | 2.04 | 2.02 | 2.05 ± 0.08 | 2.11 ± 0.06 |
3.1.5. Free energy profile of the catalytic cycle
Construction of the free energy profile for the complete catalytic cycle of hAsp implies swiching to ΔG ° values from potential energies of stationary points for chemical elementary steps. The changes in the heat capacity and the entropy contribution on going to 298 K were described by the harmonic oscillator and rigid rotor models. 37
The calculated free energy profile for the complete catalytic cycle of aspartoacylase (Fig. 20) can be used to identify the rate-limiting steps of the enzymatic reaction that make the greatest contribution to the observed kcat.

Figure 20. Free energy profile of the complete catalytic cycle of aspartoacylase. 37 The asterisk marks the initial geometry of the QM/MM simulation.
Download figure:
Standard imageThe highest activation energy (11.3 kcal mol−1) was found for the first chemical step, namely, the nucleophilic attack. In the calculation of TS1 with extended QM part, the potential energy barrier decreased from 12.5 to 10.0 kcal mol−1. The minimum-energy pathway for the first step of hydrolysis was also calculated by the QM/MM method in the tight binding approximation (SCC-DFTB) for the QM part and the CHARMM force field potentials for the MM part. 82 The barrier for the nucleophilic attack was 24 kcal mol−1; however, later the barrier was reduced to 16 kcal mol−1 by calculating the potential of mean force by the umbrella sampling approach; according to the activated complex theory, the latter value is consistent with the experimental kcat value. 42 However, the reported 37,82 descriptions of the hydrolysis mechanism cannot be considered complete, first, because of the absence of enzyme recovery steps and, second, because of limitations of the SCC-DFTB approximation for the description of proton transfer. 71
It is worth noting that for zinc-dependent proteins, the reaction barriers found by the DFT/PBE0 method are 1 – 3 kcal mol−1 lower than those found by the CCSD(T) coupled-cluster method. 83 In the general case, the calculated barriers for the nucleophilic attack reported in the literature for zinc-containing hydrolases do not exceed 13 kcal mol−1 (Ref. 83). Furthermore, the structure and the amino acid sequence of the hAsp active site are similar to those of type A carboxypeptidase. The catalytic constant of these enzymes is ∼104 s−1, which corresponds to the barrier of 11 kcal mol−1 (Ref. 84). Thus, presumably, the first step does not make a crucial contribution to the observed kcat.
The resulting reaction barriers were used to calculate the rate constants of elementary steps according to the transition state theory. 85 The overall kinetic scheme of enzymatic hydrolysis of NAA is depicted in Scheme 2.
Scheme 2
Download figure:

The steps with an activation barrier <1.7 kcal mol−1 can be described using a quasi-equilibrium approximation via equilibrium constants. The key steps of hydrolysis (ΔG ° ≥ 8 kcal mol−1) correspond to the three highest barriers (Scheme 3).
Scheme 3
Download figure:

Then the reaction rate, kcat and Km are specified by relations (2) – (4).

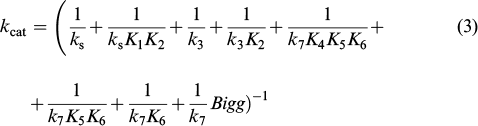

A considerable contribution to the expression for the rate constant of the catalytic process in terms of the Michaelis – Menten approach (kcat) is made only by the term 1/k3 K2 . Then the kcat and Km values are given by expressions (5) and (6).


The numerical calculation by the above relations gives kcat = 715 s−1 and Km = 1.2×104 mol litre−1, which is characteristic of hydrolases, in particular, and enzymes, in general. 86,87 The rate-limiting step can be distinguished without additional detailed analysis: the highest energy barrier (13.5 kcal mol−1) is determined by the reaction shown in Scheme 4.
Scheme 4
Download figure:

The calculated Michaelis constant is similar to the experimental one:  mol litre−1 (Ref. 33). However, the theoretical kcat value exceeds the experimental one by two orders of magnitude. This discrepancy between the theoretical and experimental values may be due to the fact that the theoretical calculation used a 'rigid' protein model based on the crystal structure. It is known that a change in the geometry of a stationary point by 0.1 Å leads to a 3 kcal mol−1 change in energy, which entails a 150-fold change in the rate constant.
mol litre−1 (Ref. 33). However, the theoretical kcat value exceeds the experimental one by two orders of magnitude. This discrepancy between the theoretical and experimental values may be due to the fact that the theoretical calculation used a 'rigid' protein model based on the crystal structure. It is known that a change in the geometry of a stationary point by 0.1 Å leads to a 3 kcal mol−1 change in energy, which entails a 150-fold change in the rate constant.
3.2. Dependence of the reaction rate on the substrate concentration
Data on the catalytic function of hAsp and the ways of its regulation derived from molecular modelling were used to formulate the overall kinetic scheme of enzymatic hydrolysis of NAA. 88,89
The ensembles of states of apo-hAsp, hAsp complex with inhibitor [hAsp:NAA(inh)] and activator [hAsp:NAA(act)] derived from molecular dynamics calculations included each 500 thousand snapshots of 500 ns MD trajectories for each system. The computed trajectories were used to construct the Markov state model (MSM). 90
The Markov state model is a kinetic model describing the probability of transitions between discrete states of the system. 91 Using MSM, it is possible to investigate the conformational transitions occurring over periods of time exceeding the lengths of trajectories. Relatively recently, the MSM approach started to be used to describe the conformational transitions and to elucidate the mechanisms of formation of the tertiary structure in protein systems. 92
The MSM calculation for hAsp systems was conducted for the Cα and Cβ atom coordinates. The system dimensionality was further decreased by time independent component analysis (tICA) followed by discretization of the sequence of states using Voronoi diagrams. The space discretization was followed by calculation of the transition probability matrix between the discrete metastable states of the system.
The MSM calculation provided independent determination of the slowest transitions in the hAsp systems. In all cases of apo-hAsp, hAsp:NAA(inh) and hAsp:NAA(act), the slowest projection derived by tICA corresponds to the transition between the open and closed gate in ASPA (Figs 21, 22).

Figure 21. Projection of six macrostates of the apo-hAsp, hAsp:NAA(inh) and hAsp:NAA(act) systems on to two slowest components of time-lagged independent component analysis. 89
Download figure:
Standard image
Figure 22. Macrostates of the Markov state model, the transitions between which are the slowest. 89
Download figure:
Standard imageThe gate in the ASPB subunit remained closed in all systems and in all macrostates of the model. The results suggest that population of the conformation corresponding to the hAsp dimer with open gates of both subunits is very low and has little effect on the catalytic properties of the enzyme. 89
The equilibrium constants for the apo-hAsp, hAsp:NAA(inh) and hAsp:NAA(act) systems were estimated from the forward and reverse rate constants for closed-gate (kc) and open-gate (ko) conformations of the ASPA (Table 5).
Table 5. Kinetic constants for conformational transitions estimated by the Markov state model. 89
| System | ko / s−1 | kc / s−1 | K |
|---|---|---|---|
| apo-hAsp | 6.7×106 | 6.1×106 | 1.1 |
| hAsp:NAA(inh) | 1.0×107 | 5.0×105 | 20 |
| hAsp:NAA(act) | 6.3×107 | 1.1×109 | 0.06 |
These results provide a semiquantitative description of the conformational dynamics of the system. In the apo-form, the open- and closed-gate conformations of ASPA are equally probable and are related as 1 : 1. Binding of NAA in the activation site shifts the equilibrium towards the open form. Conversely, the subsequent binding in the inhibition site stabilizes the closed-gate conformation of ASPA.
3.3. Kinetic model of human aspartoacylase
The kinetic approach was used to analyze the dependence of the reaction rate on the substrate concentration; the kinetic scheme is presented in Scheme 5. 89
Scheme 5
Download figure:

Each step of the proposed scheme was assumed to be equilibrium. The constant K1 describes the NAA binding in activation sites, K2 corresponds to binding in the active site, and K3 refers to binding in the inhibition site. The analytical expression for the observed reaction rate (ν) can be derived according to expressions (7) – (12).



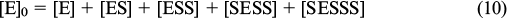
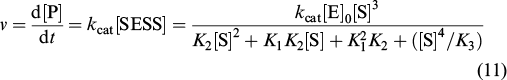
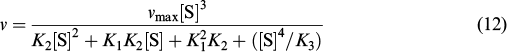
The third power of the substrate concentration in the numerator ensures the s-shape of the kinetic curve in the initial segment, while the fourth power in the denominator is responsible for decreasing activity at increased NAA concentrations. For direct comparison of the theoretical and experimental results, the points obtained by Le Coq et al. 33 were fitted to the kinetic curve derived by Khrenova et al. 89 (Fig. 23).

Figure 23. Comparison of the kinetic curve for the dependence of the aspartoacylase activity on the N-acetylaspartate concentration calculated by molecular modelling methods (blue line) with experimental data (blue dots). 33,89
Download figure:
Standard imageIt is worth noting that the proposed model describes the predominant pathway of catalysis, without denying the existence of other, unaccounted processes in this system. For example, NAA can be bound in the inhibition site in the E, ES or ESS system and so on. However, the close agreement of the computed curve and the literature data describing the experimental kinetics indicates that all key contributions to the process were included in the model.
The obtained standard free energies of NAA docking into activation and inhibition sites can be used to estimate the binding constants for these processes (Table 6).
Table 6. Equilibrium constants for the steps of the kinetic scheme for aspartoacylase calculated from experimental data and according to the proposed kinetic model. 89
| Experiment | Theory | |
|---|---|---|
| K1 = 6.4×10−5 mol litre−1 | K1 = 2.0×10−5 mol litre−1 | |

|

| |
| K2 = 4.5×10−4 mol litre−1 | − | |
| − | |
| Autodock4 | FlexX | |
| K3 = 1.5×10−3 mol litre−1 | K3 = 4.1×10−4 mol litre−1 | K3 = 3.4×10−4 mol litre−1 |

|

|
|
The energy of NAA docking into the active site was not determined, as this method is unable to take account of the effect of zinc ion located in the active site and making a considerable contribution to ES complex stabilization. The ratio of the calculated binding constants in the activation site and inhibition site was 17 – 20 (depending on the chosen value for K3), which corresponds to the experimental value of 23 for K3/K1.
4. Role of the quaternary structure in the regulation of the catalytic activity of the enzyme
According to kinetic analysis, the enzyme activity is regulated by substrate concentration in an intricate way. The dependence of the rate of hydrolysis on the NAA concentration comprises enzyme activation steps at low NAA concentrations and inhibition steps at high NAA concentrations. It is clear that the activation and inhibition effects are determined by structural changes in the protein.
According to experimental data, hAsp exists in solutions as a homodimer. For choosing the model structure for calculations, it is necessary to include the contribution of interactions between hAsp subunits (ASPA and ASPB) to stabilization of the active conformation.
The structural data suggest high mobility of the active site gate formed by loops 62 – 74 and 282 – 294 (Fig. 24 a). A superposition of the initial coordinates of the six structures of the hAsp monomer illustrates unstable arrangement of the residues Arg71 and Glu293 (see Fig. 24 b). These residues are incorporated in the opposing loops 62 – 74 and 282 – 294, which form the main gate of the hAsp active site. The position and the mobility of this chain segment dictate the accessibility of the active site for substrate binding and, hence, can directly affect the catalytic activity of hAsp.

Figure 24. Structure of the aspartoacylase transport channel. 30 – 32 (a) Positions of loops 62 – 74 and 282 – 294 relative to the active site (green surface); (b) orientation of the channel gate (Arg71 and Glu293) in the subunits of the apo-hAsp crystal structures PDB ID 2I3C and 2O53.
Download figure:
Standard imageThe ensemble of states derived from MD trajectories for six models of hAsp includes both open and closed hAsp conformers. Despite the differences in the initial geometric configurations, the equilibrium between the states is shifted towards the closed (inactive) conformation for all the MD trajectories. The closed form is stabilized by two salt bridges formed between Arg63 and Arg71 on one side and Glu285 and Glu293 on the other side. The electrostatic interactions between these amino acid residues are retained throughout all MD trajectories of the monomer.
Apart from the salt bridges, the monomer models comprise a stable interaction network between the opposite amino acid residues of the transport channel gate: Tyr64 – Glu290, Lys291 – Asp68 and Arg71 – Glu293 (Fig. 25). In the case of the dimer, these interactions are either completely absent over the 500 ns MD trajectory or their population is very low (see Fig. 25 a). Over the first 10 ns MD trajectory, one homodimer subunit is always shifted relative to the other one by ∼15°. The root-mean-square deviations of the monomers from the initial position considerably differ from each other (Fig. 26). This shift, which is subsequently retained in all MD trajectories computed in parallel, induces redistribution of the hydrogen bonds and salt bridges formed at the interface between the two subunits. 88

Figure 25. Population of the hydrogen bonds (a) formed between the opposing amino acids of the aspartoacylase transport channel (b). 88
Download figure:
Standard image
Figure 26. Shift of one subunit (ASPA) of the aspartoacylase dimer after the 13 ns MD trajectory. 88
Download figure:
Standard imageThe loss of structural symmetry in the hAsp homodimer changes the dynamic properties of the shifted subunit (ASPA). In the subunit with the least RMSD (ASPB), the distribution of distances between the centres of mass of active site gates corresponds to the closed-gate conformation, i.e., the substrate access to the ASPB active site is hampered. Conversely, in the shifted subunit (ASPA), the open form is stabilized (Fig. 27). 88

Figure 27. Distribution of distances between the centres of mass of loops 62 – 74 and 282 – 294 for the aspartoacylase monomer and ASPA and ASPB subunits of the dimer. 88
Download figure:
Standard imageThe interaction between two hAsp subunits affects the catalytic activity of the enzyme. The pocket sandwiched between the two protein (structurally identical) macromolecules is promising for allosteric binding. 88
The effect of symmetry of an enzyme system on the catalytic function is comprehensively described in modern scientific literature. Systems in which the catalytic function can be implemented only with increasing symmetry of protein complexes are encountered most often. 93 However, cases of opposite dependence are also known. 94 Apparently, the hAsp system refers to the latter case.
4.1. Allosteric inhibition site
The modern description of the effect of allostery implies, first, that the addition of a regulator shifts the equilibrium between ensembles of states; 95 second, in the general case, allosteric regulation is not always associated with a change in the macromolecular geometry. 96 The perturbation generated on the enzyme surface by a point mutation, post-translational modification or binding to an organic ligand propagates over the whole protein structure by numerous signalling pathways. The allosteric network of biological signal transmission implies the existence of more than one allosteric site. 97 Indeed, the presence of an extensive set of point mutations that affect the catalytic function of hAsp, together with high probability of influence of the interaction between ASPA and ASPB on the activity attests to the presence of numerous regulatory sites on the hAsp surface.
Human aspartoacylase is inhibited with increasing NAA concentration; hence, an allosteric inhibition site should either have lower NAA affinity than the active site or have higher activation energy.
4.1.1. Search for the allosteric inhibition site
Among the modern computer programs for the search for additional binding sites, note a large class of simple and efficient algorithms used to analyze the surface of a static system. They can be conventionally divided into programs that examine the protein surface for the presence of known structural motifs of allosteric sites and programs that determine the potential binding capacity of existing pockets by evaluating the affinity to a potential ligand. However, the structure and geometry of the pockets on the enzyme surface play a crucial role for binding to the ligand molecule. 92
The algorithm used in the Allosite software 98 correlates the amino acid composition and the surface geometry of a protein with known allosteric sites included in the Allosteric Site Database. 99 The areas selected in this way are tested for geometric matching to the hypothetical ligand molecule. 100 The application of the Allosite package identified three potential binding sites other than the ASPA and ASPB active sites. The AS1, AS2 and AS3 sites altogether occupy the whole space between the two hAsp subunits (Fig. 28).

Figure 28. Position of the allosteric site identified by means of Allosite software. 88
Download figure:
Standard imageThe identification of NAA binding site and testing of the results were performed using the SiteMap package algorithm, 101 which evaluates the interaction energy between the functional groups of the selected site and the substrate. The scoring functions of the SiteScore and DScore methods include evaluation of the site geometry, size and accessibility and take account of the negative contribution of the enhanced hydrophilicity of the detected site. Two out of the six sites found by SiteMap are also hAsp active sites. The positions of other areas coincide with AS1 and AS2: the SM1 area is also located between ASPA and ASPB, encloses SM2 and partly overlaps with AS1, being slightly inferior in the volume.
According to the results of studies of the hAsp dimer surface by two classes of algorithms, the most promising area for NAA binding is located at the interface between two subunits, which is consistent with the results of analysis of apo-hAsp dynamics. However, the detected pocket is too large for unambiguous identification of the NAA binding site. The site was identified by molecular docking carried out using Autodock4.2 software. 102
According to molecular docking results, the highest NAA affinity is inherent in the site located between ASPA and ASPB, on the ASPB surface (Fig. 29). The free binding energy was estimated as −4.7 kcal mol−1. The population of this site, according to docking, was 78%, while the next most populated site (11%) had a binding energy of −2.6 kcal mol−1. Thus, docking identified one putative site of hAsp allosteric regulation on the surface between ASPA and ASPB.

Figure 29. Position of the putative allosteric regulation site. 78
Download figure:
Standard imageIn order to confirm the possibility of spontaneous NAA binding in the detected site, the accelerated MD trajectory 103 of 50 ns length was calculated for the hAsp dimer with ten NAA molecules located at distances not less than 10 Å from the protein surface. During 35% of the simulation time, the inhibition site was linked to the substrate molecule, which supports the hypothesis of high probability of complex formation at high substrate concentrations. 88
4.1.2. Molecular dynamics of the hAsp:NAA(inh) complex
Five parallel 100 ns MD trajectories were simulated for the hAsp complex with NAA in the allosteric inhibition site. The trajectories were used to calculate the root-mean-square deviations for the loops that form the entrance of the hAsp active site. Comparison of the results with the values obtained previously for apo-hAsp showed that binding in the detected allosteric site decreases the gate mobility in the ASPA subunit, while the ASPB mobility is retained upon NAA binding. 88 The formation of the hAsp:NAA(inh) complex leads to stabilization of the closed form not only in ASPB, but also in ASPA (Fig. 30). As a result, the probability of formation of the catalytically active protein – substrate (ES) complex decreases.

Figure 30. Distribution of distances between the centres of mass of opposing loops 62 – 74 and 282 – 294 in the hAsp:NAA(inh) complex. 88
Download figure:
Standard imageThe fact that the allosteric site occurs on the surface of one monomer, while the effect is manifested on the surface of the other monomer, requires a more detailed description. The search for signalling pathways in macromolecular complexes can be carried out by a new method called dynamical network analysis. 104
4.1.3. Dynamical network analysis
Network analysis is generally used to describe and predict the dynamic properties of complex systems. It is a part of graph theory allowing not only splitting of data arrays into separate groups, but also finding the most optimal path linking any two network elements.
For the equilibrium trajectories of the apo-hAsp and hAsp:NAA(inh) systems, network analysis was carried out using the NetworkAnalyser extension of the VMD package. 105 Clustering was accomplished by the Girvan – Newman method, 106 and the signalling pathways were determined using the Floyd – Warshall algorithm. 107 Further analysis of the calculated graph was performed using the Cytoscape3 software. 108
All Cα atoms of the hAsp dimer were used as graph nodes. This approach proved to be efficient in the description of ways of signal transmission from the allosteric site to the active site. 109 The graph edges were specified by equilibrium MD trajectories of the systems. For each pair of Cα atoms, the absolute values of correlation (Cij ) were found from the results of 20 ns equilibrium MD trajectory [expression (13)].
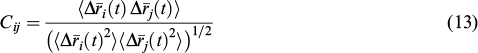
where  is the position of the Cα atom of the i th graph node and t is time.
is the position of the Cα atom of the i th graph node and t is time.
The calculated value was then substituted into the function that converted Cij to the edge length [dij , Eqn (14)].

The graph describing the system dynamics included only the edges that linked nodes separated by distances of not more than 4.5 Å throughout 75% of the trajectory length.
The use of weighted values for edges makes it possible to propose possible signalling pathways between the remote protein regions, assuming the pathways minimizing the distances in the graph space to be most probable. The total length of the signalling pathway in the network space is determined as the sum of edge lengths.
Particular communities characterized by enhanced degree of correlation of motion and close positions of nodes can be distinguished in the dynamical network of the protein. The signal transmission between particular communities involves so-called critical nodes with a high degree of degeneracy. The critical nodes transmit a large flux of information; therefore, this is the key target for the allosteric signal interruption.
4.1.4. Community analysis of apo-hAsp and hAsp:NAA(inh) networks
In the apo-hAsp system, eight communities were distinguished, three of them combining the ASPA and ASPB nodes, indicating a highly correlated motion of the two subunits.
In the hAsp:NAA(inh) complex, the number of communities decreases to seven, which may be a consequence of enhanced interactions in the system. Three communities combine the ASPA and ASPB nodes in the same way, one of them belonging to the NAA allosteric binding site. 88
Splitting of the dynamical network into communities should reflect, in particular, its catalytic properties. For instance, in apo-hAsp, two of the three communities located between ASPA and ASPB contain also amino acid residues of the monomer A active site: Glu24, Glu178 and His116. This clustering attests to enhanced influence of the interaction between the subunits on these residues. 88
Analysis of the edges formed between ASPA and ASPB in both systems confirmed the assumptions based on the calculated hydrogen bond and salt bridge populations. When NAA is bound in the allosteric inhibition site, the degree of interaction between two subunits increases and non-covalent interactions between ASPA and ASPB are reorganized. Out of the five critical nodes common to the subunits, only two nodes, Arg196 and Lys200, are retained upon binding. The number of critical nodes in ASPA doubles, with their ratio being restored as NAA is removed. 89
The distribution of amino acids of the active site gate over the communities is also different for apo-hAsp and for the inhibitor-containing system. In the apo-form, residues 62 – 74 and 282 – 294 are distributed among four communities with no common critical nodes, which attests to a reduced degree of correlation and chaotic nature of their motion.
Meanwhile, in the regulated system, the opposite loops belong to two opposing communities connected by two pairs of critical nodes.
The results of analysis of the critical nodes at the active site gate in ASPA were used to identify the key directions of loop motion: in the apo-form, the critical node Arg63 is connected by a graph edge to the critical node Ala57 located opposite to loop 282 – 294, i.e., motion of the backbone of loop 62 – 74 is directed towards the open-gate conformation (Fig. 31 a).

Figure 31. Critical nodes of loops 62 – 74 and 282 – 294 in apo-hAsp (a) and in the hAsp:NAA(inh) complex (b). 88
Download figure:
Standard imageMeanwhile, in the case of hAsp:NAA(inh), the node Arg63 is connected to the critical node Ala287, which belongs to the loop of the opposite part of the gate (see Fig. 31 b); thus, as a result of binding, motion of loop 62 – 74 is directed towards the closed-gate conformation.
4.1.5. Signal transmission pathways from the allosteric site to the active site gate
The optimal signal transmission pathways from the node most proximate to NAA in the inhibition site (Tyr289 ASPB) to the active site gate nodes (ASPA, residues 62 – 74 and 282 – 294) were calculated for the obtained graphs. All optimal pathway lengths substantially decrease upon substrate binding, while the number of pathways increases (Fig. 32). 88

Figure 32. Number of signalling pathways (N) from the ASPA gate to Tyr289 (ASPB) (a) and their length (b) in apo-hAsp and hAsp:NAA(inh). 88
Download figure:
Standard imageDissociation of hAsp:NAA(inh) induces restoration of characteristics of the apo-form. The structure perturbation caused by binding in the inhibition site on the ASPB inner surface is transmitted to the ASPA active site gate along numerous diverging pathways and shifts equilibrium towards the closed form.
In conclusion, it was found how often the signalling pathways pass through the known clinically significant point mutations of hAsp in positions 231, 285, 288 and 295. The position of the most frequent mutation of the Canavan disease (E285A) is involved in 10% of signal transmission pathways from the allosteric inhibition site to the hAsp gate. The residues Tyr231 and Tyr288 are involved in 20% of the pathways.
4.2. Allosteric activation site
The presence of self-activation effect in the hAsp – NAA system was assumed owing to the sigmoidal form of the initial part of the kinetic curve for the enzyme activity νs. substrate concentration. 33
The hAsp surface was examined for the presence of the activation binding site using the classical molecular dynamics techniques. According to the data of kinetic experiment, the self-activation effect is observed even at substrate concentrations starting from 20 μmol litre−1, i.e., the allosteric activation site should possess a higher substrate affinity than the active site and binding should occur without steric hindrance caused by neighbouring amino acid residues. Therefore, in the presence of several NAA molecules in solution, the hAsp:NAA(act) complex should form spontaneously. For the search for activation sites, the molecular dynamics trajectories of 250 ns length were calculated for the hAsp dimer with 15 NAA molecules separated from the dimer surface by not less than 15 Å. The initial geometry of hAsp corresponded to the optimized structure used in the calculations of the apo-form. After 2 ns, two NAA molecules occupied structurally symmetrical sites composed of positively charged residues Arg56, Lys59 and Lys60 at the interface between ASPA and ASPB; the populations of these sites were 93% and 98% along the trajectories for ASPA and ASPB, respectively. 89
The positions of NAA molecules in the identified binding sites were determined more accurately by molecular docking technique using the Autodock4.2 and FlexX software (Fig. 33); the estimated binding energy was −6.5 kcal mol−1.

Figure 33. Position of the potential aspartoacylase activation site. 89
Download figure:
Standard imageThe dissociation constants for the hAsp:NAA(inh) and hAsp:NAA(act) systems calculated from the docking data differ by factors of 17 – 20; hence, binding in the identified sites is preferable over binding in the inhibition site. This is in line with experimental data indicating the presence of activation effect at low NAA concentrations.
The identified allosteric sites are located on the protein surface. This position precludes the use of higher-level methods for calculation of free energy. Khrenova et al. 89 used only free energy ratios rather than absolute values, which accounts for the excellent agreement between the theoretical and experimental data.
Molecular dynamics investigation of the possible dynamic binding in the sites revealed the presence of some activation: when NAA is bound in the site on the ASPB surface, the distribution of distances between the centres of mass of loops 62 – 74 and 282 – 294, which form the active site gate, shifts towards greater values than in the apo-form, while the position of ASPB gate remains invariable (Fig. 34). 89

Figure 34. Distribution of distances between the centres of mass of opposing loops 62 – 74 and 282 – 294 in the hAsp:NAA(act) complex. 89
Download figure:
Standard imageDespite NAA binding in both structurally identical sites, the self-activation effect is again observed only for the ASPA active site. Examination of the results of network analysis showed that in the dynamical network of hAsp:NAA(act), the residue Lys60 corresponding to the ASPB activation site is located in the same community as loop 282 – 294 of the ASPA subunit, while the critical edge Lys60−Thr62, which passes through it, connects them to the amino acid residues of the site, which confirms the hypothesis of perturbation transmission from the site in ASPB to the transport channel gate in ASPA.
The activation site and the inhibition site are located at the interface between two subunits at ∼15 Å distance from each other. The close positions of the allosteric regulation sites should imply a similar nature of the signal transmitted to the active site; however, in the given system, all results obtained in the previous stages indicate the opposite. Study of the most populated signal trasmission pathways from the graph nodes corresponding to allosteric sites and their environment demonstrated that the inhibiting and activating signals are transmitted along different non-crossing pathways to the ASPA gate nodes. 89
Thus, computer simulation of the behaviour of protein globules and the relevant analysis provides an adequate picture of the processes and interprets the molecular nature of complex regulatory effects.
5. Conclusion
Aspartoacylase is a key enzyme in the human central nervous system. The high-resolution structure of the protein has now been established, the active site has been identified, and information on the post-translational protein modification has been gained. Genetic polymorphs of the protein (molecular polymorphism) were detected in the human population. Many of the polymorphs are responsible for the development of various pathologies. The most well known polymorphic modifications are associated with the Canavan disease.
The knowledge of the protein and active site structures and the use of advanced techniques of molecular mechanics, molecular dynamics and quantum mechanics – molecular mechanics combination make it possible to model the catalytic cycle, gain information about the structure of all intermediates of the catalytic event and determine the experimental characteristics for all elementary steps, including rate-limiting and superfast non-limiting steps. The investigation of aspartoacylase is an example demonstrating a fundamentally new level of molecular modelling.
Human brain aspartoacylase possesses a unique mechanism of catalytic activity regulation, including activation and inhibition by the substrate. This regulation mechanism is apparently physiologically important and determines the energetic and neurotransmitter behaviour of neurons. Description of the consequences of this type of regulation would require additional research. Furthermore, the mechanism of action of clinically significant point mutations located outside the active site remains obscure. The identical structures of active sites in the native and mutant forms suggest that their action is of allosteric nature. The mechanism of macromolecular signal transmission from the allosteric activation and inhibition sites to the enzyme catalytic site is currently known. This information was collected using the unique potential of supercomputer simulation.
This review was prepared with the partial financial support of the Russian Science Foundation (Project No. 18-13-00030).
The research is carried out using equipment of the shared research facilities of HPC computing resources at Lomonosov Moscow State University. The authors are grateful to the Joint Supercomputer Centre of the Russian Academy of Sciences for providing computing resources to perform studies the results of which are included in the review.
Footnotes
- †
All figures were drawn by the authors using data of the references indicated in the figure captions.