


Abstract
Self-assembly is a process whereby molecules organize into structures with hierarchical order and complexity, often leading to functional materials. Biomolecules such as peptides, lipids and DNA are frequently involved in self-assembly, and this leads to materials of interest for a wide variety of applications in biomedicine, photonics, electronics, mechanics, etc. The diversity of structures and functions that can be produced provides motivation for developing theoretical models that can be used for a molecular-level description of these materials. Here we overview recently developed computational methods for modeling the self-assembly of peptide amphiphiles (PA) into supramolecular structures that form cylindrical nanoscale fibers using molecular-dynamics simulations. Both all-atom and coarse-grained force field methods are described, and we emphasize how these calculations contribute insight into fiber structure, including the importance of β-sheet formation. We show that the temperature at which self-assembly takes place affects the conformations of PA chains, resulting in cylindrical nanofibers with higher β-sheet content as temperature increases. We also present a new high-density PA model that shows long network formation of β-sheets along the long axis of the fiber, a result that correlates with some experiments. The β-sheet network is mostly helical in nature which helps to maintain strong interactions between the PAs both radially and longitudinally.
Export citation and abstract BibTeX RIS
Introduction
Biological systems are a great source of inspiration for developing new materials. Hybrid materials made from peptide, DNA, lipid, and polymer building blocks provide new ways for combining unique properties based on bioinspiration. However, the properties of these materials can vary a lot depending on such factors as peptide content, DNA base-pair composition and polymer structure. Computational modeling and simulation techniques are therefore essential components of biomaterials research with numerous ways to describe phenomena that occur across a range of length and time scales.
Peptide amphiphiles (PA) are a class of molecules that self-assemble into cylindrical nanofibers, spherical micelles, or flat ribbons [1–3]. They have hydrophilic amino acid sequences attached to a hydrophobic alkyl tail and can be designed to self-assemble into cylindrical nanofibers with a broad choice of peptide sequences that form β-sheet structures. Using bottom-up self-assembly, we can design synthetic materials with specific properties and hierarchical order. While covalent bonds chemically link the hydrocarbon tail and peptide region of the PAs, intermolecular forces such as hydrogen bonding, van der Waals and electrostatic interactions hold PA chains together. These non-bonded interactions provide opportunities for the design of functional materials with different properties as they allow for reversible steps and enable the formation of complex structures. Electrostatic interactions [4], hydrophobic interactions [5], hydrogen bonding and β-sheets [6] between the PA chains all play an important role in driving self-assembly. Earlier studies focused on understanding the driving force for assembly of these PA chains into cylindrical nanofibers instead of spherical micelles. Tsonchev et al. developed a theoretical model based on packing of PAs and performed Monte Carlo simulations which showed that the inclusion of directional electrostatic interactions between the charged headgroups leads to the formation of cylindrical micelles [4]. Furthermore, Velichko et al. used a simplified coarse-grained model to investigate the influence of hydrophobic interactions and hydrogen bonding in self-assembly [5]. Additional experimental studies showed that the ability of the amino acid residues closer to the core of the PA fiber to form β-sheets plays a significant role in self-assembly of PAs into cylindrical micelles [6]. Similar studies of PA nanofiber structures have looked at other factors, such as solution pH, temperature and counterions [7,8]. Cote et al.'s coarse-grained model on the effect of pH provided insights into the role that electrostatic interaction plays in the transition between spherical micelles and nanofibers for negatively charged PAs [8].
Peptide amphiphile fibers have been of particular interest because of their various applications in the fields of regenerative medicine and therapeutics. By programming the outer residues of the peptide appropriately, it is possible to stimulate biological activity when cells are exposed to fibers. Some of the applications of these PA nanofibers have been as templates to direct mineralization of hydroxyapatite in bone [9], to promote nucleation of silver nanoparticles to create hydrogels with antimicrobial activity [10], stimulate axon regeneration in spinal cord injury in mice [11], direct cell differentiation [12], aid in controlled drug release [13,14], reduce tumor growth in cancer therapies [15,16] and target injured blood vessels [17]. There have also been applications in nonmedical areas, including their use as chromatic sensors when conjugated with polydiacetylene [18], in semiconductor synthesis by controlled mineralization of CdS [19], for light-harvesting applications by ordering metalloporphyrin arrays [20], and in ester hydrolysis as a catalyst [21].
Self-assembly of peptide amphiphiles
Theory and models developed along with the experiments have formed a framework to understand PA self-assembly. However, the earlier simple models lacked much of the detail needed for a molecular level description, and, therefore, were not useful for modeling important functions of the fibers. More recent theoretical work that has included an all-atom description of the PAs has therefore been important for capturing the process of self-assembly.
Atomistic model
Lee and coworkers were the first to describe the self-assembly of PAs into cylindrical micelles using an all-atom seeded (preformed) molecular-dynamics (MD) method [22]. In this approach, an initial structure of the PA nanofibers (with peptide sequence SLSLAAAEIKVAV) was chosen with 9 PA chains per layer arranged in a circle and 16 layers stacked with a distance of 5 Å between each other, resulting in a total of 144 PA chains for the complete structure (similar to fig. 1, as explained below). The angle between the PAs in the same layer is 40°. Each layer is stacked so that it is rotated 20° relative to the layer below.
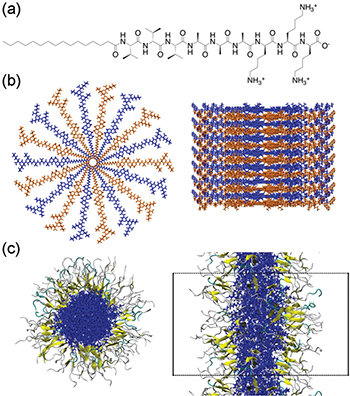
Fig. 1: (Colour online) (a) Peptide sequence used in this paper, VVVAAAKKK. (b) Initial structure of the PAs for the MD simulations based on [22]. 14 PA layers are shown in alternating blue and orange. (c) Top and side views of assembled structure at 310 K after 100 ns of simulation, respectively. The PA fiber core is shown in blue, β-sheets in yellow, turns in cyan and random coils in grey. Periodic boundaries are shown as black dotted lines. Water and ions are omitted for clarity.
Download figure:
Standard imageStarting with this seeded structure, a simulation was then defined in which the cylinder is in a box that defines an infinite periodic structure. Lee et al. found that the MD simulation stabilized in less than 40 ns, with a fiber diameter of ∼8.8 nm. On average, 14% of all PA residues formed β-sheets and the β-sheets were mostly parallel to the fiber axis. They found that the electrostatic energy between the PAs and counterions in solution as well as the van der Waals interaction between the PA chains stabilized the cylindrical fiber structure. The hydrogen bond network was most significant in the PA residues near the fiber core, and the β-sheets that formed provided stability to the cylindrical micelle.
These results are all in qualitative agreement with experiment [23], which provides validation for the model, however, quantitative comparison with experiment is not possible as structural tools for characterizing these disordered structures have limited capabilities. Another approach is to use further simulations to examine the robustness of the results to variations structural parameters. Later in this article we use this approach to explore properties of the β-sheets. These calculations follow the Lee et al. [22] seeded approach, but for a different PA sequence (Val3Ala3Lys3) that favors β-sheets. The chemical structure of the PA chain, the initial seeded structure of the fiber and the final assembled structure are shown in fig. 1. Details of the calculations are given in the Supplementary Material Supporting_Info.docx (SM). A discussion of the results is given later.
Coarse-grained model
Because all-atom models are computationally expensive to study long timescale processes such as self-assembly, Lee et al. [24] developed a coarse-grained model based on the Martini force field [25], in which a single bead represents a group of atoms, to simulate the self-assembly of PA fibers. This method allows for longer simulation times, even though much of the detail on the atomistic level is lost. Fortunately, the atomistic simulations performed previously were useful in providing the missing details of the model, such as the secondary structure of the PAs. The complete self-assembly starting from a random homogenous mixture of the PAs in water was simulated and this process was found to be complete in around 16 microseconds, as shown in fig. 2. The PA chains first formed small spherical micelles, which then merged to form a cylindrical fiber via van der Waals interactions. The final structure that formed was very similar to the atomistic result with ∼8.0 nm fiber diameter.
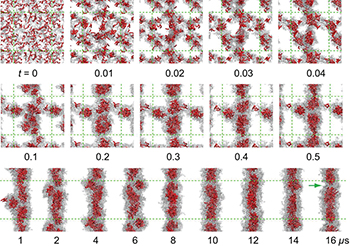
Fig. 2: (Colour online) Process of PA fiber formation from a homogeneous mixture. Hydrophobic tails are shown in red and peptides in gray. Snapshots from  to
to  (first row) show the early stage of self-aggregation. Snapshots from
(first row) show the early stage of self-aggregation. Snapshots from  to
to  are in the second row; whereas snapshots from
are in the second row; whereas snapshots from  to
to  are in the third row. Beads for water and ions are not shown for clarity. Green dotted lines represent periodic boundaries. The discontinuity of the hydrophobic core inside the fiber is indicated by a green arrow for the snapshot taken at
are in the third row. Beads for water and ions are not shown for clarity. Green dotted lines represent periodic boundaries. The discontinuity of the hydrophobic core inside the fiber is indicated by a green arrow for the snapshot taken at  . Reprinted with permission from Lee et al. [24].
. Reprinted with permission from Lee et al. [24].
Download figure:
Standard imageDynamics of the self-assembly process
Several computational studies have been performed to characterize the nature of the self-assembly process and the free-energy profile for the transition between the bound and free states of PAs in solution using steered molecular-dynamics (SMD) [26] and targeted molecular-dynamics (TMD) [27] methods. The free-energy associated with the addition of a single PA to a growing nanofiber was calculated to be enthalpically driven. Yu et al. discovered that while both electrostatic and van de Waals interactions have significant contributions to the self-assembly, the van der Waals interaction between the PA chains shows a larger effect [26]. In the next step of the analysis, Yu and coworkers looked at the complete self-assembly process and calculated the free-energy profile for transition between bound and free states of 90 PA chains [27]. In the first step of the self-assembly, PA chains randomly form aggregates as desolvation takes place. Then, they undergo conformational changes in which PAs form secondary structures including β-sheets and random coils with nearby PA chains in the aggregate. Also in this step, the PAs reorganize so that the tail and head of the PA chains are aligned to form the hydrophobic fiber core and the solvated shell. This structural change causes water to be excluded from the core of the fiber. Through this analysis, we also learn that the bound state, in which PA chains form a cylindrical nanofiber, is more stable than the free state, where PAs are randomly distributed in solution, with a difference of about 1.4 kcal/mol per PA.
Structural properties of peptide amphiphiles
The formation of β-sheets in PA nanofibers is important for biological applications where the mechanical properties of these nanomaterials play a role. Studies show that while nanofibers with weak intermolecular interactions promote cell death through disruption of cell membranes, nanofibers reinforced with β-sheets support cell viability [28]. Furthermore, fibers with most β-sheets had the highest mechanical stiffness [23], which is an important property for using PA nanofibers as scaffolds for cell growth.
When the peptide sequence contains amino acids with high β-sheet propensity, such as valine or alanine, long cylindrical nanofibers are formed. Both in experiments [23] and computer simulations [29], the valine-rich PAs showed increased β-sheet population compared to alanine-rich fibers. In one study [29], Lee et al. also showed that the two different PA sequences (PA1: Val2Ala4Glu3 and PA2: Val4Ala2Glu3) resulted in different fiber diameters (∼7.7 nm for PA1 and ∼8.0 nm for PA2). Although both fibers formed stable structures, β-sheets were not continuous along the fiber and it was not possible to observe whether the different mechanical properties of the two fibers arise from the different amount of twisted β-sheet structures as previously proposed by Heinig and Frishman [30].
The conditions in which self-assembly takes place, such as temperature, is significant for forming nanofibers with high β-sheet networks [31]. To prepare the PA nanofibers in the laboratory, Tantakitti et al. annealed the fresh PA solutions up to 80 °C during equilibration. They observed that the energy provided by annealing enabled nucleation and growth of long, monodisperse fibers [31].
We performed all-atom simulations at 310 K, 353 K and 400 K to observe the effect of temperature on the structure of the fibers. Details of the simulation protocol are included in the SM. Simulations at different temperatures were run using the seeded model shown in fig. 1. As we increase the temperature, the total percentage of β-sheets formed also increased. While 20.0 ± 0.8% of residues form β-sheets at 310 K, they amount to 25.4 ± 0.8% and 27.9 ± 1.0% at 353 K and 400 K, respectively. The distribution of β-sheets along the PA chain is of interest in understanding the effect of each amino acid on the overall structure. The PA sequence used in this study has three valine groups in the innermost section of the PA, and since valine groups favor β-sheet formation, this is expected to influence the nanofiber structure. In fig. 3 we show the percent of β-sheets formed in each amino acid residue. The highest number of β-sheets is found in the second and third valine residue of the peptide and the β-sheet formation decreases as we extend to the outer residues. Figure 3 compares the PA fibers with low and high β-sheet contents. When the temperature is increased to 400 K, the increase in β-sheet formation is not only in the valine residues, but also throughout the PA chain.
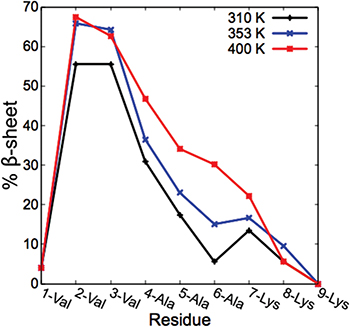
Fig. 3: (Colour online) Distribution of β-sheets along the peptide sequence at 310 K, 353 K and 400 K.
Download figure:
Standard imageAnother important consideration in modeling the self-assembly of PA fibers is fiber density in the initial seeded structure. In the original model [22], the initial fiber structure was assembled so that each stack of PAs was 5 Å apart, resulting in a fiber density of ∼19.2 PAs/nm. Although this configuration produced a stable fiber in less than 40 ns, only very short networks of β-sheets were formed. Because the experimental fiber density cannot be measured due to the limited resolution after the self-assembly takes place in solution, the validity of this model remains in question. We performed all-atom simulations (simulation details are given in the SM) using PAs with the sequence given in fig. 1(a) where we increased the fiber density to ∼27.7 PAs/nm by changing the distance between the PA stacks from 5 Å to 3.5 Å in the initial setup. This resulted in a denser structure with a larger diameter (∼9.6 nm) and longer networks of β-sheets along the long axis of the nanofiber. A total of 24.2 ± 0.5% of PA residues formed β-sheets.
The effect of temperature and fiber density on the formation of β-sheets is shown in fig. 4. With the original (5 Å) model, it takes ∼40 ns for the fibers to reach a stable structure. After the first 40 ns, the change in the number of β-sheets is small. Final assembled snapshots of the fibers at 310 K (fig. 1(c)), 353 K and 400 K (fig. 4(b)) are similar except for increased β-sheet content. At 40 K, we observe that even though there are many residues forming β-sheet structures (∼28%), these β-sheets are randomly distributed around the fiber instead of forming long networks parallel to the fiber axis. In contrast to the original model, when we increase the density of the PAs, the formation of β-sheets occurs very rapidly and remains stable over 100 ns. From the snapshots in fig. 4(b), we also see longer networks of β-sheets in the range from 2 to 9 PA chains. It is interesting to note that even though the higher-density results have only modestly higher β-sheet content than the low density at 310 K (24% vs. 20%), the extent of network formation is much higher. Similarly, the higher-density PA fibers show longer β-sheet networks compared to the original model at high temperatures that have larger β-sheet content. While most of the β-sheets in the high-density model are close to being parallel (< 25°) relative to the long axis of the fiber, some of the longer β-sheet networks are skewed. Selected PA chains are shown in fig. 5 as examples of networks formed in the high-density structure. This arrangement of β-sheets along the fiber fits the model proposed by Paramonov et al. [6] based on grazing angle and FT-IR studies. The hydrogen bonds are formed helically around the fiber core instead of completely parallel. This helps to maintain the interaction between the PAs both radially and longitudinally, and explains the elongation of the nanofibers along its main axis. Also because of this helical network, β-sheets are formed more strongly among amino acid residues that are closer to the fiber core since the shift in the radial direction causes the outer residues of the PA to be farther apart compared to the inner residues. There should, in principle, be a free-energy difference between the different density models that would determine which model is favored under equilibrium conditions, however, determining the free-energy difference is computationally challenging. Also, based on the experiments and theory in Yu et al. [32], it appears that many of the PA fibers are produced under kinetic rather than thermodynamic control.
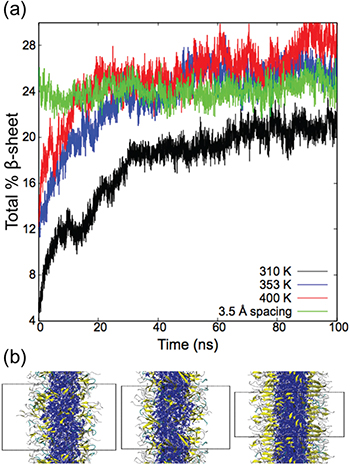
Fig. 4: (Colour online) (a) Total percentage of β-sheets formed with respect to simulation time. (b) Final assembled structures at 353 K and 400 K with the original model, and at 310 K with high PA density, respectively from left to right. The PA fiber core is shown in blue, β-sheets in yellow, turns in cyan and random coils in grey. Periodic boundaries are shown as dotted lines. Water and ions are omitted for clarity.
Download figure:
Standard image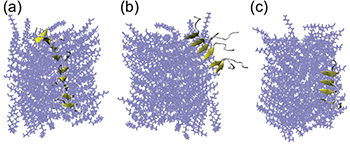
Fig. 5: (Colour online) Various lengths of β-sheets formed along the fiber. Angle between the long fiber axis and the vector formed by selected PA chains: (a) 19.3°, (b) 68.2°, (c) 17.9°. The PA fiber core is shown in blue, β-sheets are in yellow.
Download figure:
Standard imageConclusions and future directions
This paper has overviewed the development of molecular-dynamics models for the self-assembly of peptide amphiphile fiber structures in which a seeding procedure is used to drive the formation of cylindrical micelles. The resulting structures provide useful insight into what factors are important to the formation of β-sheets, and they are also useful for understanding the properties of the hydrophobic core and hydrophilic outer surface of the micelle. This information is important to understand fiber function, as the selection of the outer residues of the peptide determines the how the outer surface stimulates the biological activity when fibers come in contact with cells. Also, the hydrophobic core of the fiber can be functionalized for electrical or photonic properties, thereby allowing for device applications of the fibers.
Although the models described in this paper have been used for qualitative understanding of the fiber properties, the lack of quantitative experimental tools for characterizing fiber structures has made it hard to determine quantitatively how realistic the models are. Given this, we have focused on the sensitivity of the fiber structures (especially β-sheet properties) to the parameters of the model, including the variation of β-sheet populations with temperature, to the length of the simulations, and to the PA density. Our results show relatively weak sensitivity to temperature and simulation time, with β-sheet networks being largely converged after 40 ns simulations. However, a new result in this work is that the length of the β-sheet networks (but not the β-sheet fraction) increases significantly when density is increased. Since fiber mechanical properties are often correlated with β-sheet networks, it will be important in future work to assess if this effect of density can be used to better identify which model of these fibers (low- or high-density) is produced in the experiments as parameters such as pH and salt concentration are varied.
Acknowledgments
This work was supported as part of the Center for Bio-Inspired Energy Science, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic energy Sciences, under Award No. DE-SC0000989. We thank One-Sun Lee for help in the initial phase of this project.
Footnotes
- (a)
Contribution to Focus Issue Self-assemblies of Inorganic and Organic Nanomaterials edited by Marie-Paule Pileni.