
Abstract
The ability of surfactant-like compounds to inhibit the corrosion of aluminum in NaCl solution was systematically investigated. The basic idea of this study was to scrutinize the effect of type of backbone chain (alkyl and perfluoroalkyl), length of backbone chain (number of carbon atoms 7, 10, and 17), various anchor groups (carboxylic, thiol, and imidazole) and presence of alkylene and benzene spacers between perfluoroalkyl chain and anchor group. To tackle these effects, three model studies were designed for alkaline etched, superhydrophilic aluminum surface and then approached experimentally and by density functional theory modeling. This enabled us to decouple the adsorption affinity of selected anchor groups on the hydroxylated aluminum surface from the lateral intermolecular cohesive interactions between hydrophobic backbone chains. Fourteen compounds were used to study the changes in the surface composition, wettability and the electrochemical barrier properties. For the carboxylic anchor group, the length and type of chain are important for barrier properties and also for tuning the wettability of the surface. The addition of alkylene spacer to perfluoroalkyl chain significantly affects the properties of the modified surface. Thiol and imidazole anchor groups, however, are not efficient inhibitors regardless the type and length of backbone chains.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
This is the third of a three-part series of articles aiming to tackle different properties of organic molecules that are responsible for adsorption and formation of condensed layers, which show barrier corrosion resistance and superhydrophobicity on aluminum surfaces. To this end, we have investigated surfactant-like organic molecules, whose structures can be described as consisting of an anchor group and a backbone chain, generally designated as Cx-R', where x is the number of carbon atoms in the alkyl backbone chain and R' is the abbreviation for the anchor group. The anchor group, responsible for adsorption, points towards the metal surface, while the backbone chain protrudes outward and brings about lateral interactions between adsorbed molecules. The following aspects of such systems were addressed in the first and second part of the series: the length of the alkyl chain for a particular anchor group 1 and the effect of changing the anchor group at particular lengths of the alkyl chain. 2
The fabrication of the adsorbed organic layer on the Al surface is the same in all three parts of the series, i.e. the alkaline etched, superhydrophilic aluminum surface was modified by immersion for 30 min at room temperature in a 5 mM ethanol solution of selected organic compounds. The methodology is also the same comprising synthesis, Al surface modification, surface characterization, electrochemical measurements, immersion testing and density functional theory (DFT) modeling.
In the first part of the study 1 we investigated the effect of the length of the alkyl chain for a single carboxylic anchor group, i.e. Cx-COOH, 5 ≤ x ≤ 17. The chain length turns out to be a decisive factor for achieving barrier corrosion protection and superhydrophobicity: only Al surfaces modified by long chain carboxylic acids (x ≥ 14) acted as strong inhibitors, thereby assuring efficient and superhydrophobic barrier protection. It was proposed that adsorption proceeds via the condensation mechanism with the magnitude of adsorption energy increasing with the length of alkyl chain. Only molecules with a long alkyl chain, like octadecanoic acid, completely cover the surface due to molecular tilting and stabilization of the molecular film. 1 The importance of the long alkyl chain was further corroborated in a parallel theoretical study, 3 where we showed that organic layers thicker than about 10 Å efficiently hinder the penetration of Cl− ions toward the metal substrate, whereas for thinner organic layers the barrier for Cl− penetration decreases with decreasing film thickness.
In the second part of the study 2 we investigated the effect of the anchor group and the length of alkyl Cx-R' chain. Nine anchor groups—azide, imidazole, thiocyanate, amino, disulfide, thiol, phosphonic, carboxylic and benzoic—bonded to octyl (C8-R') and octadecyl (C18-R') alkyl chains were studied. 2 The type of anchor group is crucial for adsorption on Al. When bonded to shorter, octyl chain, azide and disulfide act as corrosion activators, carboxylic, thiocyanate, thiol, amino, benzoic and imidazole are either not inhibitors or they are weak to moderate inhibitors and the phosphonic group acts as strong inhibitor. When bonded to a longer, octadecyl alkyl chain, imidazole and thiol groups remained weak inhibitors, the phosphonic group remained a strong inhibitor, and the carboxylic acid became a strong inhibitor. In other words, thiol and imidazole groups were not protective regardless the length of alkyl chain, the carboxylic group was protective for the longer chain, and the phosphonic group was protective regardless of the length of alkyl chain. The formation of condensed, protective surface layers is followed by a change in surface wettability from superhydrophilic to superhydrophobic.
Surface analytical and DFT studies, based on the methodology from our previous publications including time-of-flight secondary ions mass spectrometry (ToF-SIMS), 4,5 and DFT modeling, 6–8 complemented electrochemical results bringing about the possibility to fully understand the mechanism of adsorption, bonding and layer formation. 1,2 X-ray photoelectron spectroscopy (XPS) was used as a finger-print for the adsorption of organic compounds and the formation of dense molecular layers on aluminum surface. Carboxylic and phosphonic anchor groups are firmly bonded to the etched Al surface, whereas the other tested anchor groups, containing nitrogen or sulfur, are not. The bonding mechanism was then analyzed using ToF-SIMS and DFT calculations: carboxylic acids are bonded with the carboxylate group to either one or two surface Al atoms, while phosphonic acids are bonded to either two or three surface Al atoms. These molecules remain on the surface for an extensive period of time (at least six months) during immersion in NaCl solution, which implies that they are strongly chemisorbed.
In the current study, which is the third part of this series, we introduced perfluoroalkyl backbone chains of different lengths and studied the effect of type and length of backbone chain, the effect of anchor group, and the effect of spacer between perfluoroalkyl chain and anchor group. The behavior of perfluoroalkyl-only (Cfx-R') and alkyl-only (Cx-R') counterparts (or antipodes) of the same chain length (x = 7, 8, 10, 17) was compared, as well as those containing perfluoroalkyl chains with alkyl (Cfx-Cy-R', where y stands for the number of C atoms in the alkylene spacer) and benzene (Cfx-Bn-R') spacers; please note that the correct chemical name for the divalent "benzene spacer" is "p-phenylene spacer", but we use the term "benzene" and the abbreviation "Bn" for two reasons: (i) to be in compliance with our previous publication 2 and (ii) because in one case also a trivalent "benzene spacer" was used. As the anchor group, a carboxylic group was selected since it was shown 2 that for chains longer than seven carbon atoms, carboxylic acids efficiently adsorb on Al surfaces. Another possibility would be to introduce the phosphonic group, which also strongly adsorbs on Al surfaces. Additionally, thiol and imidazole groups were investigated.
It seems obvious that with DFT modeling we are not able to realistically capture experimental aspects, yet with properly simplified models the three principle aspects described above can be illuminated. In particular, the strength of DFT as a first-principles quantum-mechanical method is to adequately describe bond-breaking and bond-making and to provide an unbiased description of the constituent parts of the system (metal, oxide, organic layers). It should be noted, though, that a semiempirical dispersion correction is currently utilized for the sake of computational efficiency, because first-principles non-local van der Waals density functionals are computationally more expensive.
In compliance with experimental evidence that aluminum surface is covered with a hydroxylated layer, our computational approach consists in reproducing, though with simplified model, all the interfaces present: metal/oxide, oxide/hydroxide and hydroxide/organic layer, all these layers having an impact on the chemistry and the physics of the resulting system. The role of DFT modeling in the current study is therefore to provide information on the affinity of various anchor groups to Al substrates and to what extent the type and length of the backbone affects the anchor−surface adhesion and lateral intermolecular cohesion.
Our interest in perfluoro compounds stems from academic interest to compare the behavior of alkyl (CH3(CH2)x−1 or Cx) with perfluoroalkyl (CF3(CF2)x−1 or Cfx) backbone chain. The C–F bond, with a bond strength of 485 kJ mol−1, is the strongest single bond in organic chemistry; C–H and C–C bond strengths are 411 and 346 kJ mol−1, respectively. 9 The C–F bond, with length typically of about 1.35 Å, 10 is shorter than C–C bond, which is attributed to its additional ionic character due to the partial charges on the fluorine and carbon atom. The length of C–C bond in alkanes is 1.54 Å and that of C–H is about 1.10 Å. 11 We have found no similar study that would compare the behavior of alkyl and perfluoroalkyl backbone chains and terminal (anchor) functional groups. On the other hand, perfluoro compounds are very important in everyday life. 12 Perfluoro- and polyfluoroalkyl substances (PFAS) are man-made chemicals with part or all the hydrogen atoms replaced by fluorine atoms on the carbon skeleton with a terminal functional group. Aliphatic chains are hydrophobic but due to replacement of hydrogen by fluorine the perfluoro molecule becomes also oleophobic. 13,14 The production of perfluoro compounds began in 1950s and has scaled up to various fields of industry and everyday life, e.g., stain repellent fabric, surfactants, adhesives, antifouling coatings, masking tapes, firefighting foam, food packing, etc. Given their bond strength, low surface tension, hydro- and oleophobicity and chemical inertness, these molecules have extremely high biological resistance and are difficult to break down. For this reason, PFAS can accumulate and remain in the human body and in the environment over years and decades. 12 Perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) were recognized as a major global environmental contamination and toxicologic concern. 14 PFOS was restricted in use by the Stockholm Convention on Persistent Organic Pollutants in 2009 although many purposes are still allowed. 15 The cytotoxicity of perfluorinated carboxylic acids (carbon chain from four to twelve) was reported to increase with the length of the carbon chain, with chains shorter than eight C atoms being less toxic. 16 Alternatives for long-chain eight-carbon perfluoro compounds are short-chain b PFAS such as perfluorobutanesulfonic acid or perfluorohexanoic acid. However, also short-chain PFAS may be persistent in the environment and presents an environmental threat, as recognized recently. 12,13,16
Perfluorinated compounds have been used in surface modification of metal surfaces mainly with the purpose to produce superhydrophobic surfaces. 17,18 Recently, the combination of the chemical etching process in a FeCl3 solution and chemical surface grafting by immersion in ethanol solution containing 1H,1H,2H,2H-perfluorodecyltriethoxysilane was recognized as a viable route to achieve a hierarchical surface topography and chemical bonding of silane molecules on an aluminum surface leading to (super)hydrophobic, anti-icing and self-cleaning properties. 19 2-(perfluorohexyl)ethyl phosphonic acid was considered for modification of AZ31 magnesium surfaces but was inferior to n-octadecyl phosphonic acid. 20 The investigations of perfluoro compounds for corrosion protection are rather scarce. Perfluorinated layers were synthesized from 1H,1H,2H,2H-perfluorooctyltriethoxysilane to protect a light magnesium alloy. 21,22 Fluoropolymer coatings based on the fluoroethylene vinylether alternating polymers were used as a protective coating for a bronze work-of-art. 23 Branched chain vs straight chain fluorinated surfactants were considered as protection for carbon steel and it was reported that the former may exhibit better performance. 24 These listed studies usually used one or two perfluorinated compounds and mixed them with some hybrid matrix as to form coatings. To the best of our knowledge, no study similar to the present one—aiming to investigate the adsorption of perfluoro molecules on Al surfaces as a function of backbone chain length and anchor group—was reported. We therefore believe that the current study brings new information on the use of perfluoro-based inhibitors, thus supplementing and extending our previous two studies on alkyl backbone chains and various anchor groups.
Experimental
Substrate material
Aluminum (>99.0%) 1 mm thick flat sheet, distributed by GoodFellow, England, was used as substrate. Samples were cut in a form of 1 mm thick flat discs with a diameter of 15 mm.
Organic compounds
Organic compounds used for the formation of adsorbed layers on aluminum were either purchased from commercial suppliers or synthesized in-house.
The following chemicals were purchased: octanoic acid (C7-COOH, Alfa Aesar, 98%, liquid form, colorless), undecanoic acid (C10-COOH, Fluorochem, 95%, solid form, white), octadecanoic acid (C17-COOH, Acros Organic, 97%, solid form, white), perfluorooctanoic acid (Cf7-COOH, Alfa Aesar, 95%, solid form, white), perfluoroundecanoic acid (Cf10-COOH, Aldrich, 95%, solid form, white), perfluorooctadecanoic acid (Cf17-COOH, Alfa Aesar, 97%, solid form, white) and 4-octylbenzoic acid (C8-Bn-COOH, Ark Pharm, Inc., 98%, solid form, white).
The following chemicals were synthesized in-house: 2H,2H,3H,3H-perfluoroundecanoic acid (Cf8-C2-COOH, solid form, white), 1H,1H,2H,2H-perfluorodecanethiol (Cf8-C2-SH, liquid form, colorless), 2H,2H,3H,3H,4H,4H-perfluorododecanoic acid (Cf8-C3-COOH, solid form, white), 1H,1H,2H,2H,3H,3H-perfluoroundecanethiol (Cf8-C3-SH, liquid form, colorless), 1-[3-(perfluorooctyl)propyl]imidazole (Cf8-C3-ImiH, solid form, white), 4-(perfluorooctyl)benzoic acid (Cf8-Bn-COOH, solid form, white) and 3,5-bis(perfluorooctyl)benzoic acid ((Cf8)2-Bn-COOH, solid form, white).
Structural formulae and abbreviated labels of organic compounds are presented in Fig. 1.
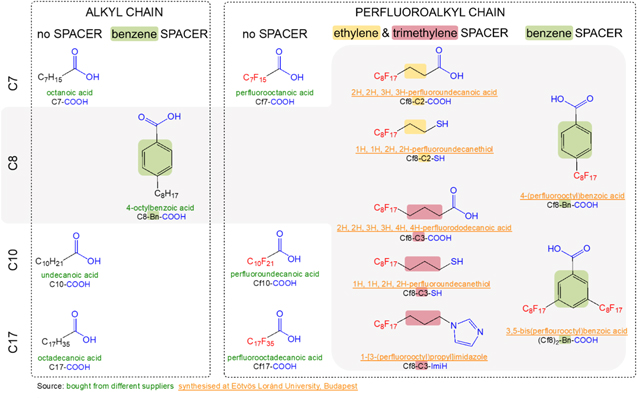
Figure 1. Skeletal and chemical formulae of alkyl and perfluoroalkyl organic compounds with different anchor groups used as potential corrosion inhibitors for etched aluminum. Compounds were either synthesized in our laboratory (colored orange) or purchased (colored green). Synthetic routes are described in Experimental (Scheme
Download figure:
Standard image High-resolution imageSynthesis of organic compounds
Seven chemicals were synthesized in-house:
a) 2H,2H,3H,3H,4H,4H-perfluorododecanoic acid (Cf8-C3-COOH) was synthesized from a 2H,2H,3H,3H,4H,4H-perfluorododecanenitrile. Mixture of 2H,2H,3H,3H,4H,4H-perfluorododecanenitrile (280 g, 0.575 mol), water (300 ml) and 98% sulfuric acid (300 ml) was stirred for 4 h at 170 °C in an oil bath (Scheme
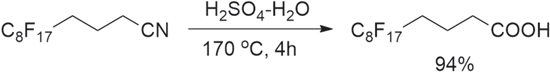
Scheme 1. Synthesis of 2H,2H,3H,3H,4H,4H-perfluorododecanoic acid (Cf8-C3-COOH).
Download figure:
Standard image High-resolution imageb) 2H,2H,3H,3H-perfluoroundecanoic acid (Cf8-C2-COOH) was synthesized from 3-(perfluorooctyl)-propanol (Scheme
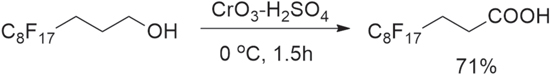
Scheme 2. Synthesis of 2H,2H,3H,3H-perfluoroundecanoic acid (Cf8-C2-COOH).
Download figure:
Standard image High-resolution imagec) 4-(Perfluorooctyl)benzoic acid (Cf8-Bn-COOH), p-C8F17C6H4CO2H, was synthesized by the copper assisted coupling reaction of C8F17I with 4-iodobenzoic acid as reported by Paciorek, et al. (Scheme
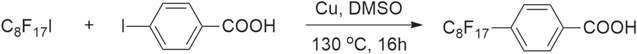
Scheme 3. Synthesis of 4-(Perfluorooctyl)benzoic acid (Cf8-Bn-COOH).
Download figure:
Standard image High-resolution imaged) 3,5-bis(perfluorooctyl)benzoic acid ((Cf8)2-Bn-COOH) was synthesized starting with the copper assisted coupling of C8F17I and methyl 3,5-dibromobenzoate, as reported by Rábai,
30
followed by the alkaline hydrolysis of the bis-fluorous methyl benzoate, as reported by Maas (Scheme

Scheme 4. Synthesis of 3,5-bis(perfluorooctyl)benzoic acid ((Cf8)2-Bn-COOH).
Download figure:
Standard image High-resolution imagee, f) 1H,1H,2H,2H-perfluorodecanethiol (Cf8-C2-SH, y = 2, where y stands for the number of C atoms in the alkylene spacer) and 1H,1H,2H,2H,3H,3H-perfluoroundecanethiol (Cf8-C3-COOH, y = 3) were synthesized from 2-(perfluorooctyl)ethyl iodide and 3-(perfluorooctyl)propyl iodide (Scheme
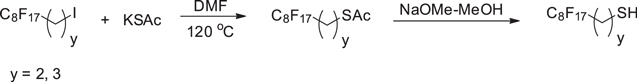
Scheme 5. Synthesis of 1H,1H,2H,2H-perfluorodecanethiol (Cf8-C2-SH, y = 2) and 1H,1H,2H,2H,3H,3H-perfluoroundecanethiol (Cf8-C3-COOH, y = 3).
Download figure:
Standard image High-resolution imageg) 1-(1H,1H,2H,2H,3H,3H-perfluoroundecyl)-imidazole (Cf8-C3-ImiH) was synthesized by the alkylating reaction of imidazole that was carried out by C8F17(CH2)3I in DMF (Scheme
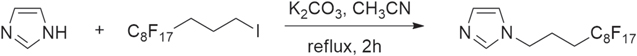
Scheme 6. Synthesis of 1-(1H,1H,2H,2H,3H,3H-perfluoroundecyl)-imidazole (Cf8-C3-ImiH).
Download figure:
Standard image High-resolution imageSubstrate pre-treatment and preparation of adsorbed organic layers
Aluminum samples were first ground under water using SiC papers up to 2400-grit (LaboPol, Struers) and etched in alkaline NaOH solution (p.a., Labochem International). 1,2,36 Briefly, 20 ml 0.1 M NaOH was heated in a flat-bottomed flask (volume 250 ml) using an ISOPAD® heater up to 90 °C. The aluminum samples were then immersed in NaOH solution and etched for 20 min at 90 °C. The samples were then cooled down for 45 min in the same solution to room temperature and rinsed by ethanol.
Organic layers were prepared by immersion of etched Al samples in ethanol solution (absolute for analysis EMSURE®) of organic chemicals at room temperature. The concentration of organic compounds was 5 mM and time of immersion was 30 min. The samples were hung in a glass vessel containing ethanol solution using a Teflon thread. After immersion, the sample was taken out, rinsed by distilled water, dried in a stream of N2 and used for further measurements.
Samples prepared by etching in NaOH and coated by immersion in ethanol solution of organic chemical are referred to as Cx-R' and Cfx-R', where Cx and Cfx denote an n-alkyl and n-perfluoroalkyl chains, respectively, consisting of x carbon atoms and R' is the abbreviation of the anchor group (Fig. 1): carboxylic (COOH), thiol (SH), and imidazole (ImiH). The length of carbon chain spans from seven to seventeen C atoms, i.e., x = 7, 8, 10, 17. In addition to pure alkyl and perfluoroalkyl chains, perfluoroalkyl chains with alkylene (Cfx-Cy-R') and benzene (Cfx-Bn-R') spacers were also studied.
Characterization methods
Electrochemical measurements
To record potentiodynamic polarization curves, a three-electrode cell (K0235 Flat Cell Kit, volume 250 ml Ametek, Berwyn, PA, USA) was used. Electrochemical measurements were performed in 0.5 M NaCl, pH = 5.8 (Honeywell Fluka, 99.5%) at room temperature.
A specimen (Al, Cx-R', Cfx-R', Cfx-Cy-R' and Cfx-Bn-R') embedded in a Teflon holder leaving an area of 1.0 cm2 exposed to the solution served as the working electrode. A silver/silver chloride (Ag/AgCl, 0.205 V vs standard hydrogen electrode) was used as the reference electrode and a platinum mesh as the counter electrode. Potentials in the text refer to the Ag/AgCl scale. Measurements were conducted using an Autolab potentiostat/galvanostat Model 204 (Utrecht, The Netherlands). Prior to measurements, the sample was allowed to rest under open circuit conditions for approximately 1 h, to reach a stable, quasi-steady state open circuit potential (Eoc) at the end of the stabilization period. Following stabilization, the potentiodynamic polarization curves were recorded using a 1 mV s−1 potential scan rate, starting 250 mV more negative with respect to Eoc, and then increased in the anodic direction. For each sample, measurements were performed at least in triplicate. Mean values with standard deviations are given in tables and a representative measurement was chosen to be presented in graphs. The corrosion potential (Ecorr) and the corrosion current density (jcorr) were obtained either from an intercept between cathodic and anodic curves or by extrapolation of the linear portion of the cathodic Tafel curve and the intersect with the line passing through Ecorr.
Immersion test
Immersion tests were carried out in 250 ml glass vials at room temperature (25 ± 2 °C). Samples were hung in the vial using a Teflon thread. The test lasted six months. After the denoted immersion time, the sample was taken out, rinsed by distilled water, dried in a stream of N2 and used for further measurements.
Wettability
Wettability of the samples was determined qualitatively. If the water drop completely spilled over surface, the sample was regarded as superhydrophilic (with high wettability). If the water drop bounced from the surface, the sample was regarded superhydrophobic (with low wettability). When the behavior of water drop was in between these extremes, the surface was regarded as hydrophilic or hydrophobic.
Surface morphology and composition
The morphology and composition of modified samples were characterized using the field-emission scanning electron microscopy (FE-SEM), JSM 7600 F, JEOL, Japan, equipped with energy dispersive X-ray spectroscopy (EDS) (Inca Oxford 350 EDS SDD); images were recorded at an energy of 5 keV in LEI (low secondary electron image) mode. Prior to analysis, the samples were sputter-coated with a thin Au layer.
Time-of-flight secondary ion mass spectrometry measurements were performed using a dual beam ToF-SIMS V spectrometer (ION-TOF GmbH, Muenster, Germany). The base pressure in the analysis chamber is maintained at less than 5.0 × 10−9 mbar in normal operating conditions. The total primary ion flux was less than 1012 ions cm−2 ensuring static conditions. A Bi+ primary ion source with a 1.2 pA current, scanned over a 100 × 100 μm2 area was used as the analysis beam. 2D spectra of negatively charged ions were recorded. Each sample was analyzed at least twice on different areas of the sample. Data acquisition and processing were performed using the IonSpec software. The exact mass values of at least five known species were used for calibration of the data.
X-ray photoelectron spectroscopy analysis (XPS) was performed using Thermo Electron Escalab 250 spectrometer. A monochromated Al Kα X-ray source (hν 1486.6 eV) was used. The base pressure in the analytical chamber was maintained at 10−9 mbar. The spectrometer was calibrated using Au 4f7/2 at 84.1 eV. The take-off angle was 90° and the analyzed area was a 500 μm diameter disk. Survey spectra were recorded with a pass energy of 100 eV at a step size of 1 eV and high resolution spectra of the C 1s, F 1s, Al 2p, O 1s, N 1s and S 2p core level regions were recorded with a pass energy of 20 eV at a step size of 0.1 eV. The values of the photoionization cross-sections (σX
) at 1486.6 eV were taken from Scofield,
37
and the inelastic mean free paths ( ) were calculated by the TPP2M formula.
38
) were calculated by the TPP2M formula.
38
Computational details
DFT calculations were performed with the PWscf code from the Quantum ESPRESSO distribution, 39,40 using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) 41 and a reparametrized D2 dispersion correction of Grimme 42 as to better describe the lateral intermolecular interactions. The reparametrization consists of setting the C6 parameter of Al to zero, because Al ions in the oxide film are practically devoid of valence electrons. This reparametrization was denoted as PBE-D0 in our previous publications. 2,6 Kohn–Sham orbitals were expanded in a plane-wave basis set up to a kinetic energy cutoff of 45 Ry (300 Ry for the charge density). Brillouin zone integrations were performed with the special point technique using a Methfessel–Paxton smearing of 0.03 Ry. 43 Molecular graphics were produced by the XCRYSDEN graphical package. 44
The model of the hydroxylated oxidized aluminum surface, designated as OH/Alx O/Al(111), is taken from our previous publications. 1,2,6,8,45 It is based on oxidizing the top side of a 6 layer slab of Al(111) with the equivalent of 2 monolayers of oxygen atoms as proposed by Lanthony et al. 46 The topmost layer of the film is then fully hydroxylated; the corresponding density of surface hydroxyls is 7.1 OH nm−2. Calculations were performed in a (4 × 4) supercell of Al(111) and a 3 × 3 × 1 k-point grid with the calculated Al bulk lattice parameter of 4.04 Å. Molecules were adsorbed on the top side of the OH/Alx O/Al(111)−(4 × 4) model and the bottommost Al(111) was kept fixed, whereas the positions of all other atoms were relaxed. The supercell dimensions were kept the same for all adsorption calculations. The supercell size along the surface normal direction was made sufficient for the thickest SAM to be separated by an about 20 Å thick vacuum region c from the adjacent slab. A dipole correction 47 was applied to cancel the artificial electric field that develops due to periodic boundary conditions imposed on the electrostatic potential. The structure of the OH/Alx O/Al(111)−(4 × 4) model is presented graphically in Fig. S1 in the Supplementary material (available online at stacks.iop.org/JES/168/071506/mmedia), whereas adsorption structures (i.e., Quantum ESPRESSO input and output files) presented herein are available at Mendeley Data. 48 Molecular adsorption was modeled at low and full monolayer coverages. Full monolayer coverage consists of four adsorbed molecules per (4 × 4) supercell, corresponding to molecular surface density of 3.54 nm−2. Low coverage was modeled by one adsorbed molecule per (4 × 4) supercell; such configurations will also be referred to as the "standalone adsorbed molecule", because at this coverage the intermolecular distances between neighboring molecules are beyond the range of chemical and London dispersion interactions.
Molecular chemisorption on OH/Alx O/Al(111)−(4 × 4) was modeled via the condensation reaction, where a molecule replaces a surface OH group by forming a water molecule as a co-product: 2,6
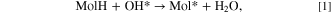
where the label MolH stands for an intact standalone molecule, Mol* represents a deprotonated molecule d adsorbed on the surface, and OH* is a surface hydroxyl group. The corresponding adsorption reaction energy (ΔEads) was calculated as:
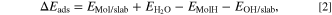
where EMol/slab and EOH/slab are the total energies of the Mol/slab adsorption system and the pristine OH-covered slab, respectively, and EMolH and  are the total energies of the inhibitor and water molecules, respectively.
are the total energies of the inhibitor and water molecules, respectively.
For a few cases we also considered plain non-dissociative molecular adsorption:
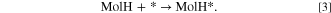
The corresponding plain adsorption energy (Eads) was calculated as:

where EMolH/OH/slab is the total energy of the adsorption system, i.e., intact MolH molecule adsorbed on the OH/Alx O/Al(111)−(4 × 4) model, designated in the subscript labels as OH/slab.
For the three considered anchor groups (carboxylic, thiol, and imidazole) the respective specific labels for MolH are R-COOH, R-SH, and R-ImiH, whereas for Mol they are R-COO, R-S, and R-Imi (note the omitted H), where R stands for backbone.
Results and Discussion
Presentation and synthesis of organic compounds
Ten perfluoro compounds were investigated (Fig. 1), among them seven were synthesized in-house (Scheme
Morphology and wettability of etched aluminum modified with perfluoroalkyl and alkyl compounds with various anchor groups
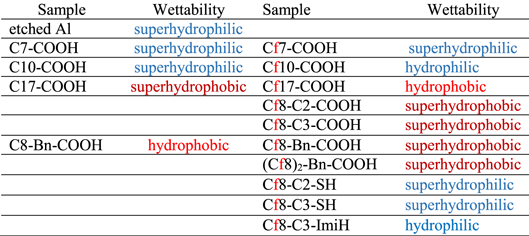
Morphology of alkaline etched Al was presented previously. 1,2 Hierarchical micro- and nanoscopically rough surface of etched Al does not change morphologically after immersion in ethanol solution of perfluoro and alkyl compounds for 30 min. The adsorbed organic layers formed are too thin to be observed by SEM; however, their formation can be confirmed indirectly through the change in wettability e of the surface (Table I). Alkaline etched Al is superhydrophilic (Table I). Among samples modified with alkyl compounds, only C17-COOH was superhydrophobic, C14-COOH was hydrophobic and those with shorter chains were superhydrophilic. f Among samples modified with perfluoroalkyl compounds, the one with long chain—Cf17-COOH—was hydrophobic, in contrast to its superhydrophobic alkyl antipode C17-COOH. Perfluoroalkyl chains with alkylene spacers, Cf8-C2-COOH and Cf8-C3-COOH, were superhydrophobic. Samples modified with backbones containing benzene instead of alkylene spacers, Cf8-Bn-COOH (14 C atoms in the backbone and spacer, x + y = 14) and (Cf8)2-Bn-COOH (x + y = 22) were also superhydrophobic. Perfluoroalkyl compounds containing SH anchor group did not change the wettability of Al, whereas samples treated with ImiH group became hydrophilic.
Table I. Wettability of the surface of alkaline etched aluminum and etched aluminum covered by an organic layer (labelled as Cx-R' and Cfx-R') where x is the number of carbon atoms in the chain (x = 7 or 17) and R' is the anchor group as defined in Fig. 1. Al was etched for 20 min in 0.1 M NaOH at 90 °C, cooled down in the same solution, and then immersed in 5 mM ethanol solution of the organic chemical.
|
Summarizing, for alkyl chains with the carboxylic anchor group, Cx-COOH, the increase in chain length for x > 12 results in progressively reduced wettability of the modified Al surface, with x = 17 even becoming superhydrophobic. The trend for counterpart perfluoroalkyl chains is similar but slower; even for Cf17 the modified surface is not superhydrophobic (Fig. 2). Perfluoroalkyl chain (Cf8) with alkylene spacer (y = 2 or 3), however, show smaller wettability than their alkyl-only or perfluoroalkyl-only antipodes which means that the addition of short alkylene spacer to moderately long (Cf10 or Cf11) hydrophilic perfluoroalkyl chain results in a superhydrophobic surface. The same effect is achieved with benzene spacer attached to one or two perfluorooctyl chains. This change in wettability with the introduction of spacers is valid only for the carboxylic anchor group, as compounds with thiol and imidazole anchor groups do not, or only insignificantly, change the wettability.

Figure 2. Wettability trends of alkaline etched Al and etched Al modified by organic compounds with alkyl-only, perfluoroalkyl-only, and perfluoroalkyl chains with alkylene or benzene spacers as a function of chain length attached to carboxylic COOH, thiol SH, and imidazole ImiH groups.
Download figure:
Standard image High-resolution imageHierarchical micro- and nanoscopically rough surface of etched Al does not change after immersion in ethanol solution of alkyl and perfluoroalkyl compounds and remained as such even after immersion in 0.5 M NaCl for six months. 1,2 Examples of samples modified with perfluoroalkyl compounds are presented in Fig. 3 for etched Al immersed in Cf8-C2-COOH, Cf8-C3-COOH, Cf8-C2-SH and Cf8-C3-SH. After six months in NaCl solution the wettability of the former two changes from superhydrophobic to hydrophobic.

Figure 3. SEM images recorded at the surface of (a, a1) alkaline etched Al and etched Al sample modified by (b) Cf8-C2-COOH, (c) Cf8-C3-COOH, (d) Cf8-C2-SH, and (f) Cf8-C3-SH after immersion in 0.5 M NaCl for 6 months. The etched Al, Cf8-C2-SH, and Cf8-C3-SH samples were superhydrophilic before and after immersion. The Cf8-C2-COOH and Cf8-C3-COOH samples were superhydrophobic before and hydrophobic after immersion. SEM images were recorded at 5 kV with magnification (a) 2,000×, (a1) 10,000×, and (b)–(e) 5,000×.
Download figure:
Standard image High-resolution imageStructural design of the study
The basic idea of this study was to investigate the effect of type of backbone chain (alkyl and perfluoroalkyl), length of backbone chain (x = 7, 10, and 17), various anchor groups (carboxylic (COOH), thiol (SH), and imidazole (ImiH)) and presence of alkylene (−(CH2)y – or Cy) and benzene (Bn) spacers between perfluoroalkyl chain and anchor group. To tackle these effects, three model studies were designed and then approached experimentally (electrochemical and surface analytical measurements) and by DFT modeling. These model studies are schematically presented in Fig. 4; for easier presentation, abbreviated labels are also shown therein.

Figure 4. Schematic presentation of the three model studies investigated in this work: (i) Model study 1 (M1) investigates the effect of type and length of alkyl and perfluoroalkyl chains at a constant COOH anchor group, (ii) Model study 2 (M2) investigates the effect of alkylene and benzene spacers for a perfluoroalkyl chain of constant length and the COOH anchor group; a comparison with pure alkyl and perfluoroalkyl chains of comparable length (from M1) is made, and (iii) Model study 3 (M3) investigates the effect of the anchor group (COOH, SH, and ImiH) for a perfluoroalkyl chain of constant length with ethylene and trimethylene spacers.
Download figure:
Standard image High-resolution imageModel study 1
The effect of type and length of the backbone chain was studied for three selected lengths of alkyl and perfluoroalkyl chains (x = 7, 10, and 17) at constant type of anchor group (COOH) (Fig. 4a).
Electrochemical properties
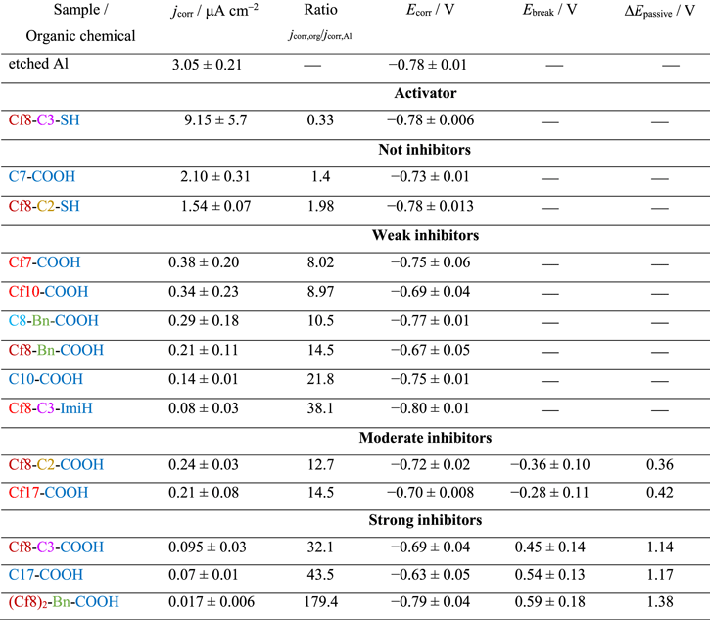
Potentiodynamic polarization curves in 0.5 M NaCl were recorded for alkaline etched Al (non-modified by organic layer), and alkaline etched Al samples immersed for 30 min in 5 mM ethanol solution of organic compounds with different lengths of alkyl and perfluoroalkyl chains and carboxylic anchor group (Fig. 5a). Electrochemical parameters deduced from polarization curves, which were recorded after 1 h under open circuit condition, are presented in Table II. The polarization curve of alkaline etched Al shows the cathodic branch related to the reduction of oxygen and the anodic branch to dissolution of Al. 49 The corrosion potential (Ecorr) was located at −0.78 V and corrosion current density (jcorr) was 3.05 μA cm−2 (Table II).

Figure 5. Potentiodynamic polarization curves recorded in 0.5 M NaCl for alkaline etched Al with and without prior immersion for 30 min in 5 mM ethanol solution of organic compounds with alkyl and perfluoroalkyl chains of different lengths (x = 7, 10, and 17) and carboxylic anchor group: (a) C7-COOH, Cf7-COOH, C17-COOH, and Cf17-COOH, and (b) C10-COOH and Cf10-COOH (Model study 1). Additionally, in (b) the effect of the ethylene spacer on perfluoroalkyl chain is presented (Cf8-C2-COOH) as to compare with the pure alkyl and perfluoroalkyl counterparts of comparable length (C10-COOH and Cf10-COOH) (Model study 2). The rest time under open circuit conditions was 1 h. The scan rate was 1 mV s−1. Electrochemical parameters are presented in Table II.
Download figure:
Standard image High-resolution imageTable II. Electrochemical parameters (corrosion current density (jcorr), corrosion potential (Ecorr), breakdown potential (Ebreak), and pseudo-passive range (ΔEpassive = ∣Ebreak − Ecorr∣)) deduced from potentiodynamic polarization curves (Figs. 5, 12, and 19) recorded for alkaline etched Al or etched Al samples covered by an organic layer with alkyl and perfluoroalkyl molecules, as defined in Figs. 1 and 4. Al was etched for 20 min in 0.1 M NaOH at 90 °C, cooled down in the same solution, and then immersed in 5 mM ethanol solution of organic chemical. Results are presented as mean-standard deviation. According to the level of protection, the organic compounds on modified Al surfaces are arbitrary classified as activators (jcorr,org > jcorr,Al), not inhibitors (jcorr,org up to 2 times smaller than jcorr,Al, no ΔEpassive), weak inhibitors (jcorr,org up to 40 times smaller than jcorr,Al, no ΔEpassive), moderate inhibitors (jcorr,org up to 15 times smaller than jcorr,Al, ΔEpassive about 0.5 V), and strong inhibitors (jcorr,org up to 180 times smaller than jcorr,Al, ΔEpassive ≥ 1.0 V).
|
Al samples modified by immersion in alkyl and perfluoro compounds show different curves than the alkaline etched Al sample, indicating that the presence of adsorbed organic layers affects the electrochemical response of Al (Fig. 5). All modified surfaces show smaller jcorr than the etched sample and act as mixed corrosion inhibitors because they caused a reduction of current density in both cathodic and anodic regions leading to an overall reduction in the corrosion current. Both alkyl and perfluoroalkyl chains with shortest x = 7 chain, C7-COOH and Cf7-COOH, are rather weak inhibitors inducing no significant change of the shape compared to that of etched sample and only a small shift in Ecorr (Fig. 5a). However, significantly smaller jcorr was observed for the perfluoro compound than for the alkyl x = 7 counterpart, i.e. 0.38 vs 2.03 μm cm−2, respectively. This is illustrated in Fig. 6, where the logarithm of jcorr is presented as a function of the number of C atoms. With prolongation of chain length to x = 10, the difference between perfluoroalkyl and alkyl compounds was reduced and they both showed similar jcorr values (0.34 and 0.14 μm cm−2, respectively), which were smaller compared to those with shorter chain length (Table II). With increasing chain length to x = 17 the corrosion resistance became strongly favorable for the alkyl chain (Fig. 5a): jcorr was by over one order of magnitude smaller than that of the etched Al sample, and, even more importantly, a broad pseudo-passive region was established with ΔEpassive g of 1.17 V. In contrast, the prolongation of the perfluoroalkyl chain did not bring such an improvement of the corrosion characteristics as for the alkyl chain; jcorr was smaller by one order of magnitude compared to the non-modified sample and Ecorr was shifted somewhat more positive but the pseudo-passive region was less extensive compared to the alkyl antipode.
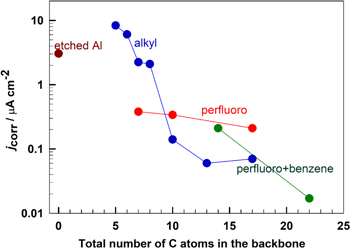
Figure 6. Corrosion current density measured for alkaline etched Al (chain length = 0) and Al modified by immersion in organic compounds of different lengths of alkyl and perfluoroalkyl chains and perfluoroalkyl chains with alkylene and benzene spacers attached to carboxylic group (chain length x = 7–22). Data were evaluated from potentiodynamic curves in Fig. 5 and Table II. Data for C5-, C6-, C8- and C13-COOH are taken from Ref. 1.
Download figure:
Standard image High-resolution imageExperimental electrochemical results show that the efficiency of barrier protection depends strongly on the chain length (as shown previously for alkyl chains 1 ) and it depends on the type of chain. At short chain lengths (x ≤ 7), alkyl and perfluoro carboxylic compounds can be characterized as not inhibitor and weak inhibitor, respectively, with the perfluoroalkyl counterpart being more efficient than the alkyl one (Fig. 6). For long chain lengths (x = 17), the trend is inversed and the alkyl counterpart acts as a strong barrier inhibitor surpassing the efficiency of the moderately strong perfluoroalkyl chain. This is reflected in a strongly reducing jcorr with increasing length of the alkyl chain (for x ≤ 10), whereas the slope of the comparative trend line for the perfluoroalkyl chain is much smaller (Fig. 6). The trends for the two counterparts intersect at approximately x = 10; for longer chains the alkyl counterparts surpass the perfluoro ones. The reduction in jcorr values and barrier character of the surface is accompanied by the establishment of broad ΔEpassive (Fig. 6, Table II).
The performance of inhibitors is often quantified in terms of inhibition efficiency (IE). The problem with IE is that it is highly non-linear, as discussed in our previous study, 50 i.e. at high values of jcorr the IE increases very rapidly for a small decrease of jcorr, whereas for IE > 90% it increases very slowly for a substantial decrease of jcorr. This is the reason that we find the jcorr,org/jcorr,Al ratio, where jcorr,org corresponds to etched Al modified by an organic layer and jcorr,Al to bare etched Al (Table II), more suitable than IE (Table SI in the Supplementary material). The modified Al surfaces are, according to experimental electrochemical data, arbitrarily classified as activators (jcorr,org > jcorr,Al), not inhibitors (jcorr,org up to 2 times smaller than jcorr,Al, no ΔEpassive), weak inhibitors (jcorr,org up to 40 times smaller than jcorr,Al, no ΔEpassive), moderate inhibitors (jcorr,org up to 15 times smaller than jcorr,Al, ΔEpassive about 0.5 V), and strong inhibitors (jcorr,org up to 180 times smaller than jcorr,Al, ΔEpassive ≥ 1.0 V). The ratios jcorr,org/jcorr,Al are given in Table II, where jcorr,Al denotes the corrosion current density measured for alkaline etched Al and jcorr,org denotes the corrosion current density measured for etched Al modified by organic chemicals in 0.5 M NaCl. Based on the proposed classification, the six organic chemicals from model study M1 are classified in the following order of increasing jcorr,org/jcorr,Al ratio and/or broadening of ΔEpassive: C7-COOH (not inhibitor) < Cf7-COOH (weak) < Cf10-COOH (weak) < C10-COOH (weak) < Cf17-COOH (moderate) < C17-COOH (strong).
XPS and ToF-SIMS analyses
High resolution C 1s and F 1s XPS spectra, recorded on the Al etched surface exposed to Cx-COOH and Cfx-COOH (x = 7, 10, or 17), are presented in Fig. 7. All C 1s spectra (Fig. 7a) evidence photopeaks at 285.0 eV associated to C–C and C–H bonds, and a second one at 289.3 eV corresponding to carboxylate anchor group. The adsorption of chemicals with an aliphatic chain and a carboxylic anchor group is strongly related to the chain length: 1 at short chain lengths such as octyl, the ratio C–C, C–H/O–C=O was much smaller than the stoichiometric one indicating that the carboxylic acid was present together with carbonaceous contamination on the surface. In contrast, for long octadecanoic chains the stoichiometric and experimental ratio was almost the same indicating that the surface is fully covered by the carboxylic acid. This is in line with electrochemical results where short aliphatic chains do not act as efficient inhibitors (Table II).

Figure 7. XPS high energy resolution spectra of (a) C 1s and (b) F 1s obtained on alkaline etched Al immersed for 30 min in 5 mM ethanol solution of Cx-COOH and Cfx-COOH (x = 7, 10, or 17).
Download figure:
Standard image High-resolution imageInhibitors with perfluoroalkyl chain (Cfx-COOH, x = 7, 10, or 17) present two peaks at 292.5 eV and 294.5 eV for CF2 and CF3 groups, respectively. The F 1 s core level presents an intense peak at 687.5 eV and a smaller one at 684.2 eV (Fig. 7b). The first one is associated to the CF2 and CF3 groups, while the second one corresponds to fluorine bonded to Al metal. 51 The Ctot/Ftot ratios, deduced from XPS intensities, are compared to the stoichiometric values (Table III). For Cfx-COOH (x = 7, 10, or 17), experimental values of Ctot/Ftot ratio are lower than stoichiometric ones, suggesting either less fluorine on the surface than expected or the presence of carbon contamination. Moreover, detection of F–Al bonds at the surface suggests that molecules dissociated via the C–F bond-cleave during (or after) adsorption. Hence these findings suggest the presence of "damaged" inhibitors on the Al etched surface, with fluorine also directly bonded to the metal.
Table III. Atomic ratios between fluorine and carbon calculated from XPS results (rXPS) compared to stoichiometric ratios (rstoich) (Figs. 7, 13, 15 and S2). The mismatch between the two values (in % units) is also given.
| Sample/Organic chemical | Ftot/Ctot stoichiometric | Ftot/Ctot experimental |
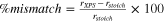
|
|---|---|---|---|
| C7-COOH | / | / | / |
| C10-COOH | / | / | / |
| C17-COOH | / | / | / |
| Cf7-COOH | 1.9 | 0.5 | 74 |
| Cf10-COOH | 1.9 | 1.4 | 26 |
| Cf17-COOH | 1.9 | 1.1 | 42 |
| Cf8-C2-COOH | 1.5 | 1.5 | 0 |
| Cf8-C3-COOH | 1.4 | 1.2 | 14 |
| C8-Bn-COOH | / | / | / |
| Cf8-Bn-COOH | 1.1 | 1.1 | 0 |
| (Cf8)2-Bn-COOH | 1.5 | 1.5 | 0 |
To confirm the presence of these inhibitors on the Al etched surface, ToF-SIMS spectra were recorded on the samples exposed to the inhibitors. An Al reference sample (alkaline etched sample non-modified by organic compound) was also analyzed by ToF-SIMS in order to identify the characteristic peaks of the inhibitors. Regarding Cx-COOH and Cfx-COOH inhibitors (x = 7, 10, or 17), the comparison between the ToF-SIMS mass spectra obtained on reference sample and on the samples exposed to inhibitors (Fig. 8) evidences the presence of each inhibitor on the Al surface. Indeed, specific and intense fragments, C7H15CO2 − (m/z = 143.1), C10H21CO2 − (m/z = 185.2), C17H35CO2 − (m/z = 283.4), C7F15CO2 − (m/z = 412.9), are assigned to C7H15CO2H (C7-COOH, 144 g.mol−1), C10H21CO2H (C10-COOH, 186 g.mol−1), C17H35CO2H (C17-COOH, 284 g.mol−1) and C7F15CO2H (Cf7-COOH, 414 g.mol−1), respectively. The detected fragments correspond to the mass of the inhibitor molecule minus one hydrogen, as commonly observed in ToF-SIMS negative polarity. However, focusing on C10F21CO2H (Cf10-COOH, 564 g.mol−1) and C17F35CO2H (Cf17-COOH, 914 g.mol−1) (Fig. 8b), the characteristic fragments observed do not correspond to the exact mass of the inhibitors (minus 1 H). Indeed, C8F13CO2 − (m/z = 387.0) is detected for the two latter inhibitors, in adequacy with their chemical formula. A heavier fragment, C16F32CO2 − (m/z = 830.9), is observed for C17F35CO2H (Cf17-COOH), i.e. the whole molecule without the terminal CF3 of the perfluoroalkyl chain (minus 1 H). This may be due to the ToF-SIMS process itself that reduces the probability of big ions emission. Note also the possibility to have fluorine directly bonded to the Al surface. Indeed, AlFn − (n = 2, 3, and 4) and AlOOFm − (m = 1 and 2) ions are observed with ToF-SIMS, when inhibitors contain the fluorinated alkyl chain (Fig. 9), confirming F–Al bond with XPS analyses. Hence fluorine can be bonded directly to the metallic substrate, as well as to the hydroxylated surface.

Figure 8. ToF-SIMS negative ion spectra obtained on alkaline etched Al surface and alkaline etched Al immersed for 30 min in 5 mM ethanol solution of Cx-COOH and Cfx-COOH (x = 7, 10, or 17).
Download figure:
Standard image High-resolution image
Figure 9. ToF-SIMS negative ion spectra obtained on alkaline etched Al surface and alkaline etched Al immersed for 30 min in 5 mM ethanol solution of Cfx-COOH.
Download figure:
Standard image High-resolution imageThus, all the inhibitors of model study M1 are detected on the Al surfaces using XPS and ToF-SIMS. As suggested in a previous study, 2 these inhibitors are bonded to the etched Al surface by the carboxylate group as monodentates or bidentates (not shown here). Esterification between the COOH group and the hydroxylated Al surface, with elimination of one water molecule, is the mechanism described for the monodentate bond. 2,6,52 However, for the bidentate bond, protonation of a hydroxyl group on the Al surface with subsequent elimination of a water molecule is required before the carboxylate can transform from monodentate to bidentate. 2,6,52 The formation of a bidentate can be therefore described as a three-step process: (1) formation of a monodentate (R–COOH + OH* → R–COO* + H2O), (2) protonation of the OH* group next to R–COO* (OH* + H+ + e− → * + H2O, where standalone * indicates a free adsorption site), and (3) formation of the second bond between COO* group and the surface (R–COO* + * → R–COO**, where suffix ** indicates bonding of COO to two surface sites, i.e., bidentate Al–O–C(R)–O–Al bonding). Another possibility is that a non-dissociated water molecule is already available at the neighboring Al ion, hence a monodentate R–COO* needs only to displace it away during the formation of the second chemical bond with the surface. It is indeed well known that not all surface Al ions of Al2O3 exhibit the same Lewis acid character and this results in a complex topology of OH and H2O adsorbed at the surface. 53,54
Computational modeling
In our previous publication we showed that the stability of self-assembled-monolayer (SAM) is due to (i) the molecule−surface adhesion and (ii) the lateral intermolecular cohesion within the SAM film. 6 The first effect is due to the anchor group and the latter effect is due to the backbone chain. We further showed that the lateral intermolecular cohesion interactions in SAM are optimized by the tilting of backbone chains which reduces the interchain distances so that they approach the optimum distance. Namely, the maximum attainable molecular surface density within SAM is given by the interplay between the steric footprint of the anchor group and the distribution of adsorption sites and it would be a coincidence if the resulting lateral distribution of the molecules would coincide with the optimum interchain distance. This principle behind the chain titling is shown schematically in Fig. 10. The distribution of adsorption sites obviously depends on the surface model and for the utilized OH/Alx O/Al(111) model the maximum surface density of carboxylic molecules within SAM is 3.54 nm−2, which results in the average intermolecular nearest-neighbor distance between adsorbed molecules of 6.5 Å (Fig. 10b), whereas the optimum interchain distance between alkyl chains is 4.2 Å, as deduced from the calculated crystal structure of polyethylene (PE) (Fig. 10c). For "fluorinated" PE (PEf), where alkyl chains are replaced by perfluoroalkyl chains, the optimum distance between chains is 5.7 Å, as deduced from the calculated crystal structure of PEf (Fig. 10d). The comparison between these distances reveals that alkyl chains need to tilt more than perfluoroalkyl chains to achieve the optimum interchain distance. A simple estimation, based on trigonometry, gives a tilt angle of about 50° and 30° (from the surface normal) for alkyl and perfluoroalkyl chains, respectively (Figs. 10e and 11), which is close to actually DFT calculated tilt angle for respective SAMs with 8 C atoms in the backbone chain (Figs. 11a, 11b).
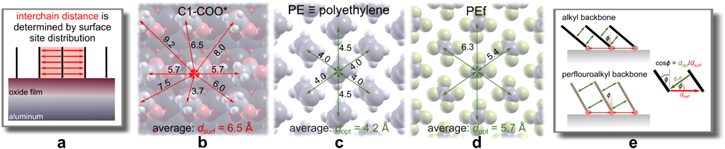
Figure 10. Schematic explanation of the chain tilting. (a) Lateral distribution of adsorbed molecules at high coverage depends on the interplay between the distribution of adsorption sites and the steric footprint of the anchor group. (b) On the utilized OH/Alx O/Al(111) model this interplay results in an average intersite distance (dsurf) between adsorbed molecules of about 6.5 Å, whereas (c,d) the optimum interchain distance (dopt) between alkyl chains is about 4.2 Å and between perfluoroalkyl chains is about 5.7 Å, as deduced from the crystal structures of polyethylene (PE) and "fluorinated" PE (PEf), respectively. (e) Because dsurf > dopt, the interchain distances are reduced by chain tilting, where the optimum tilting angle should be close to φ ≈ acos(dopt/dsurf). This implies that perfluoroalkyl chains need to tilt considerably less than alkyl chains to achieve the optimum interchain distance.
Download figure:
Standard image High-resolution image
Figure 11. (a) Dependence of the ideal chain-tilting angle, φ = acos(dopt/dsurf), on the intersite distance between adsorbed molecules for alkyl (purple curve) and perfluoroalkyl (green curve) chains. The actual DFT calculated chain-titling angles for C8-COO* and Cf8-COO* monolayers are shown by the two points and the corresponding structures are shown in (b). (c) Lateral interchain cohesion energy as a function of chain length x for alkyl (purple) and perfluoroalkyl (green) chains. Note that cohesion energy of alkyl chains displays bilinear behavior with gradual slope for chains shorter than the "switching" length (xs) of 7 C atoms and steep slope for chains longer than xs. In contrast, perfluoroalkyl chains display a linear behavior with a slope that is intermediate between those of the short and long alkyl chains.
Download figure:
Standard image High-resolution imageIn our previous publication we showed that the anchor-group adhesion and lateral interchain cohesion can be decoupled from each other and studied separately. 6 In compliance with this claim, we will show below in the model study M2 that, indeed, changing the backbone type does not directly affect the anchoring to the surface. Taking this into account, it can be then inferred that different characteristics between Cx-COOH and Cfx-COOH SAMs stem from how the backbone affects lateral cohesion in the adsorbed layer. Relying on the proposition that the anchor-group adhesion and interchain cohesion can be decoupled from each other, we utilized simplified DFT calculations of standalone SAM layers without the Al surface, where the carbon atoms of the carboxylic anchor group were fixed to their surface positions, whereas the oxygen atoms were deleted and replaced by either F or H atoms to form a methyl or perfluoromethyl group, depending on the type of the chain; this way the "standalone" SAMs were modeled by either alkane or perfluoroalkane layers with the geometry compatible to that of the respective adsorbed SAMs. The corresponding results are shown in Fig. 11c, which shows the lateral interchain cohesion as a function of the chain length for Cx- and Cfx-based SAMs at full coverage, 1 ≤ x ≤ 17. This figure reveals that the dependence on chain length is linear for perfluoroalkyl chains and bilinear for alkyl chains with slope for short chains being less steep than that for long chains. This bilinear behavior was explained in detail in our previous publication 6 and it originates from chain tilting by which the chains try to reduce the interchain distance as to optimize lateral interactions, however, for the effect to kick-in the chains need to be longer than the switching length xs (i.e., a chain length at which the slope of cohesion energy changes). For alkyl chains, which need to tilt significantly, xs = 7 C atoms, whereas for perfluoroalkyl chains, which need to tilt considerably less, xs = 1 C. This means that the tilting is fully effective already for the shortest perfluoroalkyl chain, hence for perfluoroalkyl chains the dependence of lateral interchain cohesion on the chain length is linear. Among the three slopes, shown in Fig. 11c, the slope for long alkyl chains is the steepest, followed by the slope for perfluoroalkyl chains, whereas the slope for short x < 7 alkyl chains is the least steep. That the slope of the long alkyl chains is greater than that of the perfluoroalkyl chains agrees with the cohesive energies in PE and PEf crystals, which are calculated to be −109 meV/CH2 and −90 meV/CF2, respectively.
The interplay between the three slopes—gradual slope of short alkyl chains, steep slope of long alkyl chains, and moderate slope of perfluoroalkyl chains—has interesting consequences. In particular, perfluoroalkyls are "better" for shorter chains, but for longer chains the steeper slope of alkyl chains eventually wins and net cohesion in an alkyl layer becomes stronger than that in perfluoroalkyl layer. In Fig. 11c, the crossing point is at chain length of about 12 C atoms. However, it is worth noting that the crossing point is specific to the model of the surface and it likely takes different values for different distributions of surface adsorption sites. The results presented in Fig. 11c therefore appear compatible with experimental observation that the shorter-chain Cf7-COOH compound is superior to C7-COOH and vice-versa for the longer-chain Cf17-COOH and C17-COOH (Fig. 6).
Model study 2
The effect of alkylene (−(CH2)2– or C2) and benzene (Bn) spacers was studied at a selected length of perfluoroalkyl and alkyl chains (x = 8) and constant type of anchor group (COOH) (Fig. 4b). The results for Cf8 chain and alkylene C2 spacer (Cf8-C2-COOH) were compared to pure perfluoroalkyl and alkyl chains of comparable length, i.e., Cf10-COOH and C10-COOH that were presented in model study M1 (Fig. 5b). The results for Cf8 and (Cf8)2 chains with Bn spacer (Cf8-Bn-COOH and (Cf8)2-Bn-COOH), with the total chain length of x + y = 14 and 22, respectively, were compared to pure perfluoroalkyl Cf17-COOH and alkyl C17-COOH antipodes.
Electrochemical properties
Figure 5b presents polarization curves for C10-COOH, Cf10-COOH and Cf8-C2-COOH samples aiming to tackle the effect of the C2 alkylene spacer in the perfluoroalkyl chain. Compared to "pure" C–H and C–F chains with x = 10, the replacement of −(CF2)2– with −(CH2)2– considerably improves the corrosion characteristics. Pure chains show similar curves with exponentially increasing current density in the anodic range. In contrast, chain with the C2 spacer exhibited a similar jcorr value (Table II), but the pseudo-passive range was established with a ΔEpassive of 0.38 V indicating barrier character of the modified surface. Therefore, the corrosion resistance increases in the following order Cf10-COOH < C10-COOH < Cf8-C2-COOH.
If a benzene ring was inserted instead of a short alkylene spacer (Cf8-Bn-COOH), the effect was smaller; jcorr values were similar to Cf8-C2-COOH (Fig. 12) but no pseudo-passive region was established. The effect is similar for both alkyl (C8-Bn-COOH) and perfluoroalkyl (Cf8-Bn-COOH) chains. Although the total number of C atoms in both backbone chains is 14, these modified samples did not achieve considerable improvement compared to pure alkyl and perfluoroalkyl antipodes with x = 10 (Fig. 5b). A considerable change was introduced only when using a benzene ring spacer with two side perfluorooctyl chains, i.e., (Cf8)2-Bn-COOH, which were superior to long alkyl chain (Cf17-COOH) (Fig. 12).
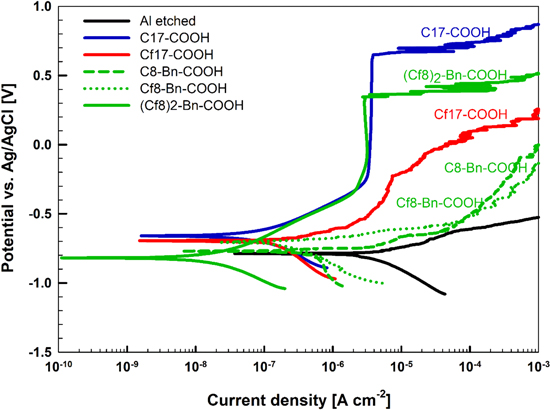
Figure 12. Potentiodynamic polarization curves recorded in 0.5 M NaCl for alkaline etched Al with and without prior immersion for 30 min in 5 mM ethanol solution of organic compounds with octyl alkyl and perfluoroalkyl chains with benzene spacer and carboxylic anchor group: C8-Bn-COOH, Cf8-Bn-COOH, and (Cf8)2-Bn-COOH (model study M2). For comparison, the curves for samples immersed in pure alkyl and perfluoroalkyl counterparts with longer chains, C17-COOH and Cf17-COOH (model study M1), are also shown. The rest time under open circuit conditions was 1 h. The scan rate was 1 mV s−1. Electrochemical parameters are presented in Table II.
Download figure:
Standard image High-resolution imageAccording to the postulated classification, the ten organic chemicals from model studies M1 and M2 are classified in the following order of increasing jcorr,org/jcorr,Al ratio and/or broadening of ΔEpassive: C7-COOH (not inhibitor) < Cf7-COOH (weak) < Cf10-COOH (weak) < C8-Bn-COOH (weak) < Cf8-Bn-COOH (weak) < C10-COOH (weak) < Cf8-C2-COOH (moderate) < Cf17-COOH (moderate) < C17-COOH (strong) < (Cf8)2-Bn-COOH (strong).
XPS and ToF-SIMS analyses
When Cy H2y (y = 2) spacer was added in the inhibitor formula between the fluorinated aliphatic chain and the carboxylic group, i.e. for Cf8-C2-COOH, XPS peaks characteristic of both molecules are observed (Fig. 13). Indeed, the carboxylate group and C–F bonds are detected on the C 1s and F 1s core level spectra at the binding energy described previously. However, there is no peak at 684.2 eV corresponding to the F–Al bond and there is no difference between the Ftot/Ctot experimental and stoichiometric ratios (Table III). XPS results show that the spacer tends to increase the stability of the molecule at the surface since the mismatch of atomic ratios between fluorine and carbon calculated from XPS results compared to stoichiometric ratios is 74% for Cf7-COOH and 0% for Cf8-C2-COOH.

Figure 13. XPS high energy resolution spectra of (a) C 1 s and (b) F 1 s obtained on alkaline etched Al immersed for 30 min in 5 mM ethanol solution of C10-COOH, Cf8-C2-COOH, or Cf10-COOH.
Download figure:
Standard image High-resolution imageWith the C2 spacer inserted between the fluorinated Cf8 aliphatic chain and the carboxylic group, the ionic species detected by ToF-SIMS on the Al surface (Fig. 14) is C8F17-C2H4-COO− (m/z = 491.1). These fragments evidence the presence of the molecule on the surface since it corresponds to C8F17-C2H4-COOH (Cf8-C2-COOH, 492 g.mol−1) minus 1 H. The inhibitor is bonded by the carboxylic group as described previously. Hence XPS and ToF-SIMS highlight the presence of these molecules, with the possibility to have F bonded to the Al surface as described for model M1 (Fig. 9). The detection of Al-F bonds by ToF-SIMS only (AlFn − and AlOOFm _) and not by XPS indicates that their number is very low (less than 1%—detection limit of the XPS).
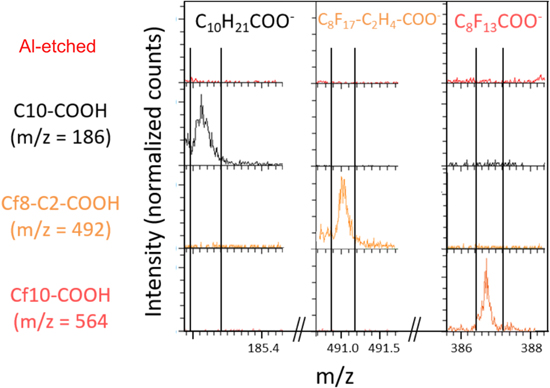
Figure 14. ToF-SIMS negative ion spectra obtained on alkaline etched Al surface and alkaline etched Al immersed for 30 min in 5 mM ethanol solution of C10-COOH, Cf8-C2-COOH, or Cf10-COOH.
Download figure:
Standard image High-resolution imageRecently, Milošev et al. 1 confirmed the affinity of aliphatic carboxylic acid with benzene derived spacer (Bn) for adsorption on aluminum. In this study, the aliphatic chain was H-free, and F saturated. Hence, changing the alkylene C2 spacer for a benzene spacer (Fig. 15) does not change previous observations and the carboxylic group is detected for C8-Bn-COOH, Cf8-Bn-COOH and (C8)2-Bn-COOH. CF2 and CF3 peaks are present for the fluorinated inhibitors, and there is no F–Al bond on the XPS spectra. Ftot/Ctot ratio are identical between stoichiometric and experimental values (Table III), meaning that these inhibitors are also bonded to the Al surface without being damaged. XPS results show that the spacer tends to increase the stability of the molecule at the surface.
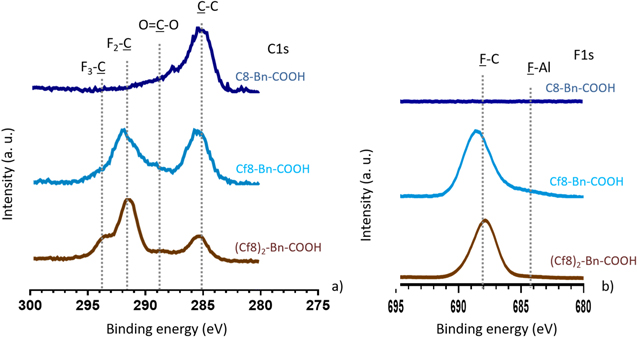
Figure 15. XPS high energy resolution spectra of (a) C 1 s and (b) F 1 s obtained on alkaline etched Al immersed for 30 min in 5 mM ethanol solution of C8-Bn-COOH, Cf8-Bn-COOH, or (Cf8)2-Bn-COOH.
Download figure:
Standard image High-resolution imageFigure 16 indicates by ToF-SIMS that C8H17-Bn-COO− (m/z = 233.2) and C8F17-Bn-COO− (m/z = 538.9), corresponding to C8H17-Bn-COOH (234 g.mol−1) and C8F17-Bn-COOH (540 g.mol−1) minus one H, are detected on the surface, ensuring that the inhibitors are bonded to the Al surface. Regarding the heavier (Cf8)2-Bn-COOH inhibitor (959 g.mol−1), the ToF-SIMS acquisition did not exceed m/z = 550. Therefore, identification of the complete molecule is not possible. However, C12F8-Bn-CO2 − (m/z = 416.0) is observed on the surface, corresponding to the majority of the molecular formula. Nevertheless, the coupling between XPS and ToF-SIMS results allows to conclude that (Cf8)2-Bn-COOH inhibitor is bonded to the surface. Therefore, all the inhibitors with carboxylic anchor group and Bn spacer are detected on the Al surfaces. As suggested previously, these inhibitors are bonded to the etched Al surface by the carboxylate group as monodentates or bidentates (not shown here). Few F bonded to the Al surface are also identified, similarly as in the model study M1 (Fig. 9). As discussed previously for model M2, the detection of Al bonded to the Al substrate by ToF-SIMS only indicates that the amount of Al-F type species is less than 1% (detection limit of the XPS).

Figure 16. ToF-SIMS negative ion spectra obtained on alkaline etched Al surface and alkaline etched Al immersed for 30 min in 5 mM ethanol solution of C8-Bn-COOH, Cf8-Bn-COOH, or (Cf8)2-Bn-COOH.
Download figure:
Standard image High-resolution imageComputational modeling
In the computational part of the model study M1, presented above, we relied on the proposition that the anchor-group adhesion and interchain cohesion can be decoupled from each other and in our previous publication 6 we indeed showed that the length of the alkyl chain does not significantly affect the molecule−surface bond of the carboxylic anchor-group, but instead determines the lateral intermolecular cohesion. Now we show that also the type of backbone chain does not significantly affect the molecule−surface bonding of the carboxylic anchor-group. To this end, we consider in Fig. 17 standalone adsorbed carboxylic acids with different types of backbone, in particular, C8–COO*, C8–Bn–COO*, Cf5–C3–COO*, and Cf8–COO* (note that all these molecules are considered to be adsorbed as carboxylates, which is why they are written as R–COO* instead of R–COOH). It is evident that differences between the reported condensation adsorption energies are not significant. Furthermore, even the standalone shortest-chain C1–COO* molecule (also shown in Fig. 17) displays a similar value. This confirms that neither the type nor the length of the organic backbone significantly affects the binding of the anchor group to the surface.
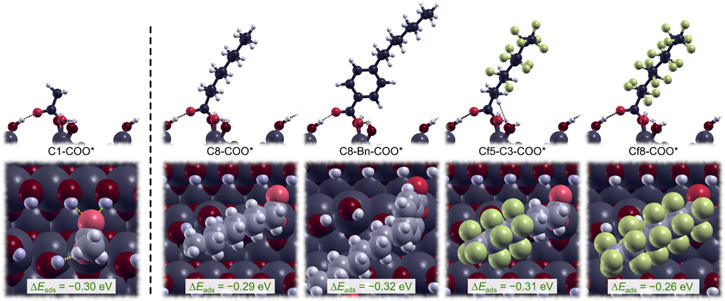
Figure 17. Side- and top-view snapshots of standalone carboxylic acids, with various backbone chains, chemisorbed on hydroxylated Al surface via condensation reaction R-COOH + OH* → R-COO* + H2O. The corresponding adsorption energies are also given. Note that the effect of the backbone type and length on the bonding of the COO* anchor group is small.
Download figure:
Standard image High-resolution imageNow we turn our attention to the full monolayer of these molecules and the corresponding condensation adsorption energies are presented in Fig. 18. The first observation is that the adsorption energies of the molecules with the longer backbones are by about 0.3 eV more exothermic than for the standalone molecules and this enhancement is due to lateral interchain cohesion. Furthermore, the adsorption energies of the longer backbones at full monolayer coverage are by about 0.4 eV more exothermic than that of the C1–COOH molecule (actually C1–COO* in the adsorbed state), which agrees with the lateral cohesion energy trend shown in Fig. 11c. In contrast to the longer backbone molecules, the adsorption energy value for the whole monolayer of C1–COO* is less exothermic than that of the standalone adsorbed C1–COO*. The reason is two-fold: (1) a standalone molecule adsorbs at the best adsorption site, h whereas in the full monolayer the molecules are adsorbed to two different adsorption sites, so called μ1 and μ2 with adsorption at μ2 being inferior to that at μ1, and (2) the lateral interactions are insignificant for the shortest chain C1–COO* and cannot surpass the inferiority of μ2-adsorption. In contrast, for the longer backbones the lateral interchain cohesion is already strong and wins over the inferiority of adsorption at μ2. This is an important result that points to a two-step regime of adsorption and SAM formation: once the molecules are adsorbed at the most reactive sites (e.g. at μ1 sites), additional molecules adsorb at less reactive sites (e.g. at μ2 sites) and are stabilized by lateral intermolecular interactions, explaining the higher surface coverage achieved for longer chains. In other words, long chains attenuate the role of the surface site specificity in adsorption, hence longer is the chain, more homogeneous is the adsorption, higher is the surface coverage, and better is the corrosion inhibition.
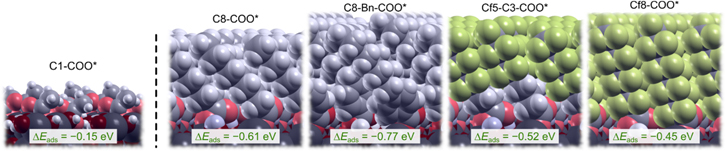
Figure 18. Analogous to Fig. 17, but for full molecular monolayers. Note that molecules with longer chains display more exothermic adsorption energies than the short-chain C1-COO* monolayer and the reason can be attributed exclusively to stronger interchain cohesion of the longer chains.
Download figure:
Standard image High-resolution imageAs for differences between the considered longer-backbone SAMs, a comparison between Figs. 17 and 18 reveals that differences in adsorption energies between various longer backbones are larger at full coverage than at low coverage and this observation can be attributed to the role of lateral intermolecular interactions. For example, SAM consisting of C8-Bn-COO* displays the most exothermic adsorption energy, which is not surprising because it displays the largest total number of C atoms in the backbone (with spacer included) among the consider molecules in Fig. 18. Note also that calculations suggest that the presence of C3 spacer strengthens the adsorption energy for Cfx-based SAMs.
Although Al substrates are currently modeled with a single surface model, the aforementioned attenuation of surface site specificity for longer backbones seems to make the current findings more generally applicable. This is important in light of the fact that alumina films on Al metal are believed to be structurally versatile. Furthermore, the tilting of the backbone chains not only strengthens the cohesion within the organic film but also makes the cohesion less susceptible to alterations of molecular coverage thus making the organic film stable for a wider range of molecular coverages (recall that the molecular density of the full monolayer coverage depends on the interplay between the distribution of adsorption sites and the steric footprint of the anchor group). These inferences are corroborated by our previous studies, 1,6 where Al substrates were modeled by two different surface models that are structurally different and are characterized by different molecular densities at full monolayer coverage.
The reason that we studied both standalone molecules and full monolayer coverage (Figs. 17 and 18) is not only to be able to disentangle the anchor−surface adhesion from the lateral intermolecular cohesion, but also to provide two limiting cases. Namely, our models of SAMs neglect the configurational entropic effects, because SAMs were modeled as totally ordered and rigid structures with fully extended trans zigzag backbone chains using structural relaxations performed at 0 K. Modeled SAM structures are therefore too ordered in comparison to real SAMs where imperfections are present. Such imperfections stem from configurational disorder of the backbones and from surface defects on Al substrates as well as imperfect molecular tiling of the SAM film (i.e., some surface sites may remain uncovered). The effects of configurational disorder and temperature on the structure of organic SAMs can be and have been addressed by force-field molecular-dynamics simulations (e.g., see Refs. 55–57). While, on the one hand, the rigidity of the currently modeled SAMs is a drawback, on the other hand, DFT is considerably superior to force-fields for the description of the anchor−surface interactions. Furthermore, it seems rather plausible that characteristics of the real SAMs should be somewhere in between the two limiting cases currently considered (fully ordered rigid SAM and standalone adsorbed molecule), most likely closer to the fully ordered rigid SAM when an organic compound prefers to form a stable SAM.
Model study 3
The effect of the anchor group (COOH, SH and ImiH) was studied for a selected length of perfluoro chains (Cf8) and C2 and C3 alkylene spacers (Fig. 4c).
Electrochemical properties
As for the role of various anchor groups with the same type of chain— a perfluoroalkyl chain with an alkylene spacer Cf17-C3-R', where R' is the COOH, SH, and ImiH anchor group—experiments reveal that the type of anchor group has a decisive effect on the electrochemical behavior (Fig. 19a). Among these three anchor groups, COOH achieved the best corrosion protection ability with a broad ΔE of 1.14 V. Compounds with imidazole ImiH and, especially, thiol SH anchor groups exhibited modest results causing only the reduction in jcorr but no change in Ecorr. The increase in current density in the anodic range is slowed down compared to the non-modified etched Al sample but no pseudo-passivity range was established.
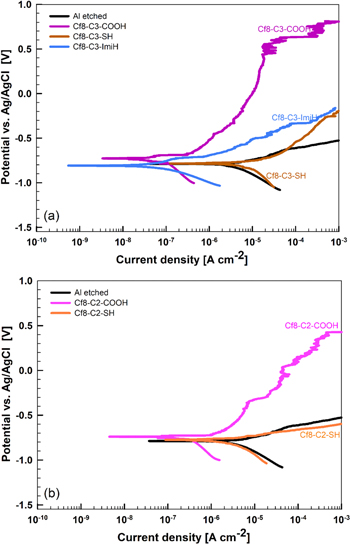
Figure 19. Potentiodynamic polarization curves recorded in 0.5 M NaCl for alkaline etched Al with and without prior immersion for 30 min in 5 mM ethanol solution of organic compounds with perfluorooctyl chain with ethylene (C2) and trimethylene (C3) spacers and different anchor groups: carboxylic COOH, thiol SH, and imidazole ImiH (model study M3). The rest time under open circuit conditions was 1 h. The scan rate was 1 mV s−1. Electrochemical parameters are presented in Table II.
Download figure:
Standard image High-resolution imageEach added C atom of the alkylene spacer is important when using a perfluoroalkyl chain and a COOH anchor group: the curves for Cf8-C2-COOH and Cf8-C3-COOH are compared in Figs. 19a and 19b. The results were considerably worse for the former (Table II), exhibiting larger jcorr (0.24 μA−2 compared to 0.095 μA cm−2) and narrower with ΔE (0.36 V compared to 1.14 V). The presented results confirm that the carboxylic anchor group attached to a perfluoroalkyl chain containing a C3 alkylene spacer surpasses the results of pure C–H and C–F chains as well as that of the mixed chain with the shorter C2 spacer.
For the thiol anchor group, no significant effect of the alkylene spacer was observed. Hence, the thiol group is a weak inhibitor for aluminum or even activator regardless the type of chain.
According to the postulated classification, the five organic chemicals from model study M3 are classified in the following order of increasing jcorr,org/jcorr,Al ratio and/or broadening of ΔEpassive: Cf8-C3-SH (activator) < Cf8-C2-SH (not inhibitor) < Cf8-C3-ImiH (weak) < Cf8-C2-COOH (moderate) < Cf8-C3-COOH (strong).
XPS and ToF-SIMS analyses
With a Cy H2y (y = 2 or 3) spacer added between the perfluoroalkyl chain and the carboxylic group, i.e. for Cf8-C2-COOH (characterized previously in Fig. 13) and Cf8-C3-COOH, characteristic XPS signals are observed (Fig. S2a). The carboxylate group and C–F bonds are detected on the C 1 s and F 1 s core level spectra at the binding energy described previously (Figs. 7, 13, 15). As observed for the inhibitor with the C2 spacer, there is no peak at 684.2 eV and there is no difference between the Ftot/Ctot experimental and stoichiometric ratios (Table III). For the inhibitor with the C3 spacer the Ftot/Ctot experimental ratio is slightly lower than the stoichiometric ratio and a component at 282 eV is observed in the C 1 s spectrum. This component is assigned to C–C, C–H bonds associated to contamination located on a part of the analyzed surface not covered by the inhibitor and presenting a lower charging effect. The ionic species detected on the Al surface by ToF-SIMS (Fig. 20a) are C8F17-C2H4-COO− (m/z = 491.1) and C8F17-C3H6-COO− (m/z = 505.1), evidencing the presence of the inhibitors C8F17-C2H4-COOH (Cf8-C2-COOH, 492 g.mol−1) and C8F17-C3H6-COOH (Cf8-C3-COOH, 506 g.mol−1) minus 1 H, respectively. XPS and ToF-SIMS results show that these molecules are present on the Al surface in the monodentate or bidentate forms (not shown here), and the spacers tend to increase the stability of molecules on the surface.
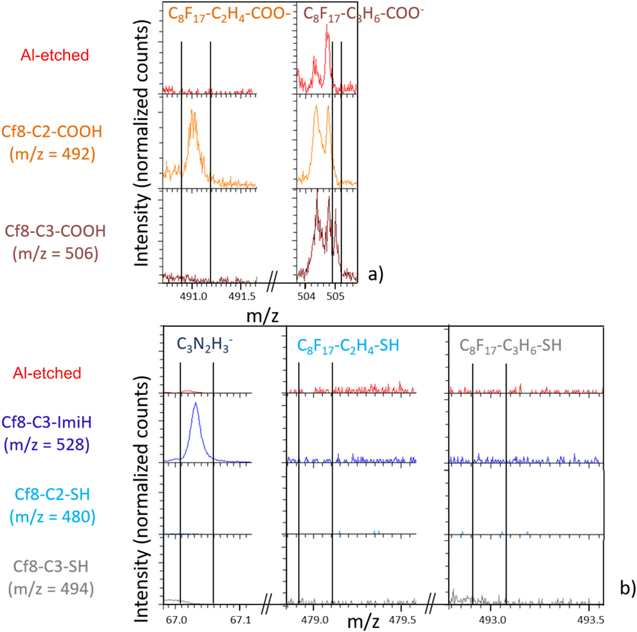
Figure 20. ToF-SIMS negative ion spectra obtained on an alkaline etched Al surface and an alkaline etched Al immersed for 30 min in 5 mM ethanol solution of Cf8-Cy chain (y = 2 or 3) with (a) carboxylic COOH and (b) thiol SH or imidazole ImiH anchor group.
Download figure:
Standard image High-resolution imageRegarding compounds with the thiol and imidazole anchor groups, i.e. Cf8-C2-SH, Cf8-C3-SH, and Cf8-C3-ImiH, no peak is detected by XPS in the F 1s spectra (Fig. S2b) suggesting that these molecules are not present on the surface.
As for Cf8-C2-SH (480 g.mol−1) and Cf8-C3-SH (494 g.mol−1) molecules, neither the whole molecule nor any fragments are detected (Fig. 20b) on the surface by ToF-SIMS, confirming the XPS results. Thiol function cannot provide strong chemisorption with such an etched Al surface. 2 For the Cf8-ImiH (528 g.mol−1), only a fragment of the molecule, corresponding to the C3N2H3 − imidazole anchor group, is detected (m/z = 67.2) (Fig. 20b). This may suggest the possibility that only imidazole anchor group bonds to the surface, without the alkyl chain.
Computational modeling
As for the comparison between the carboxylic, thiol, and imidazole anchor groups, we already showed in our previous publication 2 that C1-SH and C1-ImiH molecules cannot chemisorb via the condensation mechanism to hydroxylated Al surfaces, because the corresponding reaction energies are endothermic (and reaction free energies endergonic). Now we show that also the longer Cf8-C2 backbone chains cannot alter this trend and Fig. 21 shows that while the condensation adsorption energy for Cf8-C2-COOH is exothermic, those of Cf8-C2-SH and Cf8-C2-ImiH are endothermic.

Figure 21. Side- and top-view snapshots of standalone chemisorbed Cf8-C2-COO*, Cf8-C2-S*, and Cf8-C2-Imi* on a hydroxylated Al surface via the condensation reaction MolH + OH* → Mol* + H2O. The corresponding adsorption energies are also given. Endothermic adsorption energies of Cf8-C2-S* and Cf8-C2-Imi* indicate that chemisorption of Cf8-C2-SH and Cf8-C2-ImiH is not feasible.
Download figure:
Standard image High-resolution imageIt is worth noting that in addition to chemisorption via the condensation mechanism, molecules can weakly adsorb to hydroxylated Al surfaces also via intermolecular H-bonding interactions with the surface OH* groups and such adsorption modes are shown for standalone C1-SH and C1-ImiH molecules in Fig. S3 in the Supplementary material. It is evident from the figure that imidazole is a good H-bond acceptor, because it forms a strong hydrogen bond of about 0.5 eV with a surface OH* group, whereas C1-SH forms two weaker H-bonds of about 0.3 eV cumulative with two surface OH* groups, where the SH group acts as the H-bond donor and acceptor simultaneously. These bond strengths are about one order of magnitude weaker than the O−Al chemisorption bond between the carboxylate and the surface, whose bond strength is about 5 eV. 6 Note that in this latter case the adsorption energies ΔEads, reported in Fig. 21, do not reflect the molecule−surface bond strength, but are instead a net result of bond-breaking and bond-making during the condensation reaction. It is therefore not surprising that due to a very strong O−Al chemisorption bond carboxylates can persist for an extended period of time on the surface, whereas thiols and imidazoles cannot, which is the reason why carboxylic acids were detected by XPS and ToF-SIMS on the surface after six months in NaCl solution, whereas thiols and imidazoles were either not detected or were detected in trace amounts.
Long-term stability
Long-term stability of organic layers on etched Al modified with perfluoroalkyl compounds with different anchor groups was investigated through long-term (6 months) immersion in 0.5 M NaCl. The morphology of alkaline etched Al changed after long-term immersion in NaCl with cones progressively transformed into platelets (Figs. 3a, 3a1); the surface remained superhydrophilic. 2 In contrast, on the organically modified Al the hierarchical pattern was preserved (Figs. 3d–3e). As already noted, 2 alkaline etching in NaOH always produced a hierarchical structure but did not necessarily display the same morphology. The wettability of the initially superhydrophobic surfaces (Cf8-C2-COOH and Cf8-C3-COOH) changed to hydrophobic during immersion.
XPS spectra recorded for Cf8-C2-COOH, Cf8-C3-COOH, and (Cf8)2-Bn-COOH identified the presence of fluorine at the surface thus confirming that the molecules remained bonded to the surface even upon prolonged immersion (results not shown). However, the experimental atomic F/C ratio is smaller than prior to immersion, when it coincides with the stoichiometric value (Table III). For Cf8-C2-COOH, Cf8-C3-COOH, and (Cf8)2-Bn-COOH the experimental values after immersion were 0.32, 0.90, and 0.97, hence smaller than the stoichiometric value. This may be associated to a buildup of a thick hydrated alumina layer, covering the underlying organic layer or to the surface contamination. In addition to XPS, it was shown that the variations in durable long-term protectivity can be followed by electrochemical measurements. 50,58,59
Summary
This paper is the last in the series of five articles, 1–3,6 ,this article where we characterized different properties of organic compounds responsible for adsorption and formation of condensed layers that show barrier corrosion resistance and superhydrophobicity on aluminum surfaces. We addressed different aspects of such systems: the length of the alkyl chain for a particular anchor group, 1,6 the effect of the anchor group at a particular length of the alkyl 2 and perfluoroalkyl chains,this article and the Cl− penetration into the organic self-assembled-monolayer. 3
Archetypal structure of investigated organic compounds consists of an anchor-group (R) and a backbone (R'), as presented in Fig. 22. The stability of the corresponding organic layer on hydroxylated Al surfaces is given by the strength of the anchor-group adhesion to the surface as well as lateral cohesion between the backbones (Fig. 22a). 1,6 Among investigated inhibitors, phosphonic acids are efficient corrosion inhibitors for Al irrespective of the length of the backbone, which can be attributed to the strong adsorption affinity of the phosphonate anchor-group (Fig. 22b). 2 Carboxylic acids are also promising inhibitors, but the adsorption affinity of carboxylate anchor-group is considerably lower and the corresponding organic layer is sufficiently stabilized only with aid of long enough backbones. 1,2,6 Other investigated anchor groups do not show noticeable adsorption affinity and the corresponding compounds are inefficient for inhibiting corrosion of Al (Fig. 22b). 2 Longer backbones are beneficial for enhancing the corrosion inhibition efficiency of investigated organic compounds, however, it can be reasonably anticipated that the size of the backbone should not be too large or else the solubility of the compound would become too low and micelle formation tendency too high. According to our experimental and modelling results, for a specific type of chain, longer backbones are generally superior to shorter ones (Fig. 22c). However, the efficiency of protection depends on the type of backbone: shorter perfluoroalkyl chains (i.e. Cf8) are superior to shorter alkyl chains (i.e. C8), whereas for long backbones the alkyl chains becomes superior to perfluoroalkyl chains (i.e. C17 > Cf17).this article This "trend inversion" can be attributed to the interplay between the effective surface coverage and the lateral intermolecular interactions between backbones. For shorter chains the effective coverage is higher for perfluoroalkyl chains, whereas for longer chains the lateral cohesive interactions are superior for alkyl chains. Efficiency of shorter perfluoroalkyl chains can be substantially improved by adding either an alkylene spacer (i.e. Cf8-C3), a benzene spacer (i.e. Cf8-Bn) or an additional perfluoroalkyl chain with a benzene spacer (i.e. (Cf8)2-Bn).this article Lateral cohesive interactions are made stronger by backbone tilting, which also results in higher effective coverage for longer backbones (Fig. 22d). 1,6 Long backbones are also more effective in hindering the access of reactive species (i.e. chlorides) to the Al surface (Fig. 22e). Notice that the activation barrier for the penetration of Cl− increases with the thickness of organic layer up to about 10 Å (here a full monolayer coverage is assumed, which, according to experiments, for carboxylic acids forms only for long alkyl chains) and then it becomes almost independent on the layer thickness. 3 The role of longer backbones is therefore two-fold, i.e., to stabilize the organic layer and to effectively hinder the access to the surface of Al.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
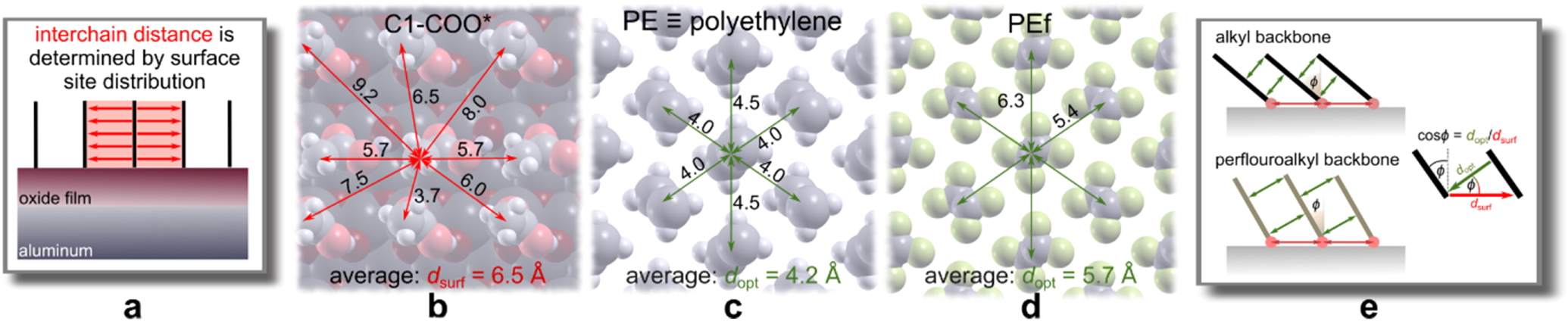{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 22. Schematic summary of integrative investigations of selected organic compounds as possible inhibitors for aluminum, 1–3,6,this article (a) archetypal structure of investigated organic compounds, (b) anchor groups, (c) backbone types ordered according to efficiency when anchored with carboxylic group, (d) lateral cohesive interactions and effective coverage for short and long backbone chains, and (e) activation barrier for penetration of Cl− as a function of organic monolayer thickness.
Download figure:
Standard image High-resolution image{kind=link}
Conclusions
- 1.Fourteen organic compounds were purposely prepared to study the effect of the type (alkyl, perfluoroalkyl, and perfluoroalkyl with alkylene or benzene spacer) and length (x = 7, 8, 10, and 17) of backbone chain and the type of anchor group (COOH, SH, and ImiH) on adsorption on a superhydrophylic, hydroxylated aluminum surface. Three model studies were designed to investigate these effects. The methodological approach consisted of four levels: (i) synthesis or purchase of appropriate organic compounds, (ii) fabrication of adsorbed organic layers on Al etched in alkaline NaOH solution; (iii) their experimental characterization using electrochemical and surface-analytical (SEM/EDS, XPS, and ToF-SIMS) techniques; and (iv) DFT modeling of adhesion and self-assembly of organic layers on oxidized Al substrates. By this integrative experimental-modeling approach applied on the three types of targeted systems—(1) Al-surface/anchor-group/alkyl-backbone, (2) Al-surface/anchor-group/perfluoroalkyl-backbone, and (3) Al-surface/anchor-group/spacer/perfluoroalkyl-backbone—we gained valuable information concerning the mechanism of interaction of various backbone chains and anchor groups with Al.
- 2.Hierarchical surface morphology and topography are necessary for the adsorption of organic molecules on aluminum. 1 We found that the adsorption bonding on the so prepared surface and, consequently, wettability and protection barrier properties are strongly dependent on the type of backbone chain and the type of anchor group. Our results further indicate that the anchor group plays a more decisive role in the formation of a stable adsorbed layer than the chain length and type.
- 3.Based on electrochemical data we classified organic chemicals into activators, not inhibitors, and weak, moderate and strong inhibitors.
- 4.Model study M1: For the carboxylic group, the length and type of chain plays a decisive role in changing the wettability and barrier properties of modified Al surface. At short chain lengths (x = 7), alkyl and perfluoroalkyl carboxylic compounds can be characterized as not inhibitors and weak inhibitors, respectively. For long chain lengths (x = 17), however, the trend is inverted and the alkyl counterpart acts as a strong barrier inhibitor surpassing the efficiency of the moderately strong perfluoroalkyl counterpart. With aid of DFT modeling this "trend inversion" is attributed to lateral intermolecular interactions between backbone chains, because for long enough chains these interactions are superior for alkyl chains compared to perfluoroalkyl chains. All tested compounds are detected on the Al surfaces using XPS and ToF-SIMS but the experimental atomic ratios indicate that stability and coverage increase with the increasing chain length, in agreement with the findings from DFT modeling, whereas ToF-SIMS experiments reveal that inhibitors are bonded to etched Al as monodentates or bidentates. ToF-SIMS analysis detected all characteristic fragments for less heavy compounds (i.e. with shorter chains) but for longer perfluoroalkyl chains heavy fragments could not be detected due to decreased probability of big ions emission. Fluorine can be also directly bonded to the hydroxylated Al surface, because Al−F bonds were detected by XPS.
- 5.Model study M2: The addition of an alkylene or a benzene spacer between perfluoroalkyl chain and carboxylic anchor group affects the wettability and barrier properties of the modified Al surface. An ethylene (–CH2CH2–) spacer between perfluorooctyl chain and COOH anchor group results in the formation of a superhydrophobic surface layer with moderate inhibitor properties. These properties surpass those of the superhydrophilic alkyl-only and perfluoroalkyl-only counterparts, which act as weak inhibitors. A benzene spacer between the anchor group and perfluorooctyl chain, however, achieved a smaller protective effect despite the surface becoming superhydrophobic. Whatever the spacer (alkylene or benzene ring) and the length of alkyl or perfluoroalkyl backbone chain, the carboxylic anchor group is bonded to the etched Al surface.
- 6.Model study M3: The perfluoroalkyl chain containing an alkylene C3 spacer is superior to the alkyl and perfluoroalkyl chains without a spacer as well as to the perfluoroalkyl chain with a shorter C2 spacer when bonded to the carboxylic anchor group. For the thiol anchor group, no significant effect of the alkylene spacer was observed. Hence, the thiol group is a weak inhibitor for aluminum or even activator regardless the type of chain, whereas the comparative chain with imidazole group acts as a weak inhibitor. XPS and ToF-SIMS results show that carboxylic molecules are present on the Al surface and the spacers tend to increase the stability of molecules at the surface. For molecules with the thiol and imidazole anchor groups attached to perfluoroalkyl chain no or almost no fluorine was detected on the surface thus suggesting that these molecules are not present on the surface (or are present only in insignificant amount). These observations are corroborated by DFT results, which show that thiol and imidazole anchor groups cannot strongly chemisorb on hydroxylated Al surfaces.
- 7.DFT calculations reveal that long backbone chains attenuate the role of the surface site specificity in adsorption. This observation is important in light of the fact that alumina films on Al metal are believed to be structurally versatile. Furthermore, the tilting of the backbone chains not only strengthens the cohesion within the organic film but also makes the cohesion less susceptible to alterations of molecular coverage. Hence the longer is the backbone chain, the more homogeneous is the adsorption, the higher is the surface coverage, and better is the corrosion inhibition. There is a drawback, though, because long backbone chains that are too long have a low molecular solubility which has adverse effect on the corrosion inhibition. 60
- 8.According to the postulated classification, the fourteen organic chemicals from the model studies M1, M2, and M3 are classified in the following order of an increasing jcorr,org/jcorr,Al ratio and/or broadening of ΔEpassive: Cf8-C3-SH (activator) < C7-COOH (not inhibitor) < Cf8-C2-SH (not inhibitor) < Cf7-COOH (weak) < Cf10-COOH (weak) < C8-Bn-COOH (weak) < Cf8-Bn-COOH (weak) < C10-COOH (weak) < Cf8-C3-ImiH (weak) < Cf8-C2-COOH (moderate) < Cf17-COOH (moderate) < < Cf8-C3-COOH (strong) < C17-COOH (strong) < (Cf8)2-Bn-COOH (strong).
Acknowledgments
This work is a part of M-ERA.NET project entitled "Design of corrosion resistant coatings targeted for versatile applications" (acronym COR_ID). The financial support of the project by MESS (Ministry of Education, Science and Sport of Republic of Slovenia; grant No. 3330-16-5000040), ANR (The French National Research Agency) and NKFIH (National Research, Development and Innovation Office, Hungary; NN 117633) is acknowledged. Région Ile-de-France is acknowledged for partial support of the ToF-SIMS equipment. Slovenian Research Agency is acknowledged for financial support (core funding No. P2-0393 and No. P1-0134) and bilateral Slovenian-French Proteus project INCOR (No. BI-FR/18-19-002). The authors thank Matjaž Dlouhy, mag. chem., for the preparation of Fig. 1. SEM/EDS analysis was performed by Barbara Kapun, BSc. We acknowledge Centre for Electron Microscopy and Microanalysis of the Jožef Stefan Institute for the use of JSM 7600 F FE-SEM.
Footnotes
- b
"Long-chain PFAS" include perfluoroalkyl carboxylic acids (PFCA) with 7 or more perfluorinated carbons, perfluoroalkane sulfonic acids (PFSA) with 6 or more perfluorinated carbons, and their precursors." Communication from the Commission to the EU Parliament, the Council, 14. 10. 2020. https://ec.europa.eu/environment/pdf/chemicals/2020/10/SWD_PFAS.pdf.
- c
The thickness of the vacuum region corresponds to the distance along the surface normal direction between the top of SAM and the bottom of the adjacent slab.
- d
More generally, Mol* stands for adsorbed molecule from which either proton or H atom was abstracted. Note that in DFT calculations one does not input any atomic charges, because electronic density is determined self-consistently. Whether H atom or proton is abstracted from a molecule, depends on a specific case. When an acidic H shifts from a molecule to OH* as to form a water molecule, it seems chemically justified to describe this as deprotonation.
- e
High wettability corresponds to hydrophilic surfaces and low wettability to hydrophobic surfaces.
- f
Data for C5-, C6-, C8- and C13-COOH are taken from Ref. 1.
- g
The region in which the current density remains constant or increases very slowly with increasing potential is defined as a pseudo-passive region; it is limited by the breakdown potential at which current density increases abruptly, i.e. ΔEpassive = ∣Ebreak − Ecorr∣. Please note that the ΔEpassive is taken arbitrarily as a passive range since it denotes the range between breakdown potential and corrosion potential, and not between breakdown potential and potential of passivation.
- h
During condensation adsorption a molecule can either substitute μ1-OH* or μ2-OH* group, where μ1-OH* bonds with its O atom to a single Al and μ2-OH* bonds to two Al ions. The substitution of the μ1-OH* (μ2-OH*) group during adsorption results in a μ1-bonded (μ2-bonded) monodentate carboxylate. In the full monolayer half of the molecules is μ1-bonded and the other half is μ2-bonded. For more details, see our previous publication. 6