
Abstract
The formation of relaxed charge-transfer (CT) excitons and its effect on photocarrier generation in a series of donor–acceptor (DA)-type polymers were examined with a focus on the two unique absorption bands that originate from low-energy charge-transfer (CT) and high-energy main-chain (MC) excitations, respectively. The photoluminescence in response to the CT excitation demonstrated Stokes shift, which is a clear evidence of the formation of bound CT excitons associated with a strong lattice relaxation. Results indicate that photocarriers are generated indirectly by CT excitation through the formation of bound electron–hole pairs, the binding energy and lifetime of which correlate with the donor–acceptor CT degree in the DA-type polymers.
Export citation and abstract BibTeX RIS
1. Introduction
Organic photovoltaic cells (OPCs) are one of the most important renewable energy devices because of their high production processability and light weight construction. Key materials for a recent major development of OPCs are donor–acceptor (DA)-type polymers whose polymer backbone consists of alternating donor and acceptor units: They allow the realization of a power conversion efficiency higher than 9%.1–7) They are featured by two distinct absorption bands in the visible and near-infrared regions. On the basis of molecular orbital calculations, it has been proposed that the near-infrared absorption band corresponds to the charge-transfer (CT) excitation between the donor and acceptor units in the polymer backbone, whereas the visible absorption band corresponds to the spatially extended main-chain (MC) excitation that is usually observed in conventional homopolymers such as poly(3-hexylthiophene-2,5-diyl) (P3HT).8,9) It is believed that the extended range of active photon energies arising from the CT absorption band is crucial for high efficiencies of DA-type polymer OPCs.10) It has recently been pointed out that the efficiency of photocarrier generation is lower for the CT absorption band than for the MC absorption band and that this might be associated with the formation of relaxed CT excitons by CT absorption.8,9,11,12) However, the experimental evidence has not yet been established to confirm the formation of relaxed CT excitons, and it has been unclear what property of DA-type polymers affects the formation of relaxed CT excitons.
In this study, we systematically investigated the formation of relaxed CT excitons in response to the near-infrared absorption band by the molecular orbital calculations and measurements of intrinsic photocurrent yields and photoluminescence lifetimes for a series of DA-type polymers: poly{[9-(1-octylnonyl)-9H-carbazole-2,7-diyl]-2,5-thiophenediyl-2,1,3-benzothiadiazole-4,7-diyl-2,5-thiophenediyl} (PCDTBT),2,3) poly{[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)oxycarbonyl]thieno[3,4-b]thiophenediyl]} (PTB7),5) and poly{2,1,3-benzothiadiazole-4,7-diyl[4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b']dithiophene-2,6-diyl]} (PCPDTBT),4) as well as for the homopolymer P3HT as a reference. From the molecular orbital calculations, it was found that these DA-type polymers exhibit CT absorption in the near-infrared range and MC excitation in the visible range, as reported for other DA-type polymers. The CT excitation of DA-type polymers exhibited a large Stokes shift in photoluminescence spectra, indicating that relaxed CT excitons are formed. We demonstrate that both the binding energy and lifetime of the relaxed CT excitons vary systematically with changes in donor–acceptor CT degree in the DA-type polymers.
2. Experimental methods
The pristine polymer films that were used as semiconductor layers were spin-coated and then annealed at 423 K for 30 min. All fabrication processes and measurements were performed under an inert atmosphere of nitrogen. In photocurrent measurements, we used lateral cells consisting of a pair of gold electrodes with a gap distance of about 150 µm on top of a semiconductor layer. To observe intrinsic photoconduction without interference from the effects of charge separation at metal–semiconductor interfaces, we restricted the area of photoillumination to the region within the semiconductor part inside the electrode gap that was subjected to an applied electric field of <60 kV cm−1. For comparison, we also measured photocurrent action spectra in the absence of a bias voltage for stack cells, each consisting of successive layers of Al, the active polymer (10–50 nm), and poly[(3,4-ethylenedioxy)thiophene-2,6-diyl]:poly(styrene sulfonate) (PEDOT:PSS) (20 nm) on a tin-doped indium-oxide (ITO)-coated glass substrate. We found that the features of the action spectra of the lateral and stack cells were similar (Fig. 1). The time decay profile of photoluminescence in the pristine DA-type polymer films was measured by using the output from the optical parametric amplifier pumped by a Ti:Al2O3 regenerative amplifier used as the excitation light source (duration: 180 fs; repetition rate: 100 kHz; output power: 18 pJ mm−2 pulse−1). A streak camera (Hamamatsu Photonics StreakScope C4334) equipped with a monochromator was employed to detect the photoluminescence and its time decay profile.
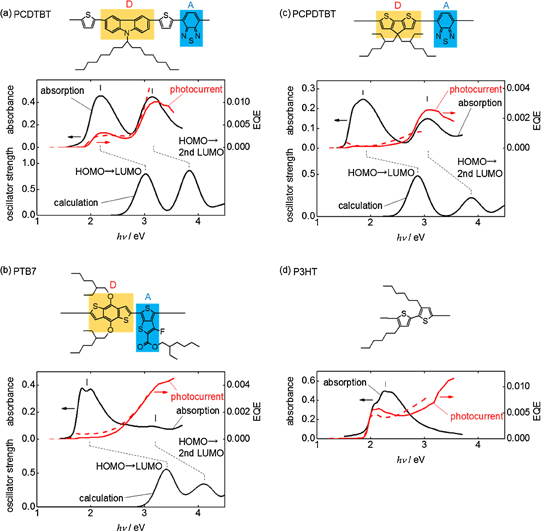
Fig. 1. Absorption, photocurrent, and calculated absorption spectra for (a) PCDTBT, (b) PTB7, (c) PCPDTBT, and (d) P3HT. The red solid and broken lines correspond to the photocurrent spectra measured for stack and lateral cells, respectively. Note that the photocurrent spectra for the lateral cells are multiplied for comparison with those for the stack cells.
Download figure:
Standard image High-resolution imageThe molecular orbital calculations were conducted using the density functional theory (DFT) and time-dependent density functional theory (TDDFT)13) coded in the software Gaussian 09.14) The calculations were carried out with the 6-31G(d) basis set at the CAM-B3LYP level15,16) for the smallest repeating unit of the DA-type polymer with its terminals at either end replaced by hydrogen atoms.
3. Results and discussion
Figure 1 shows the absorption and photocurrent action spectra of the pristine polymer films. The two unique absorption bands is clearly seen in the spectra of the DA-type polymers, especially in the cases of PCDTBT and PCPDTBT, whereas only one absorption band is found in the case of P3HT. In the action spectra of the DA-type polymers, the low-energy absorption band exhibits a lower photocurrent yield than the high-energy absorption band. This result provides evidence that the intrinsic photocarrier generation mechanism for the low-energy absorption band differs from that for the high-energy absorption band in the pristine DA-type polymers. The absorption spectra calculated using TDDFT, as presented in Fig. 1, were found to reproduce the observed spectral features and give reasonable assignment for all the DA-type polymers, i.e., the HOMO → LUMO transition for the low-energy absorption band and the HOMO → second LUMO transition for the high-energy absorption band. Figure 2 shows molecular orbitals contributing to the above transitions. As seen, LUMO is spatially localized in the acceptor unit, whereas HOMO and second LUMO are delocalized along the polymer main chain. Therefore, the HOMO → LUMO transition can be regarded as the CT excitation from the main chain to the acceptor unit, and the HOMO → second LUMO transition can be regarded as the spatially extended MC excitation as usually observed in homopolymers such as P3HT. These assignments are also supported by the difference in donor–acceptor CT degree,  , between the ground and photoexcited states: In the MC excitation, almost no charge is transferred from the donor unit to the acceptor unit, whereas a charge more than 0.1e− is transferred in the CT excitation. Note that the
, between the ground and photoexcited states: In the MC excitation, almost no charge is transferred from the donor unit to the acceptor unit, whereas a charge more than 0.1e− is transferred in the CT excitation. Note that the  value was calculated by the following procedure: Atomic charges for the ground and CT photoexcited states were calculated using DFT and TDDFT, respectively. Then, the atomic charge differences between the ground and CT photoexcited states were separately summed up for the donor and acceptor units.
value was calculated by the following procedure: Atomic charges for the ground and CT photoexcited states were calculated using DFT and TDDFT, respectively. Then, the atomic charge differences between the ground and CT photoexcited states were separately summed up for the donor and acceptor units.

Fig. 2. Molecular orbitals contributing to low-energy CT and high-energy MC absorption bands in DA-type polymers: (a) PCDTBT, (b) PTB7, and (c) PCPDTBT. The difference in donor–acceptor CT degree,  , between the ground and photoexcited states is shown for each excitation.
, between the ground and photoexcited states is shown for each excitation.
Download figure:
Standard image High-resolution imageTo investigate the origin of the low photocurrent yield for the CT excitation, we measured photoluminescence spectra of the pristine DA-type polymer films as well as that of the homopolymer P3HT as a reference. Figure 3 shows the spectra and time decay profiles of photoluminescence by the photoexcitation of the CT absorption band. As seen, the photoluminescence spectra of all the DA-type polymers showed Stokes shift in contrast to that of P3HT. This result indicates a strong exciton–phonon coupling for the CT excitation in the DA-type polymers, which leads to the formation of bound CT excitons associated with a strong lattice relaxation. This provides a reasonable explanation for why the CT excitation demonstrates a lower photocurrent yield than the MC excitation. Figure 4(a) illustrates schematically the process of photocarrier generation in pristine DA-type polymers: The photocarriers are generated directly by MC excitation but indirectly by CT excitation as a result of the formation of the relaxed CT excitons. We estimated the time scale of the relaxed CT exciton formation from the time decay profiles of photoluminescence [Fig. 3(b)]. The decay profiles include two decay components, i.e., the fast and slow decay components. The fast decay component corresponds to the relaxed CT exciton formation. By the exponential fitting of the time decay profiles, the time scale of the relaxed CT exciton formation was obtained as 80 ps for PCDTBT, 60 ps for PTB7, and 10 ps for PCPDTBT. The lifetime of the relaxed CT excitons was also estimated from the slow decay component as 710 ps for PCDTBT, 380 ps for PTB7, and 210 ps for PCPDTBT.

Download figure:
Standard image High-resolution image
Fig. 3. (a) Photoluminescence spectra measured for pristine DA-type polymer films. The black line corresponds to the photoluminescence spectra measured just after photoexcitation, whereas the red line corresponds to the photoluminescence spectra measured at 0.6, 0.7, 0.5, and 0.3 ns after photoexcitation for P3HT, PCDTBT, PTB7, and PCPDTBT, respectively. Note that the ordinate is the photoluminescence intensity normalized by the maximum peak intensity. (b) Time decay profiles of photoluminescence. The time decay profiles shown in the figure were measured at an excitation energy of 2.3 eV for P3HT and PCDTBT and 1.9 eV for PTB7 and PCPDTBT. The photoluminescence intensity was detected at photon energies of 1.7 eV for P3HT, 2.0 and 1.5 eV for PCDTBT, and 1.7 and 1.4 eV for PTB7 and PCPDTBT. Note that the ordinate is the photoluminescence intensity normalized by the maximum peak intensity.
Download figure:
Standard image High-resolution image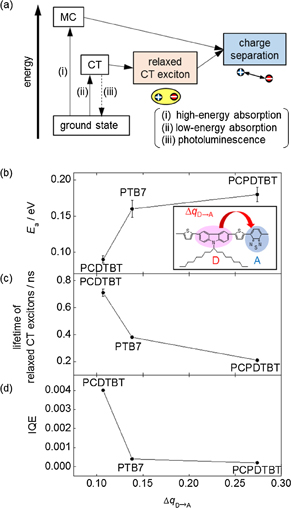
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 4. (a) Intrinsic photocarrier generation pathways for pristine DA-type polymers. (b) Activation energy, Ea, required to produce photocurrent at the photon energy of CT absorption band, (c) lifetime of relaxed CT excitons, and (d) IQE at the photon energy of CT absorption band, plotted as a function of difference in donor–acceptor CT degree,  , between the ground and CT photoexcited states.
, between the ground and CT photoexcited states.
Download figure:
Standard image High-resolution image{kind=link}
To examine the relaxed CT exciton formation in different DA-type polymers, we compared the lifetimes of the relaxed CT excitons for three DA-type polymers [Fig. 4(c)]. We also compared the internal quantum efficiencies (IQEs) and activation energies required to produce photocurrents at the photon energy of the CT absorption band in Figs. 4(d) and 4(b). The activation energies were obtained from the Arrhenius-type temperature dependence of the photocurrent yield observed in the range of 200–295 K. As seen, the lifetime decreases with an increase in  , whereas the activation energy increases with an increase in
, whereas the activation energy increases with an increase in  . Assuming that the increase in activation energy corresponds to the increase in the binding energy of relaxed CT excitons, the activation energy can be regarded as the scale of the binding energy of the relaxed CT excitons. It is considered that the increase in
. Assuming that the increase in activation energy corresponds to the increase in the binding energy of relaxed CT excitons, the activation energy can be regarded as the scale of the binding energy of the relaxed CT excitons. It is considered that the increase in  should enhance the exciton–phonon coupling, which leads to the formation of strongly bound CT excitons associated with a strong lattice relaxation. Furthermore, if the effect of lattice relaxation becomes large, the CT exciton decay should be promoted as predicted by the Marcus theory.17) Thus,
should enhance the exciton–phonon coupling, which leads to the formation of strongly bound CT excitons associated with a strong lattice relaxation. Furthermore, if the effect of lattice relaxation becomes large, the CT exciton decay should be promoted as predicted by the Marcus theory.17) Thus,  provides a reasonable explanation for the material dependence of the binding energy and lifetime of the relaxed CT excitons. In addition, the low IQE in the DA-type polymer with a large
provides a reasonable explanation for the material dependence of the binding energy and lifetime of the relaxed CT excitons. In addition, the low IQE in the DA-type polymer with a large  value is quite consistent with the large binding energy and short lifetime, as shown in Figs. 4(b) and 4(c).
value is quite consistent with the large binding energy and short lifetime, as shown in Figs. 4(b) and 4(c).
Finally, on the basis of the experimental results outlined above, we discuss the origin of the high conversion efficiency reported for DA-type polymer OPCs with a bulk heterojunction (BHJ) structure. If we assume that relaxed CT excitons must diffuse to reach the heterojunctions to generate photocarriers, the diffusion length L should be much larger than the dimensions of the polymer domain in the BHJ (∼10 nm18)) to achieve an internal quantum efficiency close to 100% reported in DA-type polymer OPCs.19) The length L was estimated to be 13 nm for PCDTBT and 2.3 nm for PCPDTBT by using the relation  , where D is the diffusion constant represented by Einstein's relation, D = μkT/e. We assumed the mobility μ value of 0.1 cm2 V−1 s−1 for PCDTBT20) and 0.01 cm2 V−1 s−1 for PCPDTBT.4) We also assumed that the diffusion time τ corresponds to the exciton lifetime estimated from the photoluminescence decay. These estimations allow us to reconsider the role of BHJs for the highly efficient generation of photocarriers. It is more appropriate to consider that the CT excitation delocalizes and can propagate coherently in the polymer domain, which results in the instantaneous generation of photocarriers at the heterojunctions before relaxed CT excitons are formed.21–23) The coherent length of unrelaxed CT excitons just after photoexcitation (<100 ps) might be a crucial parameter for understanding the relatively high quantum efficiency of DA-type polymer OPCs.
, where D is the diffusion constant represented by Einstein's relation, D = μkT/e. We assumed the mobility μ value of 0.1 cm2 V−1 s−1 for PCDTBT20) and 0.01 cm2 V−1 s−1 for PCPDTBT.4) We also assumed that the diffusion time τ corresponds to the exciton lifetime estimated from the photoluminescence decay. These estimations allow us to reconsider the role of BHJs for the highly efficient generation of photocarriers. It is more appropriate to consider that the CT excitation delocalizes and can propagate coherently in the polymer domain, which results in the instantaneous generation of photocarriers at the heterojunctions before relaxed CT excitons are formed.21–23) The coherent length of unrelaxed CT excitons just after photoexcitation (<100 ps) might be a crucial parameter for understanding the relatively high quantum efficiency of DA-type polymer OPCs.
4. Conclusions
We have studied the reason why the photocurrent yield by the low-energy CT absorption band is lower than that by the high-energy MC absorption band in pristine DA-type polymer films. We found that photocarriers are generated indirectly by CT excitation, as a result of the formation of relaxed CT excitons, which provides a reasonable explanation for the lower photocurrent yield. We also found that the binding energy of the relaxed CT excitons increases and that their lifetime decreases with an increase in the difference in donor–acceptor CT degree,  , between the ground and CT photoexcited states in the DA-type polymers. These trends were closely related to the photovoltaic characteristics; the IQE decreases with an increase in binding energy and a decrease in the lifetime. The formation of the relaxed CT excitons in DA-type polymers seems to be contradicted by the quite high internal quantum efficiency of OPCs utilizing these polymers, suggesting that the conventional photocarrier generation model based on the diffusional motion of excitons must be reconsidered. The coherent propagation of an unrelaxed exciton might be a possible origin of the efficient generation of photocarriers in DA-type polymer OPCs.
, between the ground and CT photoexcited states in the DA-type polymers. These trends were closely related to the photovoltaic characteristics; the IQE decreases with an increase in binding energy and a decrease in the lifetime. The formation of the relaxed CT excitons in DA-type polymers seems to be contradicted by the quite high internal quantum efficiency of OPCs utilizing these polymers, suggesting that the conventional photocarrier generation model based on the diffusional motion of excitons must be reconsidered. The coherent propagation of an unrelaxed exciton might be a possible origin of the efficient generation of photocarriers in DA-type polymer OPCs.
Acknowledgements
This work was supported by the New Energy and Industrial Technology Development Organization (NEDO) through its Innovative Solar Cell Program and by the Japan Society for the Promotion of Science (JSPS) through its Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program) and KAKENHI for Young Scientists (B) (24750190).