

Abstract
θ-Al13Fe4 exhibits a rich variety of crystal physics. It contains twenty crystallographically different atomic species with a diversity of chemical coordination. An understanding of its structural and physical properties is a prerequisite for controlling its formation and its use. Here we investigate systematically the intrinsic defects in θ-Al13Fe4 using a first-principles density-functional theory method. The calculations reveal that among the various intrinsic defects it is energetically favourable for Fe substitution of Al but on just three of the fifteen Al sites. This results in a new structural model, Al68Fe24 (the Roman numerals represent the Al sites) which updates the thermodynamic model, currently in use, which is associated with the formation of vacancies on some of the Al sites. The calculations demonstrate that the addition of Fe induces magnetism which gives rise to clustering. The calculations provide the dependence of the lattice parameters on Fe concentration and explain the experimental data in the literature. The information obtained here provides insight into the formation and properties of θ-Al13Fe4 and its role in the solidification of Al alloys, in determination of the microstructure and related mechanical properties of the products, and in catalysis for organic reactions.
(the Roman numerals represent the Al sites) which updates the thermodynamic model, currently in use, which is associated with the formation of vacancies on some of the Al sites. The calculations demonstrate that the addition of Fe induces magnetism which gives rise to clustering. The calculations provide the dependence of the lattice parameters on Fe concentration and explain the experimental data in the literature. The information obtained here provides insight into the formation and properties of θ-Al13Fe4 and its role in the solidification of Al alloys, in determination of the microstructure and related mechanical properties of the products, and in catalysis for organic reactions.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Our interest in θ-Al13Fe4 arises from both scientific curiosity and practical interest. Firstly, θ-Al13Fe4 has a rich variety of crystallography. It exhibits a monoclinic lattice which contains 15 crystallographically different Al types and 5 Fe species, in total 102 atoms (78 Al and 24 Fe atoms) for the chemically stoichiometric composition [1, 2]. The Fe atoms have coordination numbers ranging from 9 to 11 with Fe-Al bonds shorter than 3.0 Å. In contrast the coordination numbers of the Al atoms vary from 10 to 12, except for Al2 which has 6 bonds shorter than 3.0 Å and another 6 slightly longer bonds with lengths between 3.0 to 3.2 Å. This structure was regarded as an approximant of the corresponding quasicrystal [3, 4]. Secondly, the rich diversity of the metallic species indicates the possibility of dissolving other metallic atoms to form multicomponent crystals. Information about θ-Al13Fe4 is very helpful to understand the formation, stability and structural properties of related intermetallic compounds and their role in heterogeneous nucleation processes [5, 6]. Thirdly, iron is regarded as a harmful impurity in many Al alloys [5, 7]. Fe has a very low equilibrium solubility in crystalline Al (<0.05%) and it forms as intermetallic compounds during solidification of Al alloys [5, 6]. Fe containing intermetallic compounds, including θ-Al13Fe4, have a detrimental impact on the properties of both primary and recycled Al alloys [5, 6]. Information about θ-Al13Fe4 and related intermetallic compounds is very helpful to control the harmful Fe impurities in casting Al alloys which is becoming ever more important as the demand for recycling of Al scrap materials increases. Furthermore, it has been recently demonstrated that θ-Al13Fe4 has potential applications as a low-cost and environmentally benign catalyst for organic reactions [8].
The crystal structure of stoichiometric θ-Al13Fe4 is now well defined due to experimental and theoretical studies [1, 2, 9–13]. Early x-ray diffraction patterns suggested that this compound has an orthorhombic lattice [12]. Careful structural analysis showed twinning of crystals, which resulted in an orthorhombic pseudo-symmetry [9]. Black determined the crystallographic structure of θ-Al13Fe4 to be monoclinic with a space group C2/m (nr. 12) and a = 15.489 Å, b = 8.0831 Å, c = 12.476 Å, β = 107.72° [1]. Structural refinements from single crystal samples provided the coordinates of atoms in the unit cell [2, 13, 14].
There have been various discussions about the chemical composition and physical properties of θ-Al13Fe4 [2, 13, 14]. Grin et al reported their structural determination of θ-Al13Fe4 [2]. If all lattice sites are occupied this gives the composition Al78Fe24 i.e. Al13Fe4 and this represents the most Al rich composition of the phase according to the phase diagram. Grin et al [2] also indicated that one site, which they label as Al(2), has partial occupation of 92% or the presence of structural vacancies, although it is not clear how this is possible if the stoichiometry is exactly Al13Fe4. Their structure can be described as Al74Fe24 here Va represents vacancy, x = 0.08 and the Roman numeral II indicates the Al2 site [2]. This structural model has been widely used in an analysis of the thermal stability and phase relations of the Al-rich part of the Al-Fe system [15–19]. On the other hand Popčević et al performed single crystal structure determinations and observed no vacancy defects in the crystal [13]. Experiments also suggested that samples of θ-Al13Fe4 were magnetic [13] although the results were found to depend on composition and thermal treatments, impurities, etc. In order to address this issue, parameter-free first-principles methods may be used. Early first-principles studies focused on the quasicrystalline nature and electronic properties of θ-Al13Fe4 [4]. Attention was also paid to the stability of the phase at the stoichiometric composition [17, 20], the surface structure and the electronic and catalytic properties of this crystal [21, 22]. Moreover, the first-principles calculations revealed the non-spin-polarization solution of θ-Al13Fe4 [17, 20–23], which differs from the results of experiments [13].
here Va represents vacancy, x = 0.08 and the Roman numeral II indicates the Al2 site [2]. This structural model has been widely used in an analysis of the thermal stability and phase relations of the Al-rich part of the Al-Fe system [15–19]. On the other hand Popčević et al performed single crystal structure determinations and observed no vacancy defects in the crystal [13]. Experiments also suggested that samples of θ-Al13Fe4 were magnetic [13] although the results were found to depend on composition and thermal treatments, impurities, etc. In order to address this issue, parameter-free first-principles methods may be used. Early first-principles studies focused on the quasicrystalline nature and electronic properties of θ-Al13Fe4 [4]. Attention was also paid to the stability of the phase at the stoichiometric composition [17, 20], the surface structure and the electronic and catalytic properties of this crystal [21, 22]. Moreover, the first-principles calculations revealed the non-spin-polarization solution of θ-Al13Fe4 [17, 20–23], which differs from the results of experiments [13].
Up to now, there appears to have been no detailed crystal structure determinations on compositions away from the stoichiometry of θ-Al13Fe4 to indicate where defects arise (either from vacancy formation or from substitution). Griger et al [24] have prepared samples at different conditions and observed the variation of lattice parameters across the composition range. They reported that the lattice parameters vary linearly, and the unit cell volume decreases as the iron content increases. They explained this in terms of the substitution of Al atoms by Fe leading to a shortening of bond lengths, noting that the Goldschmidt radius of Fe is significantly lower than that of Al. In the present study we investigate, in a systematic way, intrinsic defects, including interstitials, vacancies and substitutions in and related electronic properties of θ-Al13Fe4 using a parameter-free first-principles density-functional theory (DFT) method. Our study demonstrates that there should be hardly any vacancy defects in θ-Al13Fe4 and that the dominating defect accounting for the range of homogeneity should be the replacement of Al by Fe in three of the fifteen Al sites. Based on these calculations, a new structural model has been proposed, which updates the model used hitherto for the thermodynamic properties. Magnetism and its origin in the nonstoichiometric θ-Al13Fe4 configurations are also addressed.
2. Computational details
2.1. Formation energy and defect energy
To have a measure of the relative stability of the Al13Fe4 phase, the formation energy (ΔEf) with respect to the elemental solids (α-Al, α-Fe) is defined [25, 26] as:
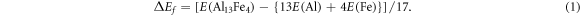
In this paper the unit of formation energy, ΔEf, will be given in eV/atom. Here E(Al13Fe4) is the calculated total valence electron energy for the compound, E(Al) and E(Fe) are the equivalent values for pure Al and Fe, respectively.
For the intermetallic structures caused by substitution, the formation energy of the defects is defined in terms of the unit cell where the formula is now Al78Fe24:
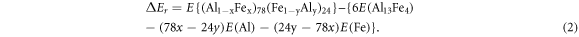
Similarly, for an intermetallic with vacancies, (Al1−xVax)78(Fe1−yVay)24, the formation energy of the defects per unit cell is defined as:
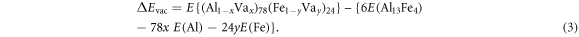
Naturally, for an intermetallic with interstitial atoms, (Al1+x)78(Fe1+y)24, the formation energy of the interstitial defects (ΔEint) per unit cell is referred as:
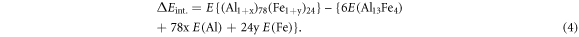
The unit for the energy of formation of the defects in equations (2)–(4) is eV per unit cell and the values are relative to the stoichiometric composition, α-Al and α-Fe.
2.2. Computational settings
The plane wave method (using VASP, Vienna ab initio Simulation Package) was used for the calculations [27]. The spin-polarized generalized gradient approximation (SP-GGA-PBE) [28] within the projector-augmented wave method [29] was employed for the exchange and correlation energy terms because the spin-polarized generalized gradient approximation describes the 3d transition metals such as Fe better than the (spin-polarized) local density approximation (LDA) [30, 31]. The cut-off energy of the wave functions was set at 550 eV and the cut-off energy of the augmentation functions was 700 eV to describe the rather localized Fe 3d orbitals. The electronic wave functions were sampled on a 4 × 8 × 6 grid with 70–100 k-points, in the irreducible Brillouin zone of θ-Al13Fe4 depending on the symmetry using the Monkhorst–Pack method [32]. Note that the Fe 4s, 4p and 3d electrons and Al 3s, 3p electrons exhibit an itinerant character in alloys and in principle belong to the whole crystal. However, we can decompose the plane waves in the atomic sphere and obtain e.g. the Fe 3d components in the spheres for both spin-up (or majority) and spin-down (minority) direction. In this way a local magnetic moment is obtained that is the difference between the spin-up electrons and spin-down electrons in the sphere. To obtain the ground state of the crystals, we performed calculations for different inputs. This avoided the possibility of our results falling into metastable solutions [25, 26, 31]. Both lattice parameters and coordinates of atoms were fully relaxed. Different k-meshes and cut-off energies were used for the waves and augmentation waves, respectively. Tests showed good convergence (<1 meV/atom).
3. Calculation results
3.1. Structure of stoichiometric θ-Al13Fe4
We first discuss the calculations for the elemental solids, α-Fe and α-Al using the settings discussed earlier. The calculations provide a lattice parameter of 4.039 Å for α-Al (face-centred cubic, fcc) and 2.830 Å for α-Fe (body-centred cubic, bcc) for 0 K. These calculated lattice parameters agree well with the experimental values, 4.0325 Å for α-Al and 2.8607 Å for α-Fe also for 0 K (within 1%) [33], respectively. Such good agreement provides confidence in the reliability of the software and the selected settings.
The calculated results (lattice parameters and formation energy) of the stoichiometric θ-Al13Fe4 crystal are summarized and compared with previous experimental and theoretical values published in the scientific literature in table 1. Figure 1 shows the structure of θ-Al13Fe4 schematically.
Table 1. Calculated and experimental results (lattice parameters, and formation energy) for θ-Al13Fe4 in comparison with previous available experimental data and theoretical calculations. The formation energy ΔEf is defined in equation (1).
| Lattice parameters (Å) | ||||||
|---|---|---|---|---|---|---|
| a | b | c | β(°) | ΔEf (eV/atom) | Method | References |
| 15.426, | 8.022, | 12.425, | 107.68 | −0.330 | DFT-PBE-GGA | This work |
| Experiment | ||||||
| −0.291 | Direct reaction calorimetry | [35] | ||||
| −0.272 | Solution calorimetry | [36] | ||||
| −0.289 | Direct reaction calorimetry | [37] | ||||
| −0.225 | CALPHAD assessment | [19] | ||||
| −0.296 | CALPHAD assessment | [16] | ||||
| −0.310 | CALPHAD | [38] | ||||
| 15.489, | 8.083, | 12.476, | 107.72 | Powder XRD | [1] | |
| 15.49, | 8.08, | 12.48, | 107.40 | Powder XRD | [4, 36] | |
| 15.509, | 8.066, | 12.469, | 107.72 | Powder XRD | [24, 37] | |
| 15.492, | 8.078, | 12.471, | 107.69 | Powder XRD | [2] | |
| 15.488, | 8.0866, | 12.4766, | 107.669 | Single crystal | [13] | |
| 15.503, | 8.063, | 12.464, | 107.71 | Powder XRD | [39] | |
| 15.423, | 8.0535, | 12.408, | 107.86 | Single crystal | [14] | |
| Theoretical | ||||||
| 15.069, | 7.864, | 12.083, | — | −0.059 | Atomist. EAM | [34] |
| — | −0.320 | DFT | [40] | |||
| — | −0.347 | DFT | [17] | |||
| 15.419, | 8.021, | 12.420, | 107.71 | −0.330 | PBE-GGA | [20] |
| 15.307, | 7.956, | 12.332, | 107.75 | −0.345 | PBEsol | |

Figure 1. Schematic structure along the [010] (a) and the [100] (b) orientation of θ-Al13Fe4. The silvery spheres represent Al and the golden spheres Fe. The red lines represent the axes of the unit cell. O represents the origin, and a, b, c the axis of the unit cell.
Download figure:
Standard image High-resolution imageAs shown in table 1, our calculated lattice parameters are in good agreement (within 1%) with available experimental values and previous first-principles calculations. However, our calculations provided rather larger differences in both lattice parameters and formation energy from data produced by the semi(empirical) embedded atom method [34]. This is due to the difficulties in deriving an EAM potential/model to describe well the intermetallic compounds and the complex interactions between atoms. We have also tested the DFT + U method (U = 2.91 eV) for θ-Al13Fe4. The calculations revealed large local magnetic moments (values ranging from 1.3 μB/Fe to 1.9 μB/Fe), being different from the weak magnetism from the experimental observations [13]. This result is in line with the previous calculations that DFT + U works better than DFT-GGA for ionic compounds, such as oxides but DFT + GGA work works better for covalent intermetallic compounds [31]. As shown in table 1, the experimental measurements of the formation energy for this compound are somewhat scattered. They are thought to be unreliable in that the chemical reactions involved may not have been carried out to completion. Our calculated formation energy is close to some of the recent thermodynamic CALPHAD assessments [16, 38], although it appears to disagree with the most recent value [19]. It is also in good agreement with the previous first-principles calculations within a few percent [17, 20, 40]. The small differences originate from different density functionals, e.g. the LDA [40], or the hybrid method (PBEsol) [20], or from different settings [17]. Our calculations confirmed that the ground state of θ-Al13Fe4 is non-magnetic in agreement with the previous first-principles studies [17, 20, 40].
3.2. Intrinsic defects in θ-Al13Fe4
To shed some light on the homogeneity range and structure of θ-Al13Fe4, we investigated different possible intrinsic defects: vacancies in the Fe or the Al sites, substitution of Fe atoms by Al and substitution of Al atoms by Fe. Test calculations of interstitial atoms in the large unoccupied sites, e.g. the Wyckoff 2a or 2b sites in θ-Al13Fe4 [2] revealed that their formation is highly improbable with formation energies over 2 eV/unit cell according to equation (4). Consequently, interstitial defects were not considered further. The calculated formation energies of the defects according to equations (2) and (3) were plotted in figure 2(a).

Figure 2. Calculated formation energies of defects (eV/cell) at 0 K relative to the stoichiometric composition, fcc Al and bcc Fe according to equations (2) and (3) (a) and the total magnetic moments (b) associated with the introduction of intrinsic defects into the unit cell of θ-Al13Fe4. The vertical dotted blue line in both pictures represents the stoichiometric composition. The dotted red curve in 2(a) connects the most stable configurations. In figure 2(b) the green squares represent the magnetic moments for configurations containing Fe on the Al7 sites. The red spheres represent anti-ferrimagnetic (AFR) ordering with the Al7 sites fully occupied by Fe atoms.
Download figure:
Standard image High-resolution imageThe calculations provide us with the following information. We use the Grin's notations for the atoms' sites throughout rest of this paper [2]:
- (i)The chemically stoichiometric composition of θ-Al13Fe4 has the lowest formation energy at 0 K;
- (ii)The formation of vacancies on the Al and/or Fe sites is associated with an increase of energy of over 1 eV/unit cell. Therefore, it is highly improbable that θ-Al13Fe4 will contain appreciable amounts of vacancies on either the Al or Fe sites even at elevated temperatures. This is in contrast to the much-quoted model of Grin et al which can be formulated as Al74Fe24(Al1−xVax)4II with x = 0.08 with the Roman numeral II designating the Al2 site [2]. However, our results agree with the single crystal structure determinations [13, 14];
- (iii)The energy costs are also too high to substitute an Al atom onto an Fe site. Therefore, a model with the formula Al78(Fe1−xAlx)24 which would allow the homogeneity range to extend towards Al rich compositions is unlikely;
- (iv)The energy costs to substitute one Fe atom onto an Al site according to the formula (Al1−xFex)78Fe24 are reasonably small in the ground state but are limited to three Al sites, Al9 (156 meV/unit cell), Al7 (181 meV/unit cell) and Al5 (188 meV/unit cell); based on the results, we propose a new model with the formula Al68Fe24
 the Roman numerals designating the corresponding Al sites.
the Roman numerals designating the corresponding Al sites. - (v)Additions of Fe induce magnetism. The magnetic moments associated with the introduction of defects are shown in figure 2(b). Overall, the magnetic moment of a composition increases with increase of Fe concentration. However, this relationship is complicated by the local magnetic moments and magnetic ordering.

Figure 2(a) also shows that the formation energies for the same chemical composition vary notably. For one Fe addition, the configuration with Fe on the Al9 site (labelled as FeIX) has the lowest formation energy (0.156 eV/unit cell). There are six configurations to add two Fe atoms into the three Al sites, among which two are noticeably more stable, that is two Fe atoms on the Al5 sites (2 FeV) and two Fe atoms on the Al7 sites (2 FeVII). There are 8 independent configurations for three Fe atoms on three Al sites. The most stable are Case I with two Fe atoms on Al7 sites and one Fe atom on an Al5 site (ΔEr = 0.451 eV/unit cell); and Case II with three Fe atoms on Al9 sites (3FeIX) with ΔEr = 0.472 eV/unit cell. It is interesting to note that, as shown in figure 2(b), for the systems with three Fe atoms, the calculations predict ferromagnetic behaviour for Case II, in contrast to ferrimagnetic (FRM) behaviour for the most stable configuration, Case I, in which the Al7 sites are fully occupied by Fe atoms. This is in contrast to a configuration with two Fe atoms on Al7 sites and one Fe atom on an Al9 site which exhibits ferromagnetic ordering but with a formation energy of 0.531 eV/unit cell, notably higher than that for Case I. In the configuration Case I, there are two Fe groups, one centred around FeVII and the other around FeV (the Roman numerals represent the Al sites). The Fe atoms in the same group display ferromagnetic ordering but they are anti-parallel to the members of the other group. As shown in figure 2(b), such magnetic behaviour also occurs for the configuration with (a) 2FeVII and 2FeV, which is the most stable, and (b) 2FeVII and 1FeV and 1FeIX. The calculations also showed that the configuration with 2FeVII and 2FeIX is ferromagnetic and has a higher formation energy. Configurations with further Fe additions are calculated to be ferromagnetic (figure 2(b)). The most stable configuration for five Fe additions is the one displaying ferromagnetic behaviour with 2FeVII and 3FeV (ΔEr = 0.901 eV/unit cell). Further addition of Fe causes the formation energies to be higher than one eV/unit cell relative to that of the stoichiometric composition and, therefore, unlikely to form even at elevated temperatures in Al alloys.
3.3. Magnetic clustering
Next, we discuss the distribution of local magnetism in the cell of the θ-Al13Fe4 crystals using the dilute replacement of Fe in the three Al sites as examples. The results are shown in table 2. Figure 3 displays a typical distribution of the spin-density for the most stable case, (one Fe at one Al9 site). Table 2 shows that the short Fe-Fe distances and related magnetic moments of the doped Fe are close to those of bulk α-Fe [41, 42]. The induced local magnetic moments at other Fe spheres range from between 0.6 and 1.2 μB/Fe.
Table 2. Calculated chemical bonds and local magnetic moments of the clustered Fe atoms which are centred around an Fe atom substituting in Al9, Al7 and Al5 sites. The species with the iron numbered as Fe_22, Fe_23, etc are labelled in parenthesis and shown in figure 4. The radius of the Wigner spheres of iron is 1.01 Å.
| Fe on Al9(site) | dFe*-Fe (Å) | M (μB) | Fe on Al7(site) | dFe*-Fe (Å) | M (μB) | Fe on Al5(site) | dFe*-Fe (Å) | M (μB) |
|---|---|---|---|---|---|---|---|---|
| Fe* (Al9) | — | 2.21 | Fe* (Al7) | — | 1.82 | Fe* (Al5) | — | 2.26 |
| Fe_9 (Fe3) | 2.48 | 1.23 | Fe_5(Fe2) | 2.56 | 0.67 | Fe9 (Fe3) | 2.52 | 0.78 |
| Fe_22(Fe5) | 2.47 | 0.76 | Fe_6(Fe2) | 2.56 | 0.67 | Fe13(Fe4) | 2.47 | 0.84 |
| Fe_23(Fe) | 2.47 | 0.76 | ||||||
| Fe_2 (Fe1) | 2.90 | 0.49 | ||||||
| Mtotal(μB/cell) | — | 5.58 | 2.86 | 5.10 |

Figure 3. Iso-surfaces of spin-density (ρ(r) = 0.007 e Å−3) for an iron cluster centred at Fe25 of the Al9 site along the (010) orientation of the lattice. The iso-surfaces are indicated by yellow spheres, the blues regions signify higher density. The gold spheres represent Fe and the silver spheres, Al atoms.
Download figure:
Standard image High-resolution imageTo obtain some further insight into the origin of magnetism introduced by the addition of Fe, we performed electronic structure calculations for spin-polarized and non-spin-polarized configurations of the system with the substitution of one Fe atom onto the Al sites. The calculations showed high stability of magnetic configurations for the substitution of Fe atoms with significant energy differences between the spin-polarized and the non-spin-polarized configurations. For example, the energy difference between the spin-polarization configuration and the non-spin-polarization configuration is 0.386 eV/unit cell for the substitution of one Fe onto the Al9 site. The calculated partial density of the 3d states of the related iron atoms is shown in figure 4.

Figure 4. The partial densities of the 3d states of the magnetic Fe atoms/ion (a) and non-spin-polarized solution (b) in the one Fe addition in the Al9 sites in θ-Al13Fe4. The labels of Fe atoms are the same as in table 2 and figure 3.
Download figure:
Standard image High-resolution imageFigure 4(b) shows that for the non-spin-polarized solution the density of the 3d states at the Fermi level is high for the related Fe atoms: ρ(EFermi) = 3.7 states/eV for Fe_25, 2.8 states/eV for Fe9, 1.5 states/eV for Fe_22 and Fe_23, and 1.6 states/eV for Fe2. The ρ(EFermi) value of Fe_25 3d states is close to that of non-spin-polarized solution of α-Fe [41–43]. It is expected that such high density of the itinerant Fe 3d states at the Fermi level will cause spin-polarization splitting which occurs in many cases according to the Stoner criteria [44, 45].
Figure 4(a) shows that the 3d states of the added Fe atom are almost fully spin-polarized: the Fe 3d states of the spin-up states are almost fully occupied while the Fermi level falls in the pseudo-gap of the Fe 3d states for the spin-down (minority) electrons. This behaviour is very similar to that of pure α-Fe [41–43]. The induced local moments are noticeably smaller, ranging from 0.49 to 1.23 (μB/unit cell) as shown in table 2. The Fe atoms which are connected to other Fe atoms that have been magnetically induced by the added Fe, also become magnetic with a smaller local moment, typically smaller than 0.35 (μB/unit cell). Figures 3 and 4 show, as an example, that the Fe10, which is directly connected to Fe9, is magnetic and has a local moment of 0.33 (μB/unit cell). Therefore, the total magnetic moment in the unit cell is different for the Fe addition on different Al sites: 5.58 μB/cell for Fe on Al9, 2.86 (μB/unit cell) for one Fe on Al7 and 5.10 (μB/unit cell) for one Fe on Al5 site (table 2).
3.4. Dependence of lattice parameters on Fe concentration
Griger et al reported their measurements for θ-Al13Fe4 of the dependence of the lattice parameters on the mole fraction [24]. These relations were considered to be helpful in order to measure the Fe concentrations of samples prepared under different conditions. Therefore, we also analysed the dependence of lattice parameters and the cell volume on the Fe concentration up to about 28.5 at% Fe. The results are plotted in figure 5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Variation of calculated lattice parameters and cell volume with Fe concentration in θ-Al13Fe4 samples.
Download figure:
Standard image High-resolution image{kind=link}
The general trend is that the length of the a-axis increases, while the lengths of b- and c-axis decrease with increase of Fe concentration (figure 5) for Fe concentrations lower than 0.29. This is in line with the experimental measurements of Griger et al [24]. However, this trend is complicated by the anomalous behaviour of configurations with Fe occupation on the Al7 sites (FeVII). In figure 5, the larger values of the length of the a-axis correspond to configurations of Fe on the Al7 sites. This is also true for other configurations containing FeVII. Furthermore, the calculations also showed that the length of the a-axis decreases with Fe concentration (XFe) when XFe is higher than 30 at%. Therefore, caution should be taken when one tries to use the dependence of lattice parameters on the Fe mole fraction as a measure of the Fe concentration of the samples. Meanwhile the b-axis of the configurations of FeVII is shorter than that of the others. As a result, the overall cell volume decreases with an increase of Fe concentration.
4. Discussion
4.1. Fe fraction and magnetism in ϑ-Al13Fe4 at elevated temperature
Our calculations showed that the stoichiometric composition of θ-Al13Fe4 has the lowest formation energy relative to the pure elements and is non-spin-polarized or non-magnetic. However, the calculations also showed that the replacement of Al atoms by Fe on three Al sites (Al5, Al7 and Al9) has only a moderate energy penalty. The defect energy cost for this type of replacement is about 0.156 eV per unit-cell to 0.188 eV per unit-cell. The defect energy increases with Fe concentration. Therefore, at low temperatures, θ-Al13Fe4 should be approximately stoichiometric at thermodynamic equilibrium. However, the addition of Fe atoms on the Al sites induces (i) an increase in the number of configurations with different Fe/Al occupation or extra freedom of atomic occupation, and (ii) magnetism of the system. According to thermodynamics, at elevated temperature, the Gibbs energy of a system can be represented as G = H – TS at the pressure p = 0 Pa. The extra freedom of occupation produces a configurational entropy, S = kB ln w, here kB is the Boltzmann constant and w is the number of independent configurations. This contribution can be calculated, e.g. for one Fe on the Al9 site (w = 4), TS = 0.120 eV for T = 1000 K or 0.168 eV for T = 1400 K. The latter value is larger than the cost of the defect energy. The calculations showed that the θ-Al13Fe4 phase with the addition of Fe develops ordered magnetic structures (ferromagnetic (FM) or ferrimagnetic (FRM)). The Fe atoms substituted into the Al sites have neighbours on the Fe sites with the Fe–Fe interatomic distances range from 2.45 to 3.12 Å. The shorter interatomic distances associated with Fe substitution on the Al 4i sites (Al5 and Al9) are approximately the same as that in α-Fe. The 3d–3d bond between Fe atoms causes high density of the itinerant Fe 3d states and therefore, spin-polarization splitting occurs. In principle the Curie–Weiss magnetic ordering contribution to the Gibbs energy of the system can be calculated since we have obtained the local magnetic ordering. However, the calculations showed Fe magnetic clustering and a variation of local magnetic moments within the Fe clusters. The variations of the Fe–Fe interatomic distances and local magnetic moments indicate a variation of Curie–Weiss transition temperatures in the Fe clusters. This is further complicated by the formation of different Fe clusters in the alloys formed at elevated temperatures (1000 to 1500 K). Therefore, the currently used models in the scientific literature [24, 41–43] cannot be applied directly to the magnetic transitions associated with Fe clustering in the Al-Fe intermetallic compounds. However, we may estimate the contributions of the Curie–Weiss magnetic ordering to the thermal stability. BCC Fe has a local moment of 2.22 μB/Fe and a Curie temperature of 1043 K. The contribution to the thermal enthalpy due to the Curie–Weiss magnetic ordering is −9180.56 J mol−1 or −95.1 meV/Fe. Our calculations showed that for one Fe on the Al9 site, there are 5 Fe atoms which are associated with magnetism with a total moment of about 5.6 μB/unit cell or about 1.1 μB/Fe. If we assume a linear relation between the magnetic moment and the enthalpy associated with magnetic ordering, then we obtain that the contribution of the magnetic ordering at elevated temperatures (ΔHmag) is about −0.234 eV/unit cell. If we apply the same approach for one Fe on an Al7 site, we obtain ΔHmag = −0.123 eV/unit cell. Therefore, at high temperature, the contribution of the extra freedoms of the irons spins (Curie–Weiss magnetic transition) further stabilizes the defective structures.
Samples of the θ- Al13Fe4 phase are generally prepared at high temperature as primary or secondary intermetallic compounds in Al alloys. At such high temperatures, the samples in thermal equilibrium should contain higher Fe concentrations in the three Al sites. The obtained composition and structure may be retained as the alloys are cooled to low temperatures. The composition and the specific location of the defects will depend strongly on the preparation conditions and thermal history. This could explain the scattering of experimental observations including the magnetic properties in the literature.
4.2. Thermodynamic model for ϑ-Al13Fe4
This work provides a new understanding of the nature of intrinsic defects in θ-Al13Fe4 which should be reflected in a new way to represent its thermodynamic properties. The model used previously in thermodynamic assessments is generally represented as (Al).6275(Fe).235(Al, Va).1375 [16–18] which, in terms of the unit cell, can be displayed as (Al)64.005(Fe)23.97(Al, Va)14.025. Clearly this provides the possibility for a much wider range of homogeneity than implied from the results of Grin et al [2], (Al)74(Fe)24(Al, Va)4 which has been used as justification for the inclusion of vacancies. This flexibility, which allows for this wider range of homogeneity, is not necessary for the Al-Fe system itself but would be necessary to model the extent of the homogeneity range of the phase in e.g. the Al-Fe-Mn system [46]. In this paper we have shown that formation of vacancies is very unfavourable energetically and that substitution of Fe onto three of the Al sites is much more plausible leading to the thermodynamic model Al68Fe24(Al, Fe)4(Al, Fe)2(Al, Fe)4. Even this description is not sufficient to represent the complexity of the phase. As shown earlier, substitution of Fe onto Al sites causes the phase to become magnetic. Simultaneous substitution of two Fe atoms on different sublattices leads to magnetic clustering and further lowering of the energy. Furthermore, at higher temperatures it might be expected that the contribution to the Gibbs energy from configurational entropy will lead to mixing on all of these three Al sites. It is therefore suggested an appropriate thermodynamic model to use for thermodynamic analyses of the θ-Al13Fe4 in ternary systems is Al68Fe24(Al, Fe)10.
5. Conclusions
We performed first-principles calculations on the structural and electronic properties of θ-Al13Fe4. Our calculations showed that at low temperature the chemically stoichiometric composition of θ-Al13Fe4 has the lowest energy of formation. It is also non-magnetic. These results agree well with the previous papers in the scientific literature. The calculations also reproduced the experimental lattice parameters in the scientific literature to within 1%. Vacancies are unlikely to be present on either Al or Fe sites. The energy associated with substitution of Fe on Al sites suggests that it is possible but limited to three sites only, Al9, Al7 and Al5. Therefore, the structures of the θ-Al13Fe4 can be described as Al68Fe24 which is in contrast to the accepted description Al74Fe24
which is in contrast to the accepted description Al74Fe24 following the work of Grin et al [2], where here the Roman numerals represent the specific Al sites, and Va indicates a vacancy. Substitution of Fe also induces magnetism, which agrees with the experimental observations. This magnetic contribution together with a configurational contribution stabilizes the defect containing θ-phase at elevated temperatures. The calculations also showed some general trends in the dependence of the lattice parameters and cell volume on Fe fraction. The magnetism of the samples with excess Fe may help to remove Fe in recycling of scrap Al alloys, and to get some insight into the catalysis of this compound for organic reactions. The obtained information is further helpful to formulate a new model to represent the thermodynamic properties of the phase for use in the critical assessment of thermodynamic and phase diagram data for Al-Fe-X ternary and multicomponent systems and the calculation of phase equilibria.
following the work of Grin et al [2], where here the Roman numerals represent the specific Al sites, and Va indicates a vacancy. Substitution of Fe also induces magnetism, which agrees with the experimental observations. This magnetic contribution together with a configurational contribution stabilizes the defect containing θ-phase at elevated temperatures. The calculations also showed some general trends in the dependence of the lattice parameters and cell volume on Fe fraction. The magnetism of the samples with excess Fe may help to remove Fe in recycling of scrap Al alloys, and to get some insight into the catalysis of this compound for organic reactions. The obtained information is further helpful to formulate a new model to represent the thermodynamic properties of the phase for use in the critical assessment of thermodynamic and phase diagram data for Al-Fe-X ternary and multicomponent systems and the calculation of phase equilibria.
Acknowledgments
EPSRC is gratefully acknowledged for supporting the EPSRC Centre under grant EP/H026177/1.