



ABSTRACT
Using quantum chemical methods, we investigate the possible outcomes of  reactions with acetylene and diacetylene molecules. We find both reactions to be exothermic reactions without barriers, yielding stable anions of the corresponding polyynes:
reactions with acetylene and diacetylene molecules. We find both reactions to be exothermic reactions without barriers, yielding stable anions of the corresponding polyynes:  and
and  . We show in this work that the computed chemical rates in the case of the formation of the
. We show in this work that the computed chemical rates in the case of the formation of the  anion would be larger than those existing for the direct radiative electron attachment (REA) process, the main mechanism generally suggested for their formation. In the case of the
anion would be larger than those existing for the direct radiative electron attachment (REA) process, the main mechanism generally suggested for their formation. In the case of the  anion, however, the present chemical rates of formation at low T are even lower than those known for its REA process, both mechanisms being inefficient for its formation under astrochemical conditions. The present results are discussed in view of their consequences on the issue of the possible presence of such anions in the ISM environments. They clearly indicate the present chemical route to
anion, however, the present chemical rates of formation at low T are even lower than those known for its REA process, both mechanisms being inefficient for its formation under astrochemical conditions. The present results are discussed in view of their consequences on the issue of the possible presence of such anions in the ISM environments. They clearly indicate the present chemical route to  formation to be inefficient at the expected temperatures of a dark molecular cloud, whereas this is found not to be the case for the
formation to be inefficient at the expected temperatures of a dark molecular cloud, whereas this is found not to be the case for the  , in line with the available experimental findings.
, in line with the available experimental findings.
Export citation and abstract BibTeX RIS
1. INTRODUCTION
The possibility of having molecular species carrying a negative charge (anionic molecules) and existing in detectable amounts in the interstellar medium (ISM), more specifically in cold dark cloud environments, has been put forward by several authors (Dalgarno & McCray 1973; Sarre 1980; Herbst 1981; Herbst & Woon 1997). They have also suggested that carbon chains and hydrocarbon radicals would have large and positive electron affinities (EAs) and could therefore lead to anion formations via the energy-releasing mechanism of radiative electron attachment (REA). The latter process would arise from the interactions of such molecules with the free electrons generated in the diffuse regions by H, He photoionization. The recombination process would then be:
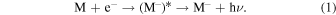
They further suggested that the above processes would be efficient for stabilizing molecular anions made up by more than four or five atoms (Herbst & Osamura 2009).
The experimental, astrochemical evidence for the presence of such species was provided later when an unidentified series of lines was detected in a radio astronomical survey of the evolved carbon star IRC+10216 by Kawaguchi et al. (1995) and was conclusively assigned to the spectrum of  by McCarthy et al. (2006) on the basis of laboratory rotational spectroscopy and independent observations toward the Taurus molecular clouds 1 (TMC-1(CP)). Additional anionic species were also observed later on by Cernicharo et al. (2007) (
by McCarthy et al. (2006) on the basis of laboratory rotational spectroscopy and independent observations toward the Taurus molecular clouds 1 (TMC-1(CP)). Additional anionic species were also observed later on by Cernicharo et al. (2007) ( ), by Remijan et al. (2007) (
), by Remijan et al. (2007) ( ), and by Thaddeus et al. (2008)
), and by Thaddeus et al. (2008)  ) in the envelope of IRC+10216.
) in the envelope of IRC+10216.  anions were also observed in the TMC-1 by Brunken et al. (2007). Additional observations of molecular negative ions were surveyed by Gupta et al. (2007), who detected
anions were also observed in the TMC-1 by Brunken et al. (2007). Additional observations of molecular negative ions were surveyed by Gupta et al. (2007), who detected  in two further sources: the pre-stellar cloud L1544 and the protostellar object L1521F.
in two further sources: the pre-stellar cloud L1544 and the protostellar object L1521F.
Since polyatomic molecules carrying a negative charge have turned out to be present under different conditions and in significant quantities (e.g., the measured column density of  in the TMC-1(CP) was found by McCarthy et al. 2006 to be 1011 cm−2) it is reasonable to expect that they also play a significant role in the chemistry of the dark molecular cloud environment and at the low temperatures assigned to these regions. Hence, it becomes relevant to investigate in more detail additional possible formation paths of such stable anionic species.
in the TMC-1(CP) was found by McCarthy et al. 2006 to be 1011 cm−2) it is reasonable to expect that they also play a significant role in the chemistry of the dark molecular cloud environment and at the low temperatures assigned to these regions. Hence, it becomes relevant to investigate in more detail additional possible formation paths of such stable anionic species.
We have previously studied, both theoretically and computationally, the general dynamics that could be driven by the free electrons available in such an environment and the important role of intermediate, metastable anions as gateways to the final, anionic species experimentally detected. Thus, the resonant attachment of slow electrons to NCCN was studied previously by our group (Sebastianelli & Gianturco 2010). We have also analyzed the possible fragmentation decays of  and
and  upon electron attachment (Sebastianelli & Gianturco 2012) as well as a detailed study of resonant electron attachment to
upon electron attachment (Sebastianelli & Gianturco 2012) as well as a detailed study of resonant electron attachment to  (Baccarelli et al. 2013). We also carried out a more extensive analysis of the dynamics of electron attachment to non-polar hydrocarbon chains like
(Baccarelli et al. 2013). We also carried out a more extensive analysis of the dynamics of electron attachment to non-polar hydrocarbon chains like  (with n from 4 to 12) at the expected low energies of planetary atmospheres and dark molecular clouds (Carelli et al. 2013). In that work we specifically showed that the attachment mechanism was driven by the prior formation of metastable anions from asymmetrically deformed non-polar polyynes that can give rise to complex intermediates of polar radical anions plus H-atom stretching:
(with n from 4 to 12) at the expected low energies of planetary atmospheres and dark molecular clouds (Carelli et al. 2013). In that work we specifically showed that the attachment mechanism was driven by the prior formation of metastable anions from asymmetrically deformed non-polar polyynes that can give rise to complex intermediates of polar radical anions plus H-atom stretching:
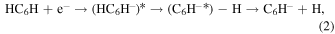
where the intermediate polar radical can initially form its closed-shell anion as a dipole-bound state (DBS) partner that can then decay into the more stable anionic valence-bound states (VBS) after internal-energy redistribution and ejection of one terminal hydrogen atom. The above chain of events is reminiscent of the indirect REA mechanism (IREA) introduced more recently by Douguet et al. (2015), where the dissipation paths of the large amount of energy that has to be released upon electron attachment in systems with large and positive EA values was suggested to occur via an electron-vibration nonadiabatic coupling mechanism. They have shown this IREA mechanism, and indeed also the REA mechanism of Equation (1), to be rather inefficient for smaller radical chains like  and
and  , thereby calling into question the efficacy of such pathways for the formation of observable quantities of the anions of such molecules. It is therefore important to better understand which other mechanisms could be responsible for the formation of the smaller C-chain anions under ISM conditions.
, thereby calling into question the efficacy of such pathways for the formation of observable quantities of the anions of such molecules. It is therefore important to better understand which other mechanisms could be responsible for the formation of the smaller C-chain anions under ISM conditions.
To this effect, we specifically explore in the present study a possible "chemical" method for the formation of the stable anions for the two smaller members of the C-bearing linear chains. One should also note here that the actual presence of the  molecular anions in ISM environments has not been confirmed as yet (Morisawa et al. 2005). We therefore investigate a chemical route to the formation of such anions by examining the efficiency of the reaction of
molecular anions in ISM environments has not been confirmed as yet (Morisawa et al. 2005). We therefore investigate a chemical route to the formation of such anions by examining the efficiency of the reaction of  with non-polar precursors like acetylene (HCCH) and diacetylene (HCCCCH) in the gas phase:
with non-polar precursors like acetylene (HCCH) and diacetylene (HCCCCH) in the gas phase:
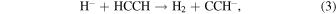
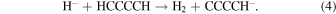
The importance of  as a chemical partner has been suggested before given its likely existence through cosmic rays that act during ion-pair reactions within the inner cores (radius ≤106 au) of the pre-stellar envelopes (Prasad & Huntress 1980), although so far little is known about the details of its actual mechanism under the above conditions (Mackay et al. 1977).
as a chemical partner has been suggested before given its likely existence through cosmic rays that act during ion-pair reactions within the inner cores (radius ≤106 au) of the pre-stellar envelopes (Prasad & Huntress 1980), although so far little is known about the details of its actual mechanism under the above conditions (Mackay et al. 1977).
Therefore, if the presence of  could be in sufficient amounts to provide a useful chemical partner, then we shall further show that both the above reactions (3) and (4) are interesting alternatives: they are markedly exothermic processes where the final products' formation involves an electron transfer (ET) mechanism during the formation of new H2 as a separate molecular product. The actual energy gains from forming anionic molecules from the two radical
could be in sufficient amounts to provide a useful chemical partner, then we shall further show that both the above reactions (3) and (4) are interesting alternatives: they are markedly exothermic processes where the final products' formation involves an electron transfer (ET) mechanism during the formation of new H2 as a separate molecular product. The actual energy gains from forming anionic molecules from the two radical  and
and  are linked to their large and positive EA values: 2.969 eV for
are linked to their large and positive EA values: 2.969 eV for  (May et al. 2008) and 3.558 eV for
(May et al. 2008) and 3.558 eV for  : both are much larger than the EA of the H atom of 0.754 eV. Furthermore, the estimated bond energies for the C–H single bonds in
: both are much larger than the EA of the H atom of 0.754 eV. Furthermore, the estimated bond energies for the C–H single bonds in  and
and  are about 5.7 eV for the former (Shi & Ervin 2000) and 5.587 eV for the latter (Mordaunt & Ashfold 1994): they have to be compared with the dissociation energy, Do value, for the H2 molecule formation: 4.478 eV (Mackay et al. 1977).
are about 5.7 eV for the former (Shi & Ervin 2000) and 5.587 eV for the latter (Mordaunt & Ashfold 1994): they have to be compared with the dissociation energy, Do value, for the H2 molecule formation: 4.478 eV (Mackay et al. 1977).
The above estimates lead reaction (3) to be exothermic by about 1.00 eV and reaction (4) by 1.69 eV. One should further note that the charge-exchange (CE) process for  formation is also strongly exothermic:
formation is also strongly exothermic:
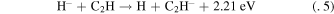
Although we will not be considering the above additional reaction in our present analysis, it has already been shown by calculations to be an inefficient path to the formation of that anion (Senent & Hochlaf 2013). In conclusion, therefore, we see that both reactions (3) and (4) would be strongly exothermic processes and likely to proceed to products even in the low-temperature conditions expected for the dark cloud environments.
In an earlier study of ours on the smaller members of the cyano derivatives,  and
and  (Satta et al. 2015) we have already indicated that the chemical nature of their reactions with
(Satta et al. 2015) we have already indicated that the chemical nature of their reactions with  , i.e., the present mechanism of an ET occurring concurrently with H2 formation, also makes the production of their anions exothermic and this occurs without an intermediate energy barrier between reactants and products. It thus becomes interesting to computationally analyze the present reactions to verify if the same behavior exists for them. In the following sections we investigate this point by carrying out accurate ab initio calculations of the reactive potential energy surfaces (RPESs) and by further modeling the ensuing reaction rates using a variational treatment discussed previously (Satta et al. 2015). In Section 2 we describe the ab initio calculations and report our findings for both the acetylene and diacetylene partners with
, i.e., the present mechanism of an ET occurring concurrently with H2 formation, also makes the production of their anions exothermic and this occurs without an intermediate energy barrier between reactants and products. It thus becomes interesting to computationally analyze the present reactions to verify if the same behavior exists for them. In the following sections we investigate this point by carrying out accurate ab initio calculations of the reactive potential energy surfaces (RPESs) and by further modeling the ensuing reaction rates using a variational treatment discussed previously (Satta et al. 2015). In Section 2 we describe the ab initio calculations and report our findings for both the acetylene and diacetylene partners with  . In the next section, Section 3, we calculate the corresponding reaction rates over a range of temperatures that represent the colder dark cloud conditions of interest in our study. In Section 4 will give our present conclusions in relation to observational studies on the existing abundances of
. In the next section, Section 3, we calculate the corresponding reaction rates over a range of temperatures that represent the colder dark cloud conditions of interest in our study. In Section 4 will give our present conclusions in relation to observational studies on the existing abundances of  and
and  .
.
2. COMPUTING THE REACTIVE POTENTIAL ENERGY LANDSCAPES
Following the computational procedure of our earlier work (Satta et al. 2015) we have begun with an analysis of reactions (3) and (4) by considering first the collinear approach between reaction partners. Additionally, given the rather "stiff" (multielectron) binding structure of the  radical fragments, e.g.,
radical fragments, e.g.,  and
and  , we decided that it would still be realistic to involve in the reaction process only the lighter H atom of the single H-C bond closest to the approaching
, we decided that it would still be realistic to involve in the reaction process only the lighter H atom of the single H-C bond closest to the approaching  : they would actually be the least strong bonds within the reactive partners of this study, hence more susceptible to reacting. Thus, we examined a reaction path essentially described by two bond changes:
: they would actually be the least strong bonds within the reactive partners of this study, hence more susceptible to reacting. Thus, we examined a reaction path essentially described by two bond changes:  , which evolve along R1 and R2 in going from reactants to products in (3 ) and (4). The angles reported in all the figures are defined in the RPESs as:
, which evolve along R1 and R2 in going from reactants to products in (3 ) and (4). The angles reported in all the figures are defined in the RPESs as:  , i.e., the angle formed between the
, i.e., the angle formed between the  partner and the nearest
partner and the nearest  bond in the linear chain. They are shown by Figure 1 together with the bonds involved in the pseudo-2D approach followed in the present analysis of low-temperature reactions.
bond in the linear chain. They are shown by Figure 1 together with the bonds involved in the pseudo-2D approach followed in the present analysis of low-temperature reactions.
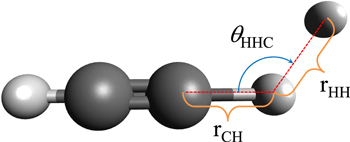
Figure 1. Graphic presentation of the three internal coordinates involved in the calculations of the reactive processes reported by Equations (3) and (4). Only the first member of the series is shown here as an example.
Download figure:
Standard image High-resolution imageThe calculations were carried out using standard quantum chemistry methods; for the sake of brevity, we refer the interested readers to any of the usual reviews on the subject (see, e.g., Fernandez-Ramos et al. 2006). We have employed the well-known density functional theory (DFT) approaches to treat the molecular electrons at each complex geometry, employing the B3LYP exchange-correlation functional and a 6-31++g** basis set expansion. All computations were performed by using the GAUSSIAN09 suite of codes (Frisch et al. 2013). The grid of radial variables was chosen to be given by a 50 × 50 set of points for which each energy value was obtained from a single-point calculation. In addition to the collinear approach, we have also examined different reactive approaches for "bent" configurations as we further discuss below when presenting our results. They correspond, as discussed below, to changes in the angle reported by Figure 1, where the collinear approach corresponds to  equal to 180o.
equal to 180o.
To search for a transition complex that might form behind a barrier between reagents and products, we report in Figures 2 and 3 the 2D maps that describe the energy evolutions along the relevant coordinates already mentioned before for reactions (3) and (4). In both figures the collinear approach is reported and we also show the energy locations of the minimum energy path (MEP) (Fernandez-Ramos et al. 2006) profiles for the two reactions.

Figure 2. Computed pseudo-2D RPES for the  reaction for the collinear approach (
reaction for the collinear approach ( ) of the
) of the  reagent to the
reagent to the  partner.The angle's definition is reported in Figure 1 and also in the main text. The corresponding minimum energy path (MEP) is shown by a red line on the energy map, going from reactants to products.
partner.The angle's definition is reported in Figure 1 and also in the main text. The corresponding minimum energy path (MEP) is shown by a red line on the energy map, going from reactants to products.
Download figure:
Standard image High-resolution imageThe calculations for both reactions clearly confirm the exothermic nature of the processes: the energy landscapes in Figures 2 and 3 indicate a downward path in energy when moving from the reagents' regions (the bottom right part of each figure) to the products' regions on the upper left parts of the maps, where the locations of two energy minima are clearly shown.

Figure 3. Same calculations for the collinear configuration of the RPES as those in Figure 2, but this time for the case of  in reaction with
in reaction with  . The relevant MEP path is also shown on the energy map by a different colored line.
. The relevant MEP path is also shown on the energy map by a different colored line.
Download figure:
Standard image High-resolution imageFurthermore, we also see from the energy features of both RPESs that no energy barriers exist when moving along the reactions' MEPs: the concurrent formation of a hydrogen molecule while the negative charge moves on to the molecular radical and stabilizes its anion is occurring with an energy gain from reactants to products without preferential conformations (i.e., transition state formation) for complexes. Thus, our calculations already show that the collinear approaches between reagents give rise to barrierless, monotonically exothermic MEPs for the reactions' partners forming the anionic polyynes.
Another pictorial way of representing the monotonic descent from reactants to products is to view them in a 3D representation. We thus report such a view in Figure 4 for both systems: the path to  formation in the upper panel and the one to
formation in the upper panel and the one to  formation in the lower panel.
formation in the lower panel.

Figure 4. 3D representation of the computed RPES for both collinear arrangements. Upper panel: the collinear reaction for HCCH; lower panel: the same collinear reaction but for HCCCCH. In both panels the lower right regions of the figures indicate the initial reactants' regions while the areas on the lower left report the final formation of the hydrogen molecules.
Download figure:
Standard image High-resolution imageThat figure also indicates that the descent into products on the left-hand side of both panels occurs at RHH distances that are fairly close to the equilibrium value in the isolated molecules: only after the formation of H2 do the reagents separate with the formations of either  or
or  . The ET process takes place as H2 is formed, whereas it is only after it that the final, closed-shell anion is separated from the new H2 molecule, hence disappearing on the back of the energy landscapes and on the left-hand sides of the maps in the figure.
. The ET process takes place as H2 is formed, whereas it is only after it that the final, closed-shell anion is separated from the new H2 molecule, hence disappearing on the back of the energy landscapes and on the left-hand sides of the maps in the figure.
To further check possible differences in the energy features as the  partner approaches either of the acetylenic partners at an angle
partner approaches either of the acetylenic partners at an angle  between the
between the  line and the
line and the  linear configurations, we report in the four panels of Figure 5 the behavior of the RPES maps for different angles of approach in both systems. In the left column we show the results for the acetylene at angles
linear configurations, we report in the four panels of Figure 5 the behavior of the RPES maps for different angles of approach in both systems. In the left column we show the results for the acetylene at angles  (upper left panel) and
(upper left panel) and  (lower left panel). The same orientations for the diacetylene molecule are given by the two panels in the right column.
(lower left panel). The same orientations for the diacetylene molecule are given by the two panels in the right column.

Figure 5. Computed pseudo-2D RPES maps for different choices of the angle  (defined in the main text). The left column refers to
(defined in the main text). The left column refers to  for
for  (above) and
(above) and  (below) whereas the same results for the
(below) whereas the same results for the  system are in the right columns. The profiles of the corresponding MEP energy behaviors are also shown by different colors in all four panels of the present figure.
system are in the right columns. The profiles of the corresponding MEP energy behaviors are also shown by different colors in all four panels of the present figure.
Download figure:
Standard image High-resolution imageFrom the panels in Figure 5 one sees that all the "bent" approaches between partners qualitatively indicate very similar behavior of the reactants: absence of any energy barriers between reagents' regions (lower right) and products' regions (upper left) and a steady energy decrease from reagents down to the region of products' formation.
A different way of analyzing the reactive maps could be obtained by selecting a slightly different presentation of the reactions' energy landscapes, as we have done in Figure 6, which refers, respectively, to the acetylene and diacetylene reactions with  , in the two panels of that figure.
, in the two panels of that figure.
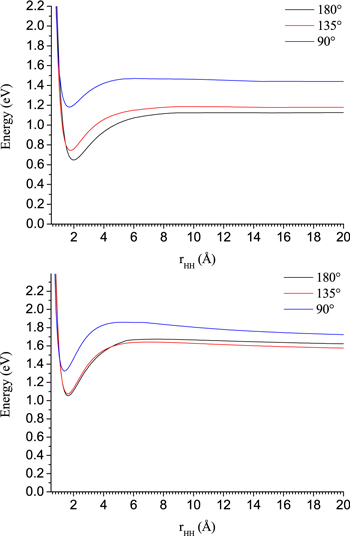
Figure 6. Computed energy profiles, at the different approaching angles of Figures 2–5, for the  reaction with
reaction with  (upper panels) and
(upper panels) and  (lower panel). The curves in the figures report the
(lower panel). The curves in the figures report the  approaching the molecular partner from larger distances (right-hand part of the figures) and exiting as H2 is formed at shorter distances: each curve refers to a different angle and each presents a minimum energy geometry of the complex (lower left side of the curves) after which the anion separates and the hydrogen molecule is formed. The coordinates of the separating products (see Figure 1) are not shown in this figure.
approaching the molecular partner from larger distances (right-hand part of the figures) and exiting as H2 is formed at shorter distances: each curve refers to a different angle and each presents a minimum energy geometry of the complex (lower left side of the curves) after which the anion separates and the hydrogen molecule is formed. The coordinates of the separating products (see Figure 1) are not shown in this figure.
Download figure:
Standard image High-resolution imageThe following should be considered when examining the data reported in both panels.
- 1.For both reactions, and for a broad range of angles of approach, we have already found (see Figure 5) that the shapes of the MEP configurations remain largely unchanged. This indicates the essential invariance of the energy profiles for many different configurations, all leading to barrierless formations of the final products.
- 2.The energy profiles related to the energy evolution of the
 partner as it gets closer to the molecular reagents indicate in all cases that at relative distances around 1.8 Å the "complex" reaches an energy minimum whereby H2 is formed and it starts to leave the reagents' area after the ET process has occurred: such a dynamical path remains essentially invariant as the reaction moves from the collinear approaches in both systems.
partner as it gets closer to the molecular reagents indicate in all cases that at relative distances around 1.8 Å the "complex" reaches an energy minimum whereby H2 is formed and it starts to leave the reagents' area after the ET process has occurred: such a dynamical path remains essentially invariant as the reaction moves from the collinear approaches in both systems. - 3.The relative energetics of the-to-H2 evolutions, however, change when the direction of approach to the molecular partners is not collinear. In both cases, in fact, the collinear approaches remain the ones leading to the most stable energy configurations of the intermediate complex with all partners close to each other: they are those yielding the largest energy release during the reactions. In other words, although all configurations indicate the reactions remain exothermic and without any energy barrier from reagents to products for all angular choices, the computational analysis shown by both panels of Figure 6 indicates that the collinear approaches remain those that afford the largest energy gains when going from reagents to the products' forming regions. The curves further show that the approaches between reacting partners occurring away from the collinear configurations follow the energy troughs into the products via a long-range energy "hump" that may hinder reactivity at lower temperatures: this feature is totally absent in the collinear approaches to products' formation.
In order to assure and verify the reliability of the results presented in Figure 6, (and indirectly the reliability of all the ones shown up to now), we have computed the potential curves at the collinear configurations using more robust levels of calculation. Namely, we have initially tested the effect of introducing the dispersion contributions onto the B3LYP functional. Then, keeping both dispersion effects and the B3LYP functional, we have further used a larger basis set: the aug-cc-pVTZ (see the definition in Fernandez-Ramos et al. 2006). We also tried to change the functional by using one often employed in the quantum chemistry literature with double hybridization, the B2PLYP (Frisch et al. 2013). Finally, always using the largest basis set defined previously as the aug-cc-pVTZ basis set, we tested an advanced post-Hartree–Fock level of calculation: we thus used the coupled-cluster method including single and double excitations with triple excitations included via perturbation theory, i.e., the CCSD(T) treatment (for implementation and definition, see Frisch et al. 2013). The largest geometry difference for the minimum of  (upper panel of Figure 6) between the shown B3LYP/6-31++g** result and all the additional calculations with the methods we described above is 0.01894 Å. Hence, starting from the position at 1.6058 Å, the maximum discrepancy is 1.19%, while for the well depth the difference is 0.01973 eV, which means that, from 0.6144 eV onward, the maximum discrepancy is 3.11%. The largest geometry difference for the minimum of the
(upper panel of Figure 6) between the shown B3LYP/6-31++g** result and all the additional calculations with the methods we described above is 0.01894 Å. Hence, starting from the position at 1.6058 Å, the maximum discrepancy is 1.19%, while for the well depth the difference is 0.01973 eV, which means that, from 0.6144 eV onward, the maximum discrepancy is 3.11%. The largest geometry difference for the minimum of the  reaction (lower panel of Figure 6) between our initial calculation and all the improved test methods listed above is 0.01913 Å. Hence, from 1.6689 Å the maximum variation is 1.16%, whereas for the depth the difference is 0.01995 eV, and starting from 0.5659 eV the maximum discrepancy is 3.38%. The above findings indicate that no substantial errors are introduced on the predicted energies at the present level of basis set choice and computational method of quality employed for our reactive energy surfaces. Thus, the ensuing predicted rates (see next Section 3) would remain within the same order of magnitude even when using other, computationally more expensive, evaluation methods.
reaction (lower panel of Figure 6) between our initial calculation and all the improved test methods listed above is 0.01913 Å. Hence, from 1.6689 Å the maximum variation is 1.16%, whereas for the depth the difference is 0.01995 eV, and starting from 0.5659 eV the maximum discrepancy is 3.38%. The above findings indicate that no substantial errors are introduced on the predicted energies at the present level of basis set choice and computational method of quality employed for our reactive energy surfaces. Thus, the ensuing predicted rates (see next Section 3) would remain within the same order of magnitude even when using other, computationally more expensive, evaluation methods.
To further illustrate the orientation features of the interactions of the acetylene and diacetylene molecules with the  partner, we report in two additional figures below the energy levels that are mapping the interaction of the anionic atom with the two molecules with
partner, we report in two additional figures below the energy levels that are mapping the interaction of the anionic atom with the two molecules with  . We employ, for better clarity, a slightly different selection of Cartesian coordinates which are briefly illustrated first in Figure 7.
. We employ, for better clarity, a slightly different selection of Cartesian coordinates which are briefly illustrated first in Figure 7.

Figure 7. Choice of Cartesian coordinates employed to represent energy map landscapes projected onto a plane, given in Figures 8 and 9. The molecular geometries of the isolated acetylene and diacetylene are selected. The (X, Y) values report the varying position of the  with respect to the fixed molecular geometry shown in the figure for the acetylene case. They are pictorially defined within the figure.
with respect to the fixed molecular geometry shown in the figure for the acetylene case. They are pictorially defined within the figure.
Download figure:
Standard image High-resolution imageThe energy maps reported in the two figures show the energetics of the "pre-reactive" situation whereby the approach of the  partner does not lead as yet to reaction but to inelastic collisions only. They do show, however, that in both systems the interaction with a negative atom is attractive mainly along the collinear approaches while becoming increasingly more repulsive as the
partner does not lead as yet to reaction but to inelastic collisions only. They do show, however, that in both systems the interaction with a negative atom is attractive mainly along the collinear approaches while becoming increasingly more repulsive as the  attacks the molecules from other directions; the presence of electron-rich, multiple bonds away from the terminal
attacks the molecules from other directions; the presence of electron-rich, multiple bonds away from the terminal  regions causes the atomic anion to sample increasingly more repulsive environments. It is the additional occurrence of the reaction that can further generate the overall attractive behaviors seen in the maps of Figures 2–6 discussed earlier.
regions causes the atomic anion to sample increasingly more repulsive environments. It is the additional occurrence of the reaction that can further generate the overall attractive behaviors seen in the maps of Figures 2–6 discussed earlier.
The marked orientational anisotropy of the non-reactive interactions between partners can be further appreciated by the different energy profiles that are reported for different orientation angles in Figures 10 and 11. One should also note here that the angles of Figures 8 through 11 have a different definition than those discussed in the previous figures and were explicitly given in Figure 7. We show there the angle between the line of the approaching atomic anion to the center-of-mass of the molecule and the principal molecular axis in each partner.

Figure 8. Energy level maps surrounding the  molecule interacting with the
molecule interacting with the  partner. The energy scale is shown by the color code on the right. The (X, Y) coordinates are those defined in Figure 7.
partner. The energy scale is shown by the color code on the right. The (X, Y) coordinates are those defined in Figure 7.
Download figure:
Standard image High-resolution imageWe report in both figures the shape of the interaction between  and either
and either  (in Figure 10) or
(in Figure 10) or  (in Figure 11), selecting specific angular "cuts" of the full energy maps shown by Figures 8 and 9 and fully defined by the coordinates of Figure 7.
(in Figure 11), selecting specific angular "cuts" of the full energy maps shown by Figures 8 and 9 and fully defined by the coordinates of Figure 7.
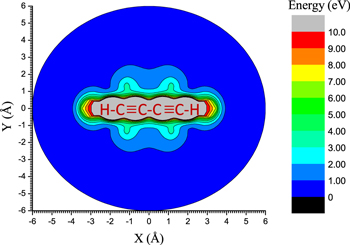
Figure 9. Same data as in Figure 8, but this time for the  molecular partner. See main text for further details.
molecular partner. See main text for further details.
Download figure:
Standard image High-resolution imageThe attractive behavior, controlled in both cases by the spherical, long-range (charge-dipole polarizability) terms, is clearly visible along the collinear direction, while the approaches at different angles cause the overall interaction to become increasingly less attractive until it gets to be fully repulsive closer to the T-shape approach of the atomic anion: the negatively charged atomic partner undergoes, in fact, net repulsive interactions as it gets closer to the electron-rich, multiple-bond regions.
In the more extended carbon chain of the diacetylene in Figure 11, we further note that for angular regions of approach around  a sort of "saddle" shape appears in the potential energy cuts: it indicates the passage between the interaction with either the triple bonds or the central single bond where the repulsion gets comparatively less strong. Since such a region does not exist for the
a sort of "saddle" shape appears in the potential energy cuts: it indicates the passage between the interaction with either the triple bonds or the central single bond where the repulsion gets comparatively less strong. Since such a region does not exist for the  partner, no similar feature appears in the cuts by Figure 10. We have examined the angular cuts over a larger number of angles than those we reported in the last two figures only to find, however, that the energy behavior moved monotonically from attractive to repulsive in going from the collinear to the T-shaped configurations. We have therefore reported only a few of them in the two figures.
partner, no similar feature appears in the cuts by Figure 10. We have examined the angular cuts over a larger number of angles than those we reported in the last two figures only to find, however, that the energy behavior moved monotonically from attractive to repulsive in going from the collinear to the T-shaped configurations. We have therefore reported only a few of them in the two figures.

Figure 10. Computed interactions between the  and the acetylene partner as a function of different angles of approach. The collinear approach is for
and the acetylene partner as a function of different angles of approach. The collinear approach is for  . The angles employed here are defined by the Cartesian coordinates reported in Figure 7.
. The angles employed here are defined by the Cartesian coordinates reported in Figure 7.
Download figure:
Standard image High-resolution imageIn conclusion, the spatial and orientational analysis of both the reactive PESs and the non-reactive interactions indicates clearly that the collinear approach of the  partner to either molecular partners provides the most facile (energetically speaking) path to the formation of the polyyne anions for the title species. The existence of strong orientational anisotropies for both initial molecules when interacting with
partner to either molecular partners provides the most facile (energetically speaking) path to the formation of the polyyne anions for the title species. The existence of strong orientational anisotropies for both initial molecules when interacting with  also could suggest that low-energy non-reactive collisions lead to rotational excitation of either molecular partner, with ensuing radiative emissions of the relevant lines from both species. This is a different aspect of the present process which we do not intend to pursue any further here, although it is interesting to mention the possibility, suggested by our calculations, of possible rotational "heating" of the initial polyacetylene partners in collisions with
also could suggest that low-energy non-reactive collisions lead to rotational excitation of either molecular partner, with ensuing radiative emissions of the relevant lines from both species. This is a different aspect of the present process which we do not intend to pursue any further here, although it is interesting to mention the possibility, suggested by our calculations, of possible rotational "heating" of the initial polyacetylene partners in collisions with  .
.
In the following section, we shall further employ the findings about the reactive mechanisms and implement a computational treatment for the final reaction rates of the processes of Equations (3) and (4). One should also keep in mind here that we are chiefly interested in modeling the chemical rates in the low-temperature regions of the dense molecular clouds. This means that both partners are expected to be internally cold and also to be translationally slow, at least within an LTE description of their conditions in the molecular clouds (Cordiner & Millar 2009).
3. EVALUATING REACTION RATES
The previous analysis has told us that the reactive paths are evolving along an exothermic process in which no barriers exist from reagents to products. Furthermore, the data discussed in the figures of the previous section tell us that the most energetically favored approach would be along the collinear MEP during which the H2 formation occurs at relatively close distances between partners (between 1.5 and 2.0 Å) and is concurrent to the ET mechanism of  formation.
formation.
From the above information, we decided that it would be reasonable to model the reaction rates by using the variational transition state theory (VTST) approach, a well-suited model for exothermic chemical reactions without a barrier (e.g., see Fernandez-Ramos et al. 2006). This is especially realistic for the cases where the relevant rates would be needed in the low-temperature regimes with internally cold reacting partners. We have recently employed the VTST treatment for obtaining the reaction rates of similar C-bearing chains which included a terminal  group (Satta et al. 2015) and found them to be in line with the expected sizes of this type of reaction.
group (Satta et al. 2015) and found them to be in line with the expected sizes of this type of reaction.
More specifically, in the absence of a potential energy barrier during the formation of the intermediate "complex" between reagents in Equations (3) and (4), employing the VTST scheme allows one to estimate the necessary transition state (TS) partition functions as being given by the direct product between the conserved modes during reactions, Qcons, and the translational mode partition function Qtrans, along which the optimal reaction bottleneck is occurring:
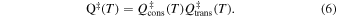
As discussed earlier, for the present reactions, the most favored exothermic path shall take place along the collinear MEP configurations so that the dominant translational mode will be along the  coordinates in both reactions. The present approach will also include the importance of quantum tunneling effects since the required passage frequency in a barrierless reaction can be taken equal to unity as the reaction enthalpy is here equal to zero (Fernandez-Ramos et al. 2006). The corresponding pre-exponential factor of the VTST approach is then given by:
coordinates in both reactions. The present approach will also include the importance of quantum tunneling effects since the required passage frequency in a barrierless reaction can be taken equal to unity as the reaction enthalpy is here equal to zero (Fernandez-Ramos et al. 2006). The corresponding pre-exponential factor of the VTST approach is then given by:
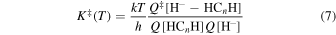
where all the required Q functions are obviously a function of T and are obtained for all the complex's geometries which evolve at different T values along the path located by the computed MEP from reagents to products. The rhs of Equation (7) is therefore also T-dependent, as it should be.
In order to employ the factor defined in Equation (7) to search for the lowest-positioned reactive path, one needs to generate the minimum values of the partition functions involved along the pseudo-2D process we are considering, i.e., along the RHH and RHC distances, for a broad range of the angle  between the reacting fragments. At all temperatures considered in the present study, the searches for minimum values of the partition functions yielded the collocation of the TS along the
between the reacting fragments. At all temperatures considered in the present study, the searches for minimum values of the partition functions yielded the collocation of the TS along the  value, a feature which thus confirms our earlier discussion of the dominance of the collinear paths to products formation in reactions (3) and (4). For a specific set of numerical examples we report in Table 1 the TS geometries obtained by variationally minimizing the partition functions of Equation (7) along the dominant pseudo-2D reaction path: in all cases the variationally attained angular value was the collinear one.The example shown is that of reaction (3).
value, a feature which thus confirms our earlier discussion of the dominance of the collinear paths to products formation in reactions (3) and (4). For a specific set of numerical examples we report in Table 1 the TS geometries obtained by variationally minimizing the partition functions of Equation (7) along the dominant pseudo-2D reaction path: in all cases the variationally attained angular value was the collinear one.The example shown is that of reaction (3).
Table 1. Computed Geometries of the Collinear, Pseudo-2D Transition State along the VTST Path from Reagents to Products, Together with the VTST Rate Constants at Various Temperatures
| T(K) | K(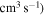
|
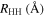
|
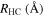
|
|---|---|---|---|
| 10.00 |
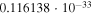
|
1.8308 | 1.0557 |
| 50.00 |
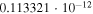
|
1.8307 | 1.0556 |
| 100.00 |
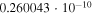
|
1.8498 | 1.0566 |
| 300.00 |
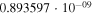
|
1.8913 | 1.0626 |
| 500.00 |
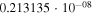
|
1.8871 | 1.0728 |
| 1000.0 |
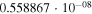
|
1.8317 | 1.0984 |
| 2000.0 |
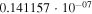
|
1.8115 | 1.1099 |
| 5000.0 |
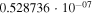
|
1.7949 | 1.1162 |
| 10000.0 |
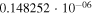
|
1.7972 | 1.1173 |
| 20000.0 |
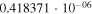
|
1.7972 | 1.1173 |
| 30000.0 |
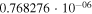
|
1.7972 | 1.1173 |
Note. The indicated data are for i  and acetylene.
and acetylene.
Download table as: ASCIITypeset image
One sees from the data reported by the table that the VTS remains essentially of very similar structures over a range of temperatures that varies from 100 K up to 30,000 K. This indicates that the absence of a barrier causes the reactions to occur at very similar geometries for all temperatures. The process takes place when  is close to the final geometry of the H − H product, while the terminal H in
is close to the final geometry of the H − H product, while the terminal H in  is not far from its value in the initial neutral molecule (see also maps of Figure 3). Considering that the physical mechanism of this exothermic reaction (with an estimated
is not far from its value in the initial neutral molecule (see also maps of Figure 3). Considering that the physical mechanism of this exothermic reaction (with an estimated  of 1.0 eV) is one in which the formation of the H2 molecule takes place as the ET occurs to the final anionic polyyne (
of 1.0 eV) is one in which the formation of the H2 molecule takes place as the ET occurs to the final anionic polyyne ( ), it stands to reason that the actual chemical forces at play would act at distances largely independent of the relative temperatures for which the rate is being considered since no barrier needs to be overcome during the final descent to products. It is also interesting to note that the reaction enthalpy given by the present calculations, (at T = 0 K), is 1.08 eV which is not far from the experimental estimates discussed earlier.
), it stands to reason that the actual chemical forces at play would act at distances largely independent of the relative temperatures for which the rate is being considered since no barrier needs to be overcome during the final descent to products. It is also interesting to note that the reaction enthalpy given by the present calculations, (at T = 0 K), is 1.08 eV which is not far from the experimental estimates discussed earlier.
The behavior of the VTS for reaction (4) is very similar to the one reported in Table 1, thus confirming the essential similarity of the mechanism involved. Also in that case the computed enthalpy at 0 K is 1.69 eV, close to the experimental estimates discussed in the Introduction. We report in Figures 12 and 13 the temperature dependence of the reaction rates for the processes of Equations (3) and (4). In each figure we show the temperature ranges covering the low-T conditions of the dense molecular clouds (Prasad & Huntress 1980).
It is well-known that in the setting-up of databases for the modeling of molecular processes in the ISM, the simple approach provided by the Langevin polarizability model is often employed for ionic reactions or for molecular processes involving ionic partners in general (e.g., see Wakelam & Herbst 2008; Walsh et al. 2009). Examples of its usage can also be found in the Ohio State University Chemical Network (OSU 2009).
In the present case, employing the dipole polarizability values for acetylene and diacetylene, the Langevin model leads to the following, temperature-independent rates:
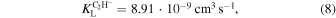
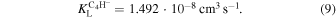
They are usually considered to provide upper bounds for the expected chemical rates of the processes considered and our quantum calculations given below indeed confirm such behavior.
From the data reported in Figure 12, for instance, we see that in the very low-T range the present rates are dramatically lower than the Langevin estimates and remain so over the whole low-T range shown in the figure. Furthermore, we also found by extending the calculations to higher temperatures (albeit not relevant for the dark molecular cloud environments) that the rate behavior above 500 K and all the way up to 30,000 K, becomes much less T-dependent and increases with T very slowly, its value finally becoming larger than the KL value at temperatures of a few thousand Kelvin.

Figure 11. Same data as those in Figure 10, but this time for the  interaction.
interaction.
Download figure:
Standard image High-resolution image
Figure 12. Computed reaction rates for the process involving the acetylene partner in reaction (3). The numerical fit for the low-T range is reported, while the T-independent line reports the value of the Langevin rate for the same reaction.
Download figure:
Standard image High-resolution imageWe now turn to the VTST calculations for the longer carbon chain: the results for the rates of formation of  are shown in Figure 13. Here again the data at the very low temperatures show a very rapid increase up to 50 K, beyond which value they become more slowly T-dependent and remain around
are shown in Figure 13. Here again the data at the very low temperatures show a very rapid increase up to 50 K, beyond which value they become more slowly T-dependent and remain around  , i.e., larger than the values obtained before for the reaction with acetylene. We also see that the Langevin estimate of these rates remains larger at all temperatures than our VTST results.
, i.e., larger than the values obtained before for the reaction with acetylene. We also see that the Langevin estimate of these rates remains larger at all temperatures than our VTST results.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 13. Same as in Figure 12, this time for reaction (4) involving diacetylene as a molecular partner.
Download figure:
Standard image High-resolution image{kind=link}
By using the analytic fitting formula employed in both cases (to be further discussed below) we can extend the VTST approach to the much higher temperatures (not shown here) and we can therefore see that the rates still exhibit a slow dependence on T and always remain lower than their Langevin estimates.
In conclusion, our VTST approach to modeling at the low temperatures the presently proposed chemical path to anions' formation indicates that the diacetylene derivative is a much more efficient partner in reaction with the hydrogen anion to form  than is the case for the smallest member of the polyynes series. The data of Table 2 clearly present the size differences between the rates involving our title systems.
than is the case for the smallest member of the polyynes series. The data of Table 2 clearly present the size differences between the rates involving our title systems.
Table 2.
Actual Numerical Values of the VTST Rates for the Reactions Involving Two Members of the Polyacetylene Series (with n = 2, 4) and the  Anion at Specific Low Temperature Values within the Expected Range of a Dark Molecular Clouds
Anion at Specific Low Temperature Values within the Expected Range of a Dark Molecular Clouds
| T(K) |
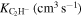
|
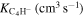
|
|---|---|---|
| 10 |
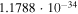
|
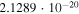
|
| 30 |
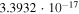
|
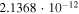
|
| 50 |
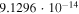
|
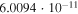
|
| 70 |
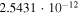
|
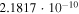
|
| 100 |
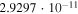
|
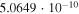
|
Download table as: ASCIITypeset image
Both sets of calculated rates have been fitted using a general formula frequently employed to describe ion–molecule reactions in the interstellar medium (e.g., see Woodall et al. 2007). The corresponding parameters are given in each of the relevant figures. The data in the table show that, for example at 30 K, the rate of formation of  with the present reaction is about five orders of magnitude larger than the rate of formation of
with the present reaction is about five orders of magnitude larger than the rate of formation of  , while at about 100 K the two rates become comparable and are both around the same, now larger rate value. This finding indeed suggests that, under cold molecular cloud conditions, the chemical route that we have investigated here becomes competitive and larger than the REA rates that we discussed below only for the longer member of the series while this chemical process is still not yet an efficient enough route to the formation of
, while at about 100 K the two rates become comparable and are both around the same, now larger rate value. This finding indeed suggests that, under cold molecular cloud conditions, the chemical route that we have investigated here becomes competitive and larger than the REA rates that we discussed below only for the longer member of the series while this chemical process is still not yet an efficient enough route to the formation of  anions. As we shall further discuss below, this point is in keeping with existing observations.
anions. As we shall further discuss below, this point is in keeping with existing observations.
4. CONCLUSIONS
In the present work we have obtained, by using an accurate ab initio description of the chemical forces at play, a realistic modeling of the formation of two members of the C-bearing molecular chains, both belonging to a larger set of chemical species discovered to be present in different environments of the ISM, especially within the cores of molecular clouds (e.g., see Cernicharo et al. 2007).
In particular, following an earlier suggestion by Cordiner & Millar (2009) whereby in the inner regions of the carbon-rich CSE stars the action of cosmic rays (CR) on H2 was assumed largely responsible for the formation of  in those regions
in those regions
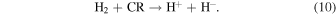
We have suggested that the stable atomic anions thus produced could be part of chemical reactions involving neutral acetylene-derivatives to form the anions of the corresponding polyynes:
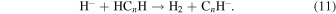
In the case of C-bearing chains with n = 2, 4 we find the process above to occur via exothermic reactions involving H2 formation acting concurrently with the ET process to the partner radicals, thereby producing the separation of final closed-shell anions. However, we also found that in the range of temperatures below about 50 K the reaction for the n = 2 member is much less efficient than the one for the n = 4 species.
A possible, qualitative explanation could come from noting that, at the lower temperatures, the residual kinetic energy needed by the product partners to escape the complexes, formed at the bottoms of either MEP energy troughs, is larger when the longer chains can help that escape by also relaxing their internal energies from their more numerous degrees of freedom, a feature also included within our VTS modeling. Thus, it would follow that fewer molecules of the smaller  partner can escape into the products' region than those from the longer diacetylene molecules, thereby yielding smaller rates at low-T for the former reaction compared to the latter. However, a more detailed quantitative explanation would have to wait for actual time-dependent reactive dynamics carried out in both systems, a study that we are currently planning in our group for the near future.
partner can escape into the products' region than those from the longer diacetylene molecules, thereby yielding smaller rates at low-T for the former reaction compared to the latter. However, a more detailed quantitative explanation would have to wait for actual time-dependent reactive dynamics carried out in both systems, a study that we are currently planning in our group for the near future.
According to Cordiner & Millar (2009) the  abundances in the inner envelopes are expected to be around
abundances in the inner envelopes are expected to be around  . It then follows that to have investigated the chemical routes of Equation (11) provides us with further options on the possible paths to formation of those anions. In the present study we have focused on the first two terms of the series,
. It then follows that to have investigated the chemical routes of Equation (11) provides us with further options on the possible paths to formation of those anions. In the present study we have focused on the first two terms of the series,  and
and  , since, for these two molecular anions the existing evaluations of the alternative route provided by the REA mechanisms as in Equation (1), have recently suggested (Douguet et al. 2015) such a mechanism to be very inefficient due to the dominance of the competitive autodetachment channels. In both cases, in fact, the estimated REA rates turned out to be around
, since, for these two molecular anions the existing evaluations of the alternative route provided by the REA mechanisms as in Equation (1), have recently suggested (Douguet et al. 2015) such a mechanism to be very inefficient due to the dominance of the competitive autodetachment channels. In both cases, in fact, the estimated REA rates turned out to be around  at temperatures of 30 K, thus indicating little importance for the REA paths to anion formation at low T. As we can see from our Table 2, therefore, our treatment of the present reaction path suggests that it is indeed a competitive option only for the formation of
at temperatures of 30 K, thus indicating little importance for the REA paths to anion formation at low T. As we can see from our Table 2, therefore, our treatment of the present reaction path suggests that it is indeed a competitive option only for the formation of  while it is instead found to be as inefficient as the REA process when it comes to the formation of the
while it is instead found to be as inefficient as the REA process when it comes to the formation of the  anion. Our present conclusions therefore confirm the fact that thus far no
anion. Our present conclusions therefore confirm the fact that thus far no  molecule has been observed in the ISM and that the data for the observation of
molecule has been observed in the ISM and that the data for the observation of  indicate that species to have very little abundance, as further discussed below.
indicate that species to have very little abundance, as further discussed below.
In terms of providing a molecular mechanism, the present findings suggest the present process to be dominated by a pseudo-2D scenario where the  interacts with one of the terminal H atoms and guides the detachment of the
interacts with one of the terminal H atoms and guides the detachment of the  final anion while the H2 molecule is formed. It is within this picture that the reactions were shown to be proceeding from reagents to products along a barrierless MEP that strongly favors the collinear arrangements of the two participating bonds, a process occurring at the bottom of their respective MEP energy troughs.
final anion while the H2 molecule is formed. It is within this picture that the reactions were shown to be proceeding from reagents to products along a barrierless MEP that strongly favors the collinear arrangements of the two participating bonds, a process occurring at the bottom of their respective MEP energy troughs.
On the strength of such findings we therefore surmise that the explored chemical routes to anion formation provide a more efficient alternative than the formation routes involving direct or indirect REA mechanisms only for the case of the  anion formation, while the
anion formation, while the  anion is still not significantly formed in this way at the expected temperatures of the dark clouds.
anion is still not significantly formed in this way at the expected temperatures of the dark clouds.
The recent observations of Cordiner et al. (2011) have provided specific indications for the  and
and  ratios (A/N ratios) over seven nearby galactic star-forming cores of molecular clouds. These measurements include four sources in which non-molecular anions have been previously detected and their findings for the
ratios (A/N ratios) over seven nearby galactic star-forming cores of molecular clouds. These measurements include four sources in which non-molecular anions have been previously detected and their findings for the  A/N ratios have turned out to be rather small (i.e., around
A/N ratios have turned out to be rather small (i.e., around  ), thus indicating rather limited permanent formation of the corresponding anion.
), thus indicating rather limited permanent formation of the corresponding anion.
Additionally, the recent observational data review regarding the possible presence of  (Morisawa et al. 2005) also came to the conclusion that there is no presence of that anion: it therefore would have to be in abundances even smaller than those for
(Morisawa et al. 2005) also came to the conclusion that there is no presence of that anion: it therefore would have to be in abundances even smaller than those for  . Our present results indeed confirm that finding: of the two members of the series discussed here, only the diacetylene derivative would have a sizeable rate of formation, whereas the acetylene derivative would not be formed in detectable amounts. In conclusion, the present computational study enables us to say that the reactions of C-bearing chains with the
. Our present results indeed confirm that finding: of the two members of the series discussed here, only the diacetylene derivative would have a sizeable rate of formation, whereas the acetylene derivative would not be formed in detectable amounts. In conclusion, the present computational study enables us to say that the reactions of C-bearing chains with the  atomic partner yield formation rates that are larger than those given by the REA formation path only in the case of the
atomic partner yield formation rates that are larger than those given by the REA formation path only in the case of the  anion, thereby adding another realistic element to the relevant chemical network for such anions.
anion, thereby adding another realistic element to the relevant chemical network for such anions.
The present work has therefore been capable of shining further light on the chemically complex network of reactions expected to control the final abundances in ISM environments of carbon-bearing chain anionic species. We hope then that it will spur additional experimental and computational studies on the complex chemical network relevant for providing the final answers to those questions.
Since the longer members of the polyynes have been found to be with larger abundances in the ISM, we intend to extend the present analysis to those members and shall report it, we hope soon, in the relevant literature.
F.A.G. and R.W. acknowledge the support of the Austrian Research Agency FWF through their Project no. P27047-N20.