


Abstract
The progress in the generation of ultrashort pulses has continuously triggered the introduction of new spectroscopic and measurement techniques which offer the opportunity to investigate unexplored research areas with unprecedented time resolution. Few-optical cycle pulses tunable from near-infrared to visible-UV allow to shed light on ultrafast electronic relaxation processes and to achieve real-time detection of molecular vibrations and structural dynamics. High-energy few-optical cycle pulses allow the efficient production of high-order harmonics up to XUV spectral region, leading to the generation of attosecond pulses shedding light on electron wave packet dynamics in complex molecules.
Export citation and abstract BibTeX RIS
Introduction
Many studies of light-matter interaction require optical pulses with ultrashort duration as well as broad frequency tunability. Both issues are of utmost importance for time-resolved optical spectroscopy and high-field physics. Several technological steps were required to advance ultrafast science from the nanosecond time scale to the femtosecond regime, and eventually to the attosecond regime. The laser provided the basis for all these advances, thanks to its ability to emit perfectly coherent light waves. In parallel, the birth of non-linear optics with the discovery of perturbative nonlinear effects [1,2] allowed to change the optical properties of the materials as a function of the laser radiation intensity. These effects provided light modulators able to vary the phase and/or amplitude of transmitted or reflected light in proportion to its intensity. Insertion of such devices in a laser oscillator allowed the eigenmodes of the laser cavity to be phase locked, turning the laser output into a regular train of light pulses with a duration inversely proportional to the laser bandwidth [3]. The continuous research effort in the development of broadband laser oscillators has allowed the generation of femtosecond pulses down to the few-optical-cycle regime [4,5] thanks to the Kerr-lens mode-locking technique [6] and to the use of chirped mirrors for dispersion control inside the laser cavity [7]. In parallel the invention of the chirped-pulse amplification technique has allowed to increase by orders of magnitude the pulse energy [8]. Nowadays, mainstream ultrashort pulse generation technology is based on Ti:sapphire oscillators operating in Kerr-lens mode-locking followed by chirped-pulse amplification, which provides highly stable and energetic femtosecond pulses. However, the frequency tunability of such sources is limited in a narrow range around the fundamental frequency (800 nm) or its second harmonic. Despite this limitation, the very high peak power and stability level of these sources enabled exploiting a series of non-linear optical effects either in the perturbative and non-perturbative regime generating an ensemble of "secondary sources" able to provide femtosecond light pulses with unprecedented tunability and duration and to enter the attosecond time regime.
Femtosecond spectroscopy nowadays is essentially based on the use of broadband optical parametric amplifiers (OPAs) as secondary sources, which exploit second-order perturbative non-linear optical effects. The principle of OPA is quite simple: in a suitable non-linear crystal, energy is transferred from a high-frequency and high-intensity beam (pump beam) to a lower-frequency, lower-intensity beam (signal beam) which is thus amplified by a proper phase matching condition. Suitably designed OPA can satisfy the phase matching condition over a very broad frequency range and can thus efficiently transfer energy from a narrowband pump pulse to a broadband signal pulse. These broadband amplifiers can be used to dramatically shorten, by more than an order of magnitude, the duration of the pump pulse [9]. One can therefore start with a femtosecond system producing relatively long pulses (100 fs) and use an OPA to shorten their duration to the sub 10 fs regime. This concept is very flexible and can be used to produce few-optical-cycle pulses over a very broad frequency range provided that proper phase matching conditions are identified [10]. Broadband OPAs and related pump-probe measurement techniques are fast enough to be able to resolve a large variety of molecular dynamics. The ability to probe elementary photophysical and photochemical relaxation processes and molecular structural changes is exploited by the fact that the temporal resolution can be pushed towards a few oscillation periods of the carrier optical wave (the period of an optical cycle in the visible and near-infrared is 2–3 fs). In addition the need to excite a system on resonance and probe optical transitions occurring at different photon energies is satisfied by the broad frequency tunability of such sources. However, the above time resolution is insufficient to capture atomic and molecular scale electronic motions.
Processes like electron tunneling in atoms, photoelectron emission from surfaces and charge oscillation in simple molecules occur on a much faster time scale requiring a sub-femtosecond time resolution. High-order harmonics, generated by focusing in noble gases powerful near-infrared few optical cycle pulses, producing coherent bursts of XUV radiation have been proven to be a valuable source of attosecond pulses [11,12]. This non-perturbative non-linear process is driven by the electric-field cycle and it is based on three steps: tunnel ionization, excursion of the quasi-free electron and its re-collision with the parent ion with emission of XUV attosecond pulses [13]. Powerful near-infrared few-optical-cycle pulses can be generated by a pulse compression technique based on the interplay of a wide spectral broadening induced by self-phase modulation in a hollow fiber filled with noble gases and an ultra-broadband group velocity dispersion setup [14,15]. The control of the field evolution of these pulses within the wave cycle permits attosecond control of the three steps involved in the emission process. So carrier envelope phase stabilized few-cycle light pulses are needed enabling the parameters of attosecond pulses to be precisely reproduced from one pulse to the next. The combination of waveform-controlled, few-cycle laser pulses and suitable gating techniques allow the reproducible generation of isolated attosecond pulses [16]. This constitutes an enabling technology for real-time access to a vast variety of electronic processes that are triggered on the attosecond time scale and may also evolve over the femtosecond time scale.
The purpose of this paper is to show two key examples of the application of femtosecond and attosecond pulses at the forefront of the laser technology and some perspectives in the field of ultrafast spectroscopy. Combining high temporal resolution with a broad spectral observation window it is possible to provide direct experimental evidence for the involvement of a conical intersection in the primary event of vision, which can be the basis for molecular structural changes in the femtosecond time scales. Then the direct measurement of the ultrafast charge dynamics in large molecules (as amino acids), initiated by attosecond pulses, provides a crucial step for the extension of attosecond methodology to complex systems. Charge fluctuations over large regions of a complex molecule can be indeed induced by attosecond pulses on a temporal scale much shorter than the vibrational response of the system.
The first step in vision: Visualizing wave packet motion through a conical intersection
Retinal proteins, also known as opsins, are a widespread family of photo-sensory proteins which are found in all domains of living organisms [17]. They are trans-membrane proteins sharing a common structure consisting of seven α-helices connected to each other by protein loops. Inside the protein pocket, the chromophore retinal is covalently attached to the seventh helix. Opsins can be broadly classified into type I or bacterial opsins, found in prokaryotes, and type-II opsins, found in animals possessing image resolving eyes. Type-I opsins are used by bacteria to harvest light energy in order to carry out metabolic processes. The prototypical one is bacteriorhodopsin, working as a pump for proton transfer across the cell membrane; the ensuing proton gradient is then converted into chemical energy. Type-II opsins are found in the photoreceptor cells (rod and cones) of the retina, where rod opsins (rhodopsins) are highly-sensitive receptors used for night and peripheral vision while cone opsins (photopsins) are pigments used for color vision.
In visual opsins the retinal pigment is in its 11-cis form. Light absorption by the retinal triggers an ultrafast isomerization to the all-trans form [18], which gives rise to a cascade of slower reactions, ultimately leading to the detachment of retinal from the protein pocket. The high quantum yield of rhodopsin isomerization (0.65) [19] and the ultrafast 200 fs production of the primary ground-state photoproduct [20] all evidence a paradoxically fast and efficient but one-way photoactivated reaction. Isomerization of rhodopsin is one of the most studied photochemical reactions. However, the structural and electronic dynamics that occur in the critical 100 fs time domain relevant to photochemical reactivity have remained experimentally unexplored.
It has been postulated theoretically that rhodopsin's unique reactivity is mediated by a conical intersection (CI) connecting the ground and excited electronic states [21,22]. CIs are ubiquitous features in theoretical descriptions of organic photochemistry and photobiology [23] and are responsible for triggering radiationless decay and efficient and ultrafast conversion of photon energy into chemical energy. CIs are collections of molecular geometries in which two (or) more electronic states are isoenergetic, forming a multi-dimensional "seam", any point of which may serve as a doorway through which a molecule may pass to reach a lower-energy electronic state. Depending on the topography around the crossing point, CIs can be classified as "peaked" or "sloped" [24]. In peaked CIs, the molecule is funneled towards the point of intersection regardless of the direction from which it approaches; in contrast, sloped topographies lead the molecule to the CI less efficiently, resulting in less efficient transitions and a wider variety of reaction products.
It has been gradually accepted that CIs are a critical part of rhodopsin photochemistry but experimental observation of CIs has remained elusive because the energy gap between the ground and excited electronic states of a molecule changes significantly over an ultrashort time scale. The direct observation of CIs thus calls for the combination of high temporal resolution and large spectral coverage to enable the real-time tracking of wave packet motion along the reaction pathway. Here we use ultra-broadband few-optical-cycle femtosecond pulses, tuneable from the visible to the near-infrared (NIR), to monitor the photoisomerization of the retinal chromophore in rhodopsin. These unique experimental capabilities allow us to directly observe, for the first time, the coherent motion of the wave packet from the Franck-Condon region along the excited-state surface, through the conical intersection all the way to the formation of the primary photoproduct [25]. Combined with highly accurate molecular dynamics, these results generate a comprehensive and dynamic picture of this important process and provide a general model for fast and efficient photochemistry mediated by CIs.
For our experiments we employ a home-built two-colour pump-probe spectrometer which provides sub-20-fs temporal resolution over a broad spectral range in the visible and infrared [26]. The photochemical reaction in purified bovine rhodopsin was initiated by excitation of the retinal chromophore with 10 fs pulses centred at 520 nm resonant with the ground-state absorption. The photo-induced dynamics were then probed by delayed pulses either in the visible wavelength region (from 500 to 720 nm, with  duration) or in the NIR (from 820 to 1020 nm, with
duration) or in the NIR (from 820 to 1020 nm, with  duration), both generated by broadband OPAs [10,26].
duration), both generated by broadband OPAs [10,26].
Figure 1(a) shows a two-dimensional differential transmission  map as a function of probe wavelength and delay. Immediately following excitation, we observed a positive signal (blue in the figure) with maximum intensity at
map as a function of probe wavelength and delay. Immediately following excitation, we observed a positive signal (blue in the figure) with maximum intensity at  , which can be assigned to stimulated emission (SE) from the excited reactant state. The SE signal rapidly shifts to the red while losing intensity and disappearing to wavelengths longer than 1000 nm within
, which can be assigned to stimulated emission (SE) from the excited reactant state. The SE signal rapidly shifts to the red while losing intensity and disappearing to wavelengths longer than 1000 nm within  . At this time, the
. At this time, the  signal changes sign and turns into a weak photoinduced absorption (PA) signal (red in the figure), which first appears at 1000 nm and then gradually blue shifts and increases in intensity. For delays longer than 200 fs, the PA signal stabilizes to a long-lived band peaking at 560 nm, indicating the presence of the all-trans photoproduct. Time traces at selected probe wavelengths are shown in fig. 1(b), highlighting the red shift of the SE signal and the subsequent blue shift of the PA.
signal changes sign and turns into a weak photoinduced absorption (PA) signal (red in the figure), which first appears at 1000 nm and then gradually blue shifts and increases in intensity. For delays longer than 200 fs, the PA signal stabilizes to a long-lived band peaking at 560 nm, indicating the presence of the all-trans photoproduct. Time traces at selected probe wavelengths are shown in fig. 1(b), highlighting the red shift of the SE signal and the subsequent blue shift of the PA.

Fig. 1: (Colour on-line) Wave packet dynamics through the rhodopsin conical intersection. Experimental (a) and simulated (c) differential transmission  map as a function of probe delay and wavelength in the visible and NIR spectral regions. Gray lines are a guide to the eye highlighting the shifts of the SE and PA signals in time. Experimental (b) and simulated (d) time traces at selected probe wavelengths (as indicated).
map as a function of probe delay and wavelength in the visible and NIR spectral regions. Gray lines are a guide to the eye highlighting the shifts of the SE and PA signals in time. Experimental (b) and simulated (d) time traces at selected probe wavelengths (as indicated).
Download figure:
Standard imageTo extract a dynamic model of the photoinduced process from these data we simulated the transient signals (S1 → S0 SE, S1 → S2 PA and S0 → S1 PA) by using scaled complete active space-self consistent field (CASSCF) transition energies over hybrid quantum-mechanical (QM, CASSCF)/molecular-mechanical (MM, Amber) trajectories [27] following the evolution of the opsin-embedded chromophore from the excited to the ground electronic state. This approach approximates full non-adiabatic complete active space with second-order perturbation theory (CASPT2) dynamics [28] that are currently possible only for smaller systems [29]. Virtual spectroscopies and dynamics (figs. 1(c) and (d)) agree almost quantitatively with the experimental results. The same holds for the 61% photo-isomerization quantum yield and  average S1 → S0 hopping time extracted from the simulations, thus further supporting the reliability of the theoretical approach.
average S1 → S0 hopping time extracted from the simulations, thus further supporting the reliability of the theoretical approach.
The ultrafast complex spectral evolution observed both experimentally and theoretically can be understood qualitatively with the help of fig. 2. Here, the motion of the wavepacket is depicted along the ground- and excited-state potential energy surfaces of the rhodopsin chromophore as a function of the isomerization coordinate. The wave packet initially created in the Frank-Condon region of the excited state of the 11-cis reactant rapidly evolves along the reaction pathway towards the CI, and the SE progressively shifts to the red as the band gap between the excited and ground states narrows. Near the CI region, which is reached in  according to both experiments and simulations, the SE signal vanishes as the two surfaces approach each other and the transition dipole moment decreases. Following the "jump" to the hot ground state of the photoproduct, a symmetric PA signal is formed. This PA band rapidly shifts to the blue as the surfaces move away from each other energetically and the wave packet relaxes to the bottom of the photoproduct well, reflecting the redistribution of the excess energy deposited in the molecule and the final torsional movement to the all-trans configuration.
according to both experiments and simulations, the SE signal vanishes as the two surfaces approach each other and the transition dipole moment decreases. Following the "jump" to the hot ground state of the photoproduct, a symmetric PA signal is formed. This PA band rapidly shifts to the blue as the surfaces move away from each other energetically and the wave packet relaxes to the bottom of the photoproduct well, reflecting the redistribution of the excess energy deposited in the molecule and the final torsional movement to the all-trans configuration.
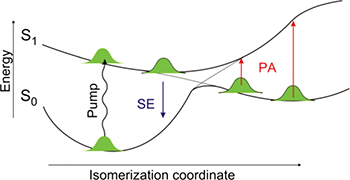
Fig. 2: (Colour on-line) Isomerization potential energy surfaces of rhodopsin. Sketch of the ground- and excited-state potential energy surfaces of the chromophore in rhodopsin as a function of the isomerization coordinate. It shows that stimulated emission (SE) from the excited state of the parent molecule and photo-induced absorption (PA) from the hot photoproduct can monitor the wave packet dynamics through the CI.
Download figure:
Standard imageTaken together, these data allow us to finally expose the complete molecular dynamics of rhodopsin isomerization and thereby understand how it achieves its unique reaction speed. The structural evolution from the reactant towards the CI is restricted almost exclusively to the atoms in the centre of the molecule. This behaviour is promoted by the tight-binding pocket provided by the protein to the chromophore, which restricts the possible motion at its ends through non-covalent interactions. Therefore, retinal can use all of the incident photon energy to drive minimal atomic displacements in the relevant region and reach the CI region within 80 fs resulting in a very fast reaction speed. Once the local isomerization has taken place, the overall highly strained structure can then relax in the photoproduct well to result in the more trans-like structure, which completes the primary isomerization reaction in rhodopsin [30].
These observations are consistent with a CI having a strongly peaked topography, leading to a very efficient ballistic transfer of the molecular wave packet from the 11-cis reactant to the all-trans photoproduct and to the very high quantum efficiency of the visual response. The present combined experimental-computational approach can be extended to investigate CIs with different topography, such as those in rhodopsin mutants or analogs (as 9-cis isorhodopsin [31]).
Electron dynamics in complex molecules initiated by attosecond pulses
Following the successful development of isolated attosecond laser pulses less than a decade ago, their use in studying atomic photoexcitation and photoionization and electron dynamics in solids has raised the prospects that molecular science may similarly benefit from the introduction of attosecond techniques. The time scale for the motion of atoms associated with chemical transformations is necessarily in the femtosecond domain, but the electronic rearrangement that accompanies the sudden removal or excitation of a selected electron is intrinsically faster. The application of attosecond techniques to molecules offers the possibility of investigating primary relaxation processes, which involve electronic and nuclear degrees of freedom and their coupling [32].
The first pump-probe measurement in molecules with sub-femtosecond temporal resolution has been reported in 2010 by Sansone et al. [33]. The process of charge localization after prompt ionization induced by isolated attosecond pulses was investigated in H2 and D2 molecules, by using attosecond pump pulses and 6 fs NIR pulses (with 740 nm central wavelength) with stable carrier-envelope phase (CEP). In this experiment the pump pulse ionizes the molecule, thus initiating a rearrangement of the remaining electron; the molecule dissociates during the interaction with the probe pulse. Upon measuring the kinetic energy and the angular distribution of the ionic fragment (H+ in H2 and D+ in D2), as a function of the temporal delay between pump and probe pulses, it was possible to follow and control the migration process of the electron remaining on the neutral fragment. Control of photo-ionization of D2 and O2 molecules has been achieved by using attosecond pulse trains [34,35].
Particularly interesting is the possibility to investigate the ultrafast electron dynamics in complex molecules (e.g., biologically relevant molecules), where sudden ionization by attosecond pulses may produce ultrafast charge migration along the molecular skeleton, which can precede nuclear rearrangement. This behavior has been predicted in theoretical calculations by various authors [36,37], whose work was stimulated by pioneering experiments performed by Weinkauf, Schlag, and co-workers on fragmentation of peptide chains [38]. This electron dynamics, evolving on an attosecond or few-femtosecond temporal scale, can determine the subsequent relaxation pathways of the molecule. The process is induced by the prompt generation of an electronic wave packet, which moves across the molecular chain and induces a site selective reactivity, which is related to charge localization in a particular site of the molecule. Although picosecond and femtosecond pulses are suitable for the investigation of nuclear dynamics, in order to study the electron wave packet dynamics in the outer-valence molecular orbitals relevant to most chemical and biological systems, attosecond pulses are required.
A typical experimental setup for pump-probe measurements with attosecond temporal resolution is shown in fig. 3. Visible/near-infrared (VIS/NIR) pulses, with 4 fs duration, 2 mJ energy and stable CEP, obtained by using the hollow-fiber compression technique [12], are divided in two parts by a beam splitter. The transmitted pulses are focused in a cell filled with gas (argon, krypton or xenon) for the production of XUV radiation by high-order harmonic generation (HHG). Isolated attosecond pulses, with energy up to a few nanojoules, can be produced by employing various temporal gating techniques (e.g., polarization gating and ionization gating) [39]. The VIS/NIR beam reflected by the beam splitter is collinearly recombined with the XUV beam by using a mirror with a central hole. The temporal delay between the attosecond pump pulses and the femtosecond VIS/NIR probe pulses can be adjusted with attosecond resolution by using a piezoelectric translation stage. Pump and probe pulses are then collinearly focused by a toroidal mirror in the interaction region, where the molecular sample under investigation can be placed.
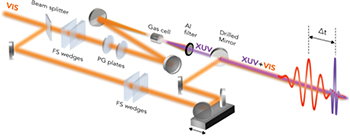
Fig. 3: (Colour on-line) Attosecond experimental setup. FS wedges: ultrathin fused silica wedges for fine dispersion compensation; PG plates: birefringent plates for polarization gating; Al filter: 100 nm thick aluminum filter to block fundamental radiation and to compensate for the intrinsic chirp of attosecond pulses.
Download figure:
Standard imageThe first application of attosecond pulses (in the form of short trains of pulses) to the investigation of ultrafast dynamics in a biologically relevant molecule, the amino acid phenylalanine, was reported in 2012 by Belshaw et al. [40]. In this case a femtosecond dynamics was evidenced, which was explained in terms of ultrafast electron transfer along the molecular structure. An amino acid consists of a central carbon atom (α carbon), linked to an amine (-NH2) group, a carboxylic group (-COOH), a hydrogen atom and a side chain, specific for each amino acid. Phenylalanine is an aromatic α-amino acid, where the side chain is a methylene (-CH2-) group terminated by a phenyl ring.
More recently, clear evidences of charge migration in the same amino acid molecule were reported [41]. The parent and fragment ions produced by the interaction of the molecules with the pump and probe pulses were collected by a linear time-of-flight device for mass analysis. Ionization induced by the attosecond pulse occurred in a sufficiently short time interval to exclude substantial electron rearrangement during the excitation process. The yield for the production of doubly charged immonium ions (whose structure is ++NH2–CH-R) was measured as a function of pump-probe delay. The temporal evolution of the immonium yield was characterized by a relaxation time of about 25 fs, which can be associated to a charge transfer process. Upon increasing the temporal resolution of the measurement by reducing the delay step between pump and probe pulses from 3 fs to 0.5 fs, a clear oscillation of the dication yield was evidenced. Figure 4 displays a 30 fs wide zoom of the exponential decay. The corresponding fitting curve (shown as solid red line in fig. 4), which closely follows the measured points, is given by the sum of an exponential function (with a time constant of 25 fs) and two sinusoidal functions, with periods of 4.3 fs and 3.4 fs, obtained from the Fourier transform of the experimental data. This ultrafast temporal evolution can be associated with electronic processes, which precede any relevant nuclear motion, thus representing the first experimental evidence of charge migration in a biologically relevant molecule.

Fig. 4: (Colour on-line) Generation yield of doubly charged immonium ion as a function of pump-probe delay. The red line is the fitting curve discussed in the text. Error bars show the standard error of the results of four measurements.
Download figure:
Standard imageTheoretical calculations to describe the hole dynamics induced by an attosecond pulse similar to that used in the experiment have been performed [41]. Due to the large bandwidth of the pulse, a manifold of ionization channels are open, thus leading to a superposition of many cationic states, i.e., to an electronic wave packet. For all open channels, the ionization amplitudes have been quantitatively determined. The evolution of the electronic wave packet has then been described by using a standard time-dependent density matrix formalism.
The results of the numerical simulations clearly show the production of an ultrafast electron dynamics, characterized by oscillation frequencies in excellent agreement with the experimental results. This is evidenced in fig. 5, which displays snapshots of the relative hole density calculated at three different pump-probe delays.
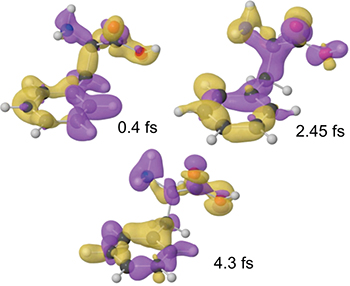
Fig. 5: (Colour on-line) Snapshots of the calculated electron dynamics for three particular pump-probe delays. The figure shows the variation of the hole density with respect to its time-averaged value (yellow and purple correspond to positive and negative values, respectively).
Download figure:
Standard imageThe ability to initiate and observe purely electronic dynamics in an amino acid represents a crucial step forward in attosecond science. The experimental observation of charge migration on an electron temporal scale can open new perspectives in the understanding of the physical processes governing electron transport mechanisms of biological signals. For instance, how the ultrafast motion of a hole in DNA created by a high-energy particle might initiate cell necrosis or mutation. Ultrafast charge migration is also the first step in many biological processes following a sudden energy deposition, either from light or electron and ion bombardment, like in photo- and hadron-therapy.
Perspectives
Major achievements in the study of atomic and molecular dynamics have been along with the development of appropriate ultrafast spectroscopic tools. For femtosecond spectroscopy further development of OPAs with broad gain bandwidths is the main perspective by selecting appropriate non-linear crystals and phase matching conditions in order to cover almost continuously the frequency range from the visible to the mid-infrared. We envision several applications for these tunable ultrabroadband pulses for two-color pump-probe spectroscopy on solid-state and molecular systems, with an unprecedented combination of temporal resolution and spectral coverage. In the attosecond science the currently available technology is limited by the low energy of the isolated XUV pulses, which poses constraints on the applications. The development of laser systems for the generation of high-energy, few-optical-cycle pulses with controlled electric field and continuous progress in the generation of XUV pulses from gas and solid targets excited by high-intensity laser pulses, promise to offer new possibilities for the generation of high-energy attosecond pulses. These developments would allow the possibility to perform attosecond-pump/attosecond-probe experiments and the investigation of non-linear processes in the XUV spectral region. Moreover, particularly promising will be the application of attosecond pulses to the investigation of multielectron systems with the possibility to directly measure and control ultrafast charge migration process in complex molecules opening the way to the emergence of attochemistry, with important consequences both for fundamental research and technology.
Acknowledgments
We acknowledge the contribution of D. Brida, F. Calegari, C. Manzoni, D. Polli, G. Sansone and A. Trabattoni. We acknowledge the contribution of various research groups. In particular, F. Frassetto and L. Poletto (IFN-CNR, Padova, Italy), D. Ayuso, A. Palacios and F. Martín (Universidad Autonoma de Madrid, Spain), M. Garavelli (Università di Bologna, Italy), P. Kukura (Oxford University, UK), R. A. Mathies (UC Berkeley, USA), S. De Camillis and J. B. Greenwood (Queen's University, Belfast, UK), P. Decleva (Università di Trieste, Italy). We acknowledge the support from the European Research Council under the ERC Advanced grants No. 227355 ELYCHE and No. 291198 STRATUS.