
Crystal plasticity is mediated through dislocations, which form knotted configurations in a complex energy landscape. Once they disentangle and move, they may also be impeded by permanent obstacles with finite energy barriers or frustrating long-range interactions. The outcome of such complexity is the emergence of dislocation avalanches as the basic mechanism of plastic flow in solids at the nanoscale. While the deformation behavior of bulk materials appears smooth, a predictive model should clearly be based upon the character of these dislocation avalanches and their associated strain bursts. We provide here a comprehensive overview of experimental observations, theoretical models and computational approaches that have been developed to unravel the multiple aspects of dislocation avalanche physics and the phenomena leading to strain bursts in crystal plasticity.
Focus on MMM 2016
Guest Editors
Eric Cancès École des Ponts ParisTech, France
Benoit Devincre CNRS/ONERA, France
Olivier Politano Université de Bourgogne, France
David Rodney Université Claude Bernard Lyon 1, France
François Willaime CEA, DEN, France
Scope
This focus collection reflects the important domains of research presented during the Multiscale Materials Modeling (MMM) 2016 conference, including recent developments in numerical modelling of materials devoted to processes involving multiple scales. In particular, this collection emphasizes the importance of tackling the physical, mathematical, and computational challenges presented by the need for quantitative analysis of large and complex systems as well as the rapidly increasing amount of data coming from experiments.
Articles submitted for inclusion in the MMM 2016 focus collection were subject to the standard MSMSE editorial standards and peer review process.
Preface
Topical Review
Papers
The core structure of  -type screw dislocations in hexagonal close packed titanium is investigated computationally using periodic supercells with quadrupolar configurations in combination with density functional theory (DFT) and a modified embedded atom method (MEAM) classical potential. Two arrangements of the quadrupolar supercell configurations are examined, and within each arrangement two initial dislocation positions are compared. (Meta)stable pyramidal and prismatic dislocation core structures exist within both DFT and MEAM methods, and the relaxed structure from a given configuration resulting from our anisotropic elasticity theory solution depends only on the assumed initial dislocation positions. Within DFT we find the ground state core structure to be spread on the pyramidal plane. We find that it is necessary to include the semi-core 3p electrons as valence states in the DFT calculations in order to converge the ground state dislocation core configuration and difference in energy between structures. In terms of k-point sampling, it is found that at least a
-type screw dislocations in hexagonal close packed titanium is investigated computationally using periodic supercells with quadrupolar configurations in combination with density functional theory (DFT) and a modified embedded atom method (MEAM) classical potential. Two arrangements of the quadrupolar supercell configurations are examined, and within each arrangement two initial dislocation positions are compared. (Meta)stable pyramidal and prismatic dislocation core structures exist within both DFT and MEAM methods, and the relaxed structure from a given configuration resulting from our anisotropic elasticity theory solution depends only on the assumed initial dislocation positions. Within DFT we find the ground state core structure to be spread on the pyramidal plane. We find that it is necessary to include the semi-core 3p electrons as valence states in the DFT calculations in order to converge the ground state dislocation core configuration and difference in energy between structures. In terms of k-point sampling, it is found that at least a  k-point mesh is necessary to converge the dislocation core structure for a supercell one Burgers vector deep. Use of higher k-point densities or inclusion of additional semi-core electronic states as valence electrons results in the same core structure. With the MEAM potential considered in this work, we find the ground state core configuration to be spread predominantly on the prismatic plane, in contrast with the DFT results.
k-point mesh is necessary to converge the dislocation core structure for a supercell one Burgers vector deep. Use of higher k-point densities or inclusion of additional semi-core electronic states as valence electrons results in the same core structure. With the MEAM potential considered in this work, we find the ground state core configuration to be spread predominantly on the prismatic plane, in contrast with the DFT results.
A combination of dynamic transmission electron microscopy (DTEM) experiments and CALPHAD-informed phase-field simulations was used to study rapid solidification in Cu–Ni thin-film alloys. Experiments—conducted in the DTEM—consisted of in situ laser melting and determination of the solidification kinetics by monitoring the solid–liquid interface and the overall microstructure evolution (time-resolved measurements) during the solidification process. Modelling of the Cu–Ni alloy microstructure evolution was based on a phase-field model that included realistic Gibbs energies and diffusion coefficients from the CALPHAD framework (thermodynamic and mobility databases). DTEM and post mortem experiments highlighted the formation of microsegregation-free columnar grains with interface velocities varying from ∼0.1 to ∼0.6 m s−1. After an 'incubation' time, the velocity of the planar solid–liquid interface accelerated until solidification was complete. In addition, a decrease of the temperature gradient induced a decrease in the interface velocity. The modelling strategy permitted the simulation (in 1D and 2D) of the solidification process from the initially diffusion-controlled to the nearly partitionless regimes. Finally, results of DTEM experiments and phase-field simulations (grain morphology, solute distribution, and solid–liquid interface velocity) were consistent at similar time (μs) and spatial scales (μm).
We compare Rényi entropy-based mesoscale approaches for characterizing 2D polycrystalline network topology and geometry, based on the grain number of sides and grain areas, respectively. We study the effect of microstructure disorder on mechanical properties such as elastic and damage response by performing simulations of quasi-static uniaxial compression loading tests on an idealized material using grain-level micro-mechanical discrete element model. While not comprehensive enough to make general conclusions, this study allows us to make observations about the sensitivity of mechanical parameters such as Young's modulus, proportional limit, first yield stress, toughness and amount of microstructure damage to different entropy measures.
Atomistic tight-binding based simulations are widely used to study transition metal alloys properties. However, they still require to be improved if one aims at modelling segregation and ordering phenomena in the case of magnetic materials, since they generally rely on local charge neutrality rules per site, valence orbital and element, but not per spin! We propose here a strategy to overcome this difficulty, that we illustrate in two magnetic systems of particular interest: CoPt and FeNi alloys.
Metastable configurations in condensed matter typically fluctuate about local energy minima at the femtosecond time scale before transitioning between local minima after nanoseconds or microseconds. This vast scale separation limits the applicability of classical molecular dynamics (MD) methods and has spurned the development of a host of approximate algorithms. One recently proposed method is diffusive MD which aims at integrating a system of ordinary differential equations describing the likelihood of occupancy by one of two species, in the case of a binary alloy, while quasistatically evolving the locations of the atoms. While diffusive MD has shown itself to be efficient and provide agreement with observations, it is fundamentally a model, with unclear connections to classical MD. In this work, we formulate a spin-diffusion stochastic process and show how it can be connected to diffusive MD. The spin-diffusion model couples a classical overdamped Langevin equation to a kinetic Monte Carlo model for exchange amongst the species of a binary alloy. Under suitable assumptions and approximations, spin-diffusion can be shown to lead to diffusive MD type models. The key assumptions and approximations include a well-defined time scale separation, a choice of spin-exchange rates, a low temperature approximation, and a mean field type approximation. We derive several models from different assumptions and show their relationship to diffusive MD. Differences and similarities amongst the models are explored in a simple test problem.
A highly efficient simulation model for 2D and 3D grain growth was developed based on the level-set method. The model introduces modern computational concepts to achieve excellent performance on parallel computer architectures. Strong scalability was measured on cache-coherent non-uniform memory access (ccNUMA) architectures. To achieve this, the proposed approach considers the application of local level-set functions at the grain level. Ideal and non-ideal grain growth was simulated in 3D with the objective to study the evolution of statistical representative volume elements in polycrystals. In addition, microstructure evolution in an anisotropic magnetic material affected by an external magnetic field was simulated.
Using ab initio density functional theory calculations, we investigate the effect of interstitial carbon solutes on  screw dislocations in non-magnetic body-centered cubic transition metals from group 5 (V, Nb, Ta) and group 6 (Mo, W). The two groups are found to display different solute–dislocation interaction behaviors. Group 6 shows a core reconstruction similar to that previously reported in Fe(C): the dislocation adopts a hard-core configuration with the carbon atoms at the center of regular trigonal prisms formed by the metal atoms. The solute–dislocation interaction energies are strongly attractive, ranging from −1.3 to −1.9 eV depending on the metal and the carbon–carbon distance. By way of contrast, the configuration of lowest energy in group 5 consists of the dislocation in its easy core and the carbon atom in a fifth nearest neighbor octahedral site. The configuration is attractive, but less than in group 6. We show that this group dependence is consistent with the carbon local environment in the stable stoichiometric carbide structures, namely cubic NaCl-type for group 5 and hexagonal WC-type for group 6: in both cases the carbon atoms are at the center of octahedra and prisms respectively.
screw dislocations in non-magnetic body-centered cubic transition metals from group 5 (V, Nb, Ta) and group 6 (Mo, W). The two groups are found to display different solute–dislocation interaction behaviors. Group 6 shows a core reconstruction similar to that previously reported in Fe(C): the dislocation adopts a hard-core configuration with the carbon atoms at the center of regular trigonal prisms formed by the metal atoms. The solute–dislocation interaction energies are strongly attractive, ranging from −1.3 to −1.9 eV depending on the metal and the carbon–carbon distance. By way of contrast, the configuration of lowest energy in group 5 consists of the dislocation in its easy core and the carbon atom in a fifth nearest neighbor octahedral site. The configuration is attractive, but less than in group 6. We show that this group dependence is consistent with the carbon local environment in the stable stoichiometric carbide structures, namely cubic NaCl-type for group 5 and hexagonal WC-type for group 6: in both cases the carbon atoms are at the center of octahedra and prisms respectively.
In this paper we propose a novel approach to accelerate the multi-scale simulation of metal plasticity. In macroscopic zones of nearly homogeneous strain responses, the evolution of plastic anisotropy at each finite element integration point can be approximated from the properties at a representative point within a zone. We show how these zones can be identified by a clustering algorithm and can be utilised to reduce the computational cost of the simulation. We present and analyse the results obtained for two test cases.
Structure maps predict the crystal structure of a compound from the knowledge of constituent elements and chemical composition. We recently developed a highly predictive, three-dimensional structure map for stoichiometric binary sp-d-valent compounds. Here we show that the descriptors of this structure map are transferable to off-stoichiometric compounds with similar predictive power. We furthermore demonstrate that the descriptors are suitable for ternary prototypes. In particular, we construct a three-dimensional structure map for 129 prototypical crystal structures for ternary compounds. The crystal structure is predicted correctly with a probability of 78%. With a confidence of 95% the correct crystal structure is among the three most likely crystal structures predicted by the structure map.
Molten salts have been proposed as heat carrier media in the nuclear and concentrating solar power plants. Due to their high melting temperature, solidification of the salts is expected to occur during routine and accidental scenarios. Furthermore, passive safety systems based on the solidification of these salts are being studied. The following article presents new developments in the modeling of eutectic molten salts by means of a multiphase, multicomponent, phase-field model. Besides, an application of this methodology for the eutectic solidification process of the ternary system LiF–KF–NaF is presented. The model predictions are compared with a newly developed semi-analytical solution for directional eutectic solidification at stable growth rate. A good qualitative agreement is obtained between the two approaches. The results obtained with the phase-field model are then used for calculating the homogenized properties of the solid phase distribution. These properties can then be included in a mixture macroscale model, more suitable for industrial applications.
In this paper, we study the dependence of neighboring grain orientation on the local stress state around a deformation twin in a hexagonal close packed (HCP) crystal and its effects on the resistance against twin thickening. We use a recently developed, full-field elasto-visco-plastic formulation based on fast Fourier transforms that account for the twinning shear transformation imposed by the twin lamella. The study is applied to Mg, Zr and Ti, since these HCP metals tend to deform by activation of different types of slip modes. The analysis shows that the local stress along the twin boundary are strongly controlled by the relative orientation of the easiest deformation modes in the neighboring grain with respect to the twin lamella in the parent grain. A geometric expression that captures this parent-neighbor relationship is proposed and incorporated into a larger scale, mean-field visco-plastic self-consistent model to simulate the role of neighboring grain orientation on twin thickening. We demonstrate that the approach improves the prediction of twin area fraction distribution when compared with experimental observations.
Welding is one of the most wide-spread processes used in metal joining. However, there are currently no open-source software implementations for the simulation of microstructural evolution during a weld pass. Here we describe a Potts Monte Carlo based model implemented in the SPPARKS kinetic Monte Carlo computational framework. The model simulates melting, solidification and solid-state microstructural evolution of material in the fusion and heat-affected zones of a weld. The model does not simulate thermal behavior, but rather utilizes user input parameters to specify weld pool and heat-affect zone properties. Weld pool shapes are specified by Bézier curves, which allow for the specification of a wide range of pool shapes. Pool shapes can range from narrow and deep to wide and shallow representing different fluid flow conditions within the pool. Surrounding temperature gradients are calculated with the aide of a closest point projection algorithm. The model also allows simulation of pulsed power welding through time-dependent variation of the weld pool size. Example simulation results and comparisons with laboratory weld observations demonstrate microstructural variation with weld speed, pool shape, and pulsed-power.
In this paper, we use a multi-levels modeling approach to describe the elaboration of directly integrated energetic materials. The deposition of copper oxide on aluminum substrate is described. Atomic scale calculations are first conducted to identify local mechanisms involved during the growth of CuO on Al(111). These atomic scale data are then used to parameterize a macroscopic code, inspired on a kinetic Monte Carlo methodology dedicated to simulate vapor like deposition process. The objective is to establish the link between the microstructure of materials and the way they are achieved, i.e. the process parameters such as temperature and gas pressure. This work is conducted in the context of the integration of nano-structured energetic thermites used as micro energy source in microelectronic devices. We show that the temperature of the deposition process appears as the driving parameter to tailor the thickness of interfacial layers.
In the current work we present an extensive study on the impact of short- and long-range interactions between solids and liquids on the hydrodynamic and lubrication behaviour of a tribological system. We have implemented a coarse grain molecular dynamics description of two ionic liquids (ILs) as lubricants which are confined by two infinitely long flat iron solids and which are subjected to a shearing flow. The impact of surface polarizability and molecule geometry on the ion arrangement under shearing has been studied in detail. The results have revealed two regimes of lubrication, with a liquid phase being present under low normal loads, while solidification of the ILs, accompanied by a steep rise of normal forces and significant wall slip is observed at small plate-to-plate distances.
Composite materials consisting of hard particles in a ductile metallic matrix are of major interest since their strength and deformability can be dramatically changed by varying volume fraction, size and shape of the particles. Understanding dislocation motion in composite materials as the cause of plastic deformation therefore is an important task. Recently, advanced dislocation-based continuum theories of plasticity have been developed for performing meaningful averages over systems of straight and curved dislocation lines in a continuum approach. In this paper, we focus on a single slip heterogeneous microstructure and investigate how the dislocation interactions can be represented in an averaged dislocation density based continuum description. The representation of strong dislocation density gradients is discussed in the context of a formulation, which aims at a coarse-grained resolution. We introduce a set of dislocation density evolution equations which account for the formation and dissolution of dislocation dipoles. By applying the model to a composite structure, we demonstrate that the dislocation density based description can well describe the physical processes in the microstructure and a comparison to discrete dislocation dynamics simulations shows good agreement for the relaxation behavior of the considered composites.
Structural, thermodynamic, atomic and thermal transport properties of solid and liquid phases of the Ni–Al system were studied by means of MD simulations using three embedded-atom method (EAM) potentials developed by Mishin and colleagues (Mishin et al 2002 Phys. Rev. B 65 224114; Mishin 2004 Acta Mater. 52 145167; Purja Pun and Mishin 2009 Phil. Mag. 89 32453267). The extracted properties (lattice parameter, enthalpy, heat capacity, mass diffusivity and thermal conductivity) were compared with experimental data. The limitations of EAM potentials for studying different aspects of reactivity were assessed for each potential separately.
Using the framework of density functional theory, the structural and energetic response of two face-centred cubic (fcc) Al grain boundaries (GBs) to combined tension and shear loadings has been examined. It is shown that tension will serve to inhibit the Σ5 [100] 36.87° twist GB response to shear in a mixed-mode loading scenario, by increasing the difference in structural environments for inequivalent atoms at the GB plane. We propose that the presence of such atoms, rather than the full structural details of the GB structure, is instrumental in triggering this tension–shear interplay. As support for this hypothesis, we compute the Σ3 [-110] (111) 60° symmetric tilt GB mixed-mode loading response. Here, all atoms at the GB plane are equivalent, and the qualitative shear energy variation is unaffected by tension. Our findings indicate that general fcc Al GBs may display a stronger shear energy variation at larger levels of tension, contrasting general expectations. The implications to GB breakage are discussed.
In this study, sequential sputter deposition, diffusion bonding and focused ion beam milling are used to fabricate sapphire micropillars encapsulating a thin single crystal niobium film. A distinct Bauschinger effect is observed during the cyclic axial compression of the samples. Plain strain discrete dislocation plasticity is used to interpret the experimental results obtained for the encapsulated film-micropillar geometry. The simulations show that the experimental samples correspond to a saturated source density regime, producing the maximum Bauschinger effect for the chosen mean nucleation strength. Next, the source density and mean nucleation strength are shown to have a coupled effect on the size of the Bauschinger effect, understood in terms of the differing number of pile-ups occurring per source in the film. The coupled effect is found to be represented by the density of dislocations annihilated upon unloading: a consistent linear relationship is observed between the size of the Bauschinger effect and the annihilated dislocation density over the entire source density and nucleation strength parameter space investigated. It is found that different film orientations fulfil the same linear relationship, whereas changing the film thickness causes the slope of the linear trend to vary suggesting a length-scale dependence on reverse plasticity. Finally, all results are found to be unified by a power-law relationship quantifying the Bauschinger effect of the form  where it is argued that the number of dislocations undergoing reverse glide in the confined film is represented by
where it is argued that the number of dislocations undergoing reverse glide in the confined film is represented by  , the mean free path of dislocations by l and the effect of hardening processes by the exponent n. The net reverse glide is thus represented by
, the mean free path of dislocations by l and the effect of hardening processes by the exponent n. The net reverse glide is thus represented by  which can be used as a measure of the Bauschinger effect.
which can be used as a measure of the Bauschinger effect.
Knowledge of the deformation mechanisms of (Mg,Fe)2SiO4 olivine is important for the understanding of flow and seismic anisotropy in the Earth's upper mantle. We report here a numerical modelling at the atomic scale of dislocation structures and slip system properties in Mg2SiO4 forsterite. Our study focuses on screw dislocations of [100] and [001] Burgers vectors. Computations are performed using the so-called THB1 empirical potential set for Mg2SiO4. Results of dislocation core structures highlight the primary importance of the (010) plane for [100] slip dislocations. For [001] dislocations, we confirm the occurrence of a stable narrow core that evolves into transient planar configurations to glide in (100) and (010). Such configurations suggest a locking–unlocking mechanism.
Different molecular dynamics methods like the direct method, the Green–Kubo (GK) method and homogeneous non-equilibrium molecular dynamics (HNEMD) method have been widely used to calculate lattice thermal conductivity ( ). While the first two methods have been used and compared quite extensively, there is a lack of comparison of these methods with the HNEMD method. Focusing on the underlying computational parameters, we present a detailed comparison of the GK and HNEMD methods for both bulk and vacancy Si using the Stillinger–Weber potential. For the bulk calculations, we find both methods to perform well and yield
). While the first two methods have been used and compared quite extensively, there is a lack of comparison of these methods with the HNEMD method. Focusing on the underlying computational parameters, we present a detailed comparison of the GK and HNEMD methods for both bulk and vacancy Si using the Stillinger–Weber potential. For the bulk calculations, we find both methods to perform well and yield  within acceptable uncertainties. In case of the vacancy calculations, HNEMD method has a slight advantage over the GK method as it becomes computationally cheaper for lower
within acceptable uncertainties. In case of the vacancy calculations, HNEMD method has a slight advantage over the GK method as it becomes computationally cheaper for lower  values. This study could promote the application of HNEMD method in
values. This study could promote the application of HNEMD method in  calculations involving other lattice defects like nanovoids, dislocations, interfaces.
calculations involving other lattice defects like nanovoids, dislocations, interfaces.
This work develops a method for calibrating a crystal plasticity model to the results of discrete dislocation (DD) simulations. The crystal model explicitly represents junction formation and annihilation mechanisms and applies these mechanisms to describe hardening in hexagonal close packed metals. The model treats these dislocation mechanisms separately from elastic interactions among populations of dislocations, which the model represents through a conventional strength-interaction matrix. This split between elastic interactions and junction formation mechanisms more accurately reproduces the DD data and results in a multi-scale model that better represents the lower scale physics. The fitting procedure employs concepts of machine learning—feature selection by regularized regression and cross-validation—to develop a robust, physically accurate crystal model. The work also presents a method for ensuring the final, calibrated crystal model respects the physical symmetries of the crystal system. Calibrating the crystal model requires fitting two linear operators: one describing elastic dislocation interactions and another describing junction formation and annihilation dislocation reactions. The structure of these operators in the final, calibrated model reflect the crystal symmetry and slip system geometry of the DD simulations.
Experiments and atomic-scale simulations suggest that the transmission of plasticity carriers in deforming amorphous–crystalline nanolaminates is mediated by the biphase interface between the amorphous and crystalline layers. In this paper, we present a micromechanics model for these biphase nanolaminates that describes defect interactions through the amorphous–crystalline interface (ACI). The model is based on an effective-temperature framework to achieve a unified description of the slow, configurational atomic rearrangements in both phases when driven out of equilibrium. We show how the second law of thermodynamics constrains the density of defects and the rate of configurational rearrangements, and apply this framework to dislocations in crystalline solids and shear transformation zones (STZs) in amorphous materials. The effective-temperature formulation enables us to interpret the observed movement of dislocations to the ACI and the production of STZs at the interface as a 'diffusion' of configurational disorder across the material. We demonstrate favorable agreement with experimental findings reported in (Kim et al 2011 Adv. Funct. Mater. 21 4550–4), and demonstrate how the ACI acts as a sink of dislocations and a source of STZs.
The Green's function molecular dynamics (GFMD) method for the simulation of incompressible solids under normal loading is extended in several ways: shear is added to the GFMD continuum formulation and Poisson numbers as well as the heights of the deformed body can now be chosen at will. In addition, we give the full stress tensor inside the deformed body. We validate our generalizations by comparing our analytical and GFMD results to calculations based on the finite-element method (FEM) and full molecular dynamics simulations. For the investigated systems we observe a significant speed-up of GFMD compared to FEM. While calculation and proof of concept were conducted in two-dimensions only, the methodology can be extended to the three-dimensional case in a straightforward fashion.
Fitted interatomic potentials are widely used in atomistic simulations thanks to their ability to compute the energy and forces on atoms quickly. However, the simulation results crucially depend on the quality of the potential being used. Force matching is a method aimed at constructing reliable and transferable interatomic potentials by matching the forces computed by the potential as closely as possible, with those obtained from first principles calculations. The potfit program is an implementation of the force-matching method that optimizes the potential parameters using a global minimization algorithm followed by a local minimization polish. We extended potfit in two ways. First, we adapted the code to be compliant with the KIM Application Programming Interface (API) standard (part of the Knowledgebase of Interatomic Models project). This makes it possible to use potfit to fit many KIM potential models, not just those prebuilt into the potfit code. Second, we incorporated the geodesic Levenberg–Marquardt (LM) minimization algorithm into potfit as a new local minimization algorithm. The extended potfit was tested by generating a training set using the KIM environment-dependent interatomic potential (EDIP) model for silicon and using potfit to recover the potential parameters from different initial guesses. The results show that EDIP is a 'sloppy model' in the sense that its predictions are insensitive to some of its parameters, which makes fitting more difficult. We find that the geodesic LM algorithm is particularly efficient for this case. The extended potfit code is the first step in developing a KIM-based fitting framework for interatomic potentials for bulk and two-dimensional materials. The code is available for download via https://www.potfit.net.
Related Papers
Efficient time integration is a necessity for dislocation dynamics simulations of work hardening to achieve experimentally relevant strains. In this work, an efficient time integration scheme using a high order explicit method with time step subcycling and a newly-developed collision detection algorithm are evaluated. First, time integrator performance is examined for an annihilating Frank–Read source, showing the effects of dislocation line collision. The integrator with subcycling is found to significantly out-perform other integration schemes. The performance of the time integration and collision detection algorithms is then tested in a work hardening simulation. The new algorithms show a 100-fold speed-up relative to traditional schemes. Subcycling is shown to improve efficiency significantly while maintaining an accurate solution, and the new collision algorithm allows an arbitrarily large time step size without missing collisions.
First-principles calculations are used to characterize the bulk elastic properties of cubic and tetragonal phase metal dihydrides, 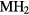 {
{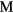 = Sc, Y, Ti, Zr, Hf, lanthanides} to gain insight into the mechanical properties that govern the aging behavior of rare-earth di-tritides as the constituent 3H, tritium, decays into 3He. As tritium decays, helium is inserted in the lattice, the helium migrates and collects into bubbles, that then can ultimately create sufficient internal pressure to rupture the material. The elastic properties of the materials are needed to construct effective mesoscale models of the process of bubble growth and fracture. Dihydrides of the scandium column and most of the rare-earths crystalize into a cubic phase, while dihydrides from the next column, Ti, Zr, and Hf, distort instead into the tetragonal phase, indicating incipient instabilities in the phase and potentially significant changes in elastic properties. We report the computed elastic properties of these dihydrides, and also investigate the off-stoichiometric phases as He or vacancies accumulate. As helium builds up in the cubic phase, the shear moduli greatly soften, converting to the tetragonal phase. Conversely, the tetragonal phases convert very quickly to cubic with the removal of H from the lattice, while the cubic phases show little change with removal of H. The source and magnitude of the numerical and physical uncertainties in the modeling are analyzed and quantified to establish the level of confidence that can be placed in the computational results, and this quantified confidence is used to justify using the results to augment and even supplant experimental measurements.
= Sc, Y, Ti, Zr, Hf, lanthanides} to gain insight into the mechanical properties that govern the aging behavior of rare-earth di-tritides as the constituent 3H, tritium, decays into 3He. As tritium decays, helium is inserted in the lattice, the helium migrates and collects into bubbles, that then can ultimately create sufficient internal pressure to rupture the material. The elastic properties of the materials are needed to construct effective mesoscale models of the process of bubble growth and fracture. Dihydrides of the scandium column and most of the rare-earths crystalize into a cubic phase, while dihydrides from the next column, Ti, Zr, and Hf, distort instead into the tetragonal phase, indicating incipient instabilities in the phase and potentially significant changes in elastic properties. We report the computed elastic properties of these dihydrides, and also investigate the off-stoichiometric phases as He or vacancies accumulate. As helium builds up in the cubic phase, the shear moduli greatly soften, converting to the tetragonal phase. Conversely, the tetragonal phases convert very quickly to cubic with the removal of H from the lattice, while the cubic phases show little change with removal of H. The source and magnitude of the numerical and physical uncertainties in the modeling are analyzed and quantified to establish the level of confidence that can be placed in the computational results, and this quantified confidence is used to justify using the results to augment and even supplant experimental measurements.