

Abstract
Liver transplant is the only curative treatment option for patients with end-stage liver failure, however there are few donor livers available for transplant. Tissue engineering of a human liver would potentially solve the problem of escalating donor shortages. A major challenge presents itself in the form of the hepatic extracellular matrix (ECM); a finely controlled in vivo niche which supports hepatocytes and plays a critical role in the development of liver disease. Polymers and decellularized tissues each provide some of the necessary biological cues for the hepatocytes, however, neither alone has proved sufficient. Equally, the ability to fine tune the microenvironment using bioactive molecules presents researchers with the opportunity to create personalised niches for hepatocytes, representing both normal and diseased phenotypes. This study combines cell derived ECM with a fibronectin vector and electrospun scaffolding techniques to produce a platform for creating customisable ECM microenvironments for hepatocytes (Abstract image). The resulting poly-L-lactic acid-extracellular matrix (PLA-ECM) scaffolds were validated using HepG2 hepatocytes. As expected, statistically significant mechanical differences were observed between the synthetically derived ECM (SD-ECM) scaffolds and normal ECM (N-ECM) scaffolds, confirming that the ECM has been altered by the fibronectin producing vector. The PLA-ECM scaffolds maintained hepatocyte growth and function and influence the gene expression of key hepatic genes. Furthermore, immunohistochemistry showed SD and N-ECMs differed in ratios of Collagen I, Laminin and Fibronectin. Our results demonstrate that hybrid PLA-ECM scaffolds and the synthetic production of ECM provide a viable, translatable platform for customising microenvironments for hepatocytes. This technology offers a potential solution to current obstacles in regenerative medicine, disease modelling and whole organ tissue engineering.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
In the US alone, as of August 20th 2017, the Organ Procurement and Transplantation Network (OPTN) estimates 14 158 patients are waiting for a donor liver. With a lack of viable new pharmaceuticals, liver transplant remains the sole treatment option for end-stage disease patients [1]. Liver disease is an increasing burden on the health of the Western world, with both incidence and mortality increasing rapidly since 1970 [2]. Demand for donor livers far outstrips supply, and the incidence of liver disease shows no signs of slowing down [3].
Tissue engineers seek to solve this problem by engineering liver 'organoids'; laboratory created organs which can function as a liver in vivo [4–7]. The 3D environment exerts extensive influence on the behaviour and function of hepatocytes [6, 8]. With this in mind, tissue engineers employ scaffold manufacturing technologies to create structures which encompass key characteristics of the native 3D ECM [9–14]. Several different methods of creating a scaffold are in use, and they can be made from a myriad of substances; both natural and synthetic [15], and enhanced with bio-decoration methods [8]. There has been particular focus on decellularization, which provides an ECM bioscaffold with the 3D site-specific vasculature required for hepatocyte function upon their repopulation of the organ [16]. Decellularized organs have been repopulated with hepatocytes and endothelial cells which subsequently survive and exhibit some level of function, clearly demonstrating the importance of the ECM in supporting hepatocyte survival and phenotype [17–21]. However, decellularization requires a human or animal source of whole, undamaged organs and while research is showing great promise, the field is fragmented and to date no scaffold has been created which allows hepatocytes to function as well as in vivo [22, 23].

Figure 1. Methodology used to biofuctionalize electrospun scaffolds with synthetically derived ECM.
Download figure:
Standard image High-resolution imageTable 1. Advantages of a combinatorial approach to tissue engineering of liver environments.
| Polymer scaffolds | Decellularized tissues | Vector technology | Combinatorial approach | ||||
|---|---|---|---|---|---|---|---|
| ✓ | Reproducible | ✓ | Provides biochemical cues of ECM | ✓ | Customizable protein profile | ✓ | Mechanically & proteomically customizable |
| ✓ | Mechanically customizable | ✓ | Vasculature exists within tissue | ✓ | Rapid production of desired proteins | ✓ | Cell lines used—donors avoided |
| ✗ | Does not provide complex biochemical cues of ECM | ✗ | Limited donors & donor safety | ✗ | Vector safety concerns | ✓ | Vectors removed—safety concerns abated |
Synthetic biology and genetic engineering are vital tools for tissue engineers and have been used to alter gene expression, enhance intracellular imaging and study fundamental processes in hepatocytes [24–26]. Recently, synthetic biology tools have been successfully employed to direct both stem cell lineage and fate in 3D constructs [27, 28], in the manipulation of biofilms for cell culture [29] and in the production of therapeutic proteins in in vivo systems [30, 31] demonstrating the ability of synthetic biology techniques to manipulate 3D environments and thus their potential benefit to the field of tissue engineering. Groundbreaking CRISPR [32–34] and minicircle vector [35–37] technologies have made the idea of therapeutic gene editing in humans a viable reality, however concerns exist regarding the safety and subsequently, the translatability of such tools with regards to patient treatment. Heavily publicised and tragic events such as the 1999 death of 18 year old Jesse Gelsinger [38] and the 2002 clinical trial in which four children developed leukaemia following gene therapy for their Severe Combined Immune Deficiency disease [39] have led to obvious worries regarding the safety of gene therapies. The inflammatory and immune modulating effects of damage-associated molecular patterns (DAMPs) such as foreign DNA fragments [40] are well documented, however clinical use of decellularized ECM in exogenic and allogenic form has demonstrated great promise, giving no immune response and showing regenerative potential. This paves the way for using synthetic biology to tissue engineer the ideal ECM environment. The ideal ECM would utilise synthetic biology and genetic technologies to their utmost, considerable, potential but remove any risk from the tools used; such as the genetically modified cells themselves (table 1).
With the potential of synthetic biology for manipulation of protein production in mind, we set out to address this problem; developing a novel bio-active hybrid scaffold which possesses the potential for customization of the ECM microenvironment (Figure 1). To date, no bioengineers have combined the promising fields of scaffold manufacture, decellularized tissue and synthetic biology. Here we report the first use of a sacrificial, transfected cell line to bio-functionalise an electrospun polymer scaffold for liver tissue engineering. We have successfully decellularized the bio-functionalised scaffold, and validated the platform using cells representative of the liver, HepG2s.
Table 2. Electrospinning parameters.
| Volume per hour | Total volume | Mandrel:needle distance | Positive charge | Negative charge | Mandrel rotation | Needle movement |
|---|---|---|---|---|---|---|
| 0.5 ml | 10 ml | 14 cm | 16 kV | −3 kV | 250 rpm | 100 mm s−1 |
Materials and methods
Electrospinning
A 10% wt/vol solution of poly-L-lactic acid (Goodman) and hexafluoroisopropanol (Manchester Organics) was dissolved overnight at room temperature with agitation. Solutions were placed into a 10 ml syringe and pumped using syringe pump EP-H11 (Harvard Apparatus) into the EC-DIG electrospinning system (IME technologies) via a 27G bore needle under the following parameters; table 2.
The mandrel was coated in non-stick aluminium foil for collecting the electrospun fibres. The sheets of electrospun fibres were allowed to dry overnight in a fume hood when the electrospinning session was completed. The average fibre size was 1.48 μm as calculated by ImageJ plugin 'DiameterJ' [41].
Scaffold preparation
10 mm discs of scaffold were cut from the dry fibre sheet. The scaffolds were soaked in 70% isopropyl alcohol for 10 min, rinsed three times in phosphate buffered saline for 15 min each and allowed to dry completely at room temperature. Scaffolds were placed into an antibiotic/antimycotic treatment solution of Dulbeccos Minimal Essential Media supplemented with 100 U/ml penicillin, 100 μg ml−1 streptomycin, 0.25 μg ml−1 Fungizone® (amphotericin B) Anti-Anti solution (Gibco) for 1 h.
Initial layer cell seeding and culture
Scaffolds were removed from the antibiotic/antimycotic treatment solution and rinsed three times for 15 min each in complete media; Dulbeccos Minimal Essential Media supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg ml−1 streptomycin (Gibco). They were then placed into a fresh 48 well tissue culture plate.
5637 human urinary bladder epithelials (ATCC) were cultured and expanded as per supplier recommendations, using the media described above. Cells for scaffold seeding were trypsinized using 0.25% Trypsin-EDTA (Gibco) from tissue culture flasks and counted using the trypan blue exclusion method. 1 × 105 cells at passage 23 were suspended in 100 μl of complete media and seeded directly on to the scaffolds. The cells were allowed to incubate in this small volume on the scaffolds for 2 h, before an additional 400 μl of complete media was added.
Media was changed after 24 h using standard methods and subsequently changed every 48 h. Controls were scaffold only, i.e. not seeded with an initial cell layer and a 'normal' initial layer i.e. untransfected cells. Initial layers of cells were cultured for 7 days at 37 °C and 5% CO2 in a humidified incubator.
Fibronectin vector
Vectors were obtained from the DNASU plasmid repository. In brief, the human fibronectin gene (FN1) was placed into a retroviral expression vector, PJ1520. The insert sequence was verified by sequence analysis and restriction enzyme digest by DNASU. The vector was obtained in DH5-alpha T1 phage resistance E. coli glycerol stock. We cultured the E. coli under selective conditions; 100 μg ml−1 ampicillin, 34 μg ml−1 chloramphenicol and 7%wt/vol sucrose in LB media. Plasmid extractions were performed using Cambridge Bioscience's Zyppy™ plasmid extraction kit following manufacturer's methods.
Transfections
Transfections were performed using Invitrogen Lipofectamine 3000®. Following titration experiments, a concentration of 1 μg plasmid DNA and 0.75 μl lipofectamine reagent per scaffold was chosen. The initial layer of 5637 epithelials was cultured on the scaffolds under standard conditions for 7 days. Transfection was performed on the 7th day. Scaffold-cell constructs were then placed into selective media containing 150 μg ml−1 puromycin. The cell-scaffold constructs were cultured under selection for a further 7 days to allow production of the vector derived fibronectin before being decellularized.
Decellularization
Decellularization was performed under sterile conditions at room temperature (19–22 °C) and with agitation. Scaffolds were placed into 50 ml falcon tubes and placed on a rotator at 20RPM. Scaffolds were washed with phosphate buffered saline (PBS) for 15 min and then rinsed in 10 mM tris buffered saline (TBS) for 15 min.
The scaffolds were submerged in a 0.1% vol/vol Triton X-100 (Sigma-Aldrich) 1.5 M potassium chloride (Acros Organics) 50 mM TBS for 4 h. They were rinsed for 15 min in 10 mM TBS before being submerged in fresh 10 mM TBS overnight.
Scaffolds were given a final rinse in 10 mM TBS for 15 min before being incubated in complete media for 15 min and then transferred to new 48 well plates for seeding.
Functional layer cell seeding and culture
HepG2 cells were trypsinized using standard methods from tissue culture flasks and counted using the trypan blue exclusion method. 1 × 105 cells at passage 17 were suspended in 100 μl of complete media and seeded directly on to the scaffolds. The cells were allowed to incubate in this small volume on the scaffolds for 2 h, before an additional 400 μl of complete media was added.
Media was changed after 24 h and changed every 48 h after the initial 24 h adherence and recovery period. This functional layer (FL) of cells was cultured using standard methods for either 3 or 5 days at 37 °C and 5% CO2 in a humidified incubator.
Live/dead® viability/cytotoxicity assay
To determine cellular viability, cell/scaffold constructs were incubated with 10 μm calcein and 2 μm ethidium ho-modimer-1 (Ethd-1) for 30 min as part of the two colour live/dead assay (Molecular Probes). Calcein is actively converted to calcein-AM in living cells, which then appear green when excited during fluorescence microscopy. Ethd-1 only accumulates in dead cells, which subsequently appear red. The method allows differentiation between dead and viable cells. The scaffolds were rinsed three times in CaCl2/MgCl2 free PBS to remove excess dye and placed onto a standard microscope slide with a 25 mm glass coverslip (VWR). All images were captured using a Zeiss Axio Imager fluorescent microscope (COIL, University of Edinburgh) at 40x magnification and post processed using ImageJ.
CellTiter-blue® cell viability assay
The assay was performed according to manufacturer's instruction (Promega). For each condition group, n = 5. Importantly, cell/scaffolds constructs were moved into fresh 48 well plates to prevent reading activity from tissue culture plastic bound cells. Measurements were read in a Modulus™ II microplate reader at an excitation wavelength of 525 nm and emission wavelength of 580–640 nm and reported as fluorescence.
Albumin quantification
A bromocresol green (BCG) albumin assay (Sigma) was used to quantify serum albumin produced by the HepG2 functional cell layer over 24 h at 3 and 5 day timepoints. The assay was performed according to manufacturer's instructions and results read at an absorbance of 620 nm in a Modulus™ II microplate reader.
Picogreen® DNA quantification
The Quant-IT™ Picogreen® dsDNA assay kit (Life Technologies™) was employed to establish the efficiency of the decellularization method in removing cellular material and to estimate cell number on the cell/scaffold constructs. The assay was performed according to manufacturer instructions. In brief, constructs (n = 5) were digested in a solution of CaCl2 and MgCl2 free PBS (Sigma), containing 2.5 U/ml papain extract (Sigma) 5 mM cysteine-HCl (Sigma) and 5 mM EDTA (Sigma) and incubated for 48 h at 60 °C. Picogreen solution was added to the digests and fluorescent intensity measurements read in a Modulus™ II microplate reader at an excitation wavelength of 480 nm and emission wavelength of 510–570 nm. A standard λ dsDNA curve of graded known concentrations was used to calibrate fluorescence intensity versus dsDNA concentration.
Sectioning and staining
The samples were rinsed three times in PBS (Gibco) for 15 min each, then fixed in 4% v/v formalin buffered in saline for 15 min at room temperature. After rinsing with fresh PBS, constructs were embedded in low melting temperature polyester wax (Electron Microscopy Supplies) using methods adapted from Steele et al (2014). In brief, samples are dehydrated through 70%–100% ethanol, then incubated in 50:50 ethanol:wax overnight at 45 °C overnight with agitation. The samples were moved into 100% wax for 3 h at 45 °C and then fresh 100% wax for 1 h at 45 °C. Samples were embedded and allowed 72 h to fully cure before sectioning. Immunohistochemical staining was undertaken using antibodies for Collagen I (Stratech), Laminin (Stratech) and Fibronectin (Sigma). All images were captured using a Zeiss Axio Imager system (Centre Optical Instrumentation Laboratory, University of Edinburgh) at 40x magnification and post processed using ImageJ.
Scanning electron microscopy
SEM was used to characterise the scaffold architecture. Samples were rinsed three times in PBS for 15 min each, then fixed in 2.5% v/v glutaraldehyde (Fisher Scientific) in 0.1 M phosphate buffer (PB) (pH 7.4) at 4 °C overnight. They were then rinsed three times in 0.1 M PB before being post-fixed in 1% v/v osmium tetroxide (Electron Microscopy Supplies) buffered with 0.1 M PB. Samples were again rinsed three times in 0.1 M PB and dehydrated through an ethanol gradient (30%–100%). They were dried by placing them in hexamethyldisilazane (HDMS, Sigma) which was allowed to evaporate off at room temperature overnight. We mounted the samples onto SEM chucks using double sided carbon tape and coated them with a thin layer of gold and palladium alloy (Polaron Sputtercoater).
All images were captured at 5 kV using a Hitach S-4700 SEM (BioSEM, University of Edinburgh).
Mechanical testing
Nanoindentation experiments were undertaken to establish the dynamic properties of scaffolds and decellularized ECM/scaffold constructs using the Keysight/Agilent 5200 nano indenter testing system.
Scaffolds and constructs were subject to indentation by a DCM II actuator flat-ended cylindrical punch (D = 100 μm) using a max load of 1g-f. All nanoindentation experiments were carried out on fresh, hydrated (suspended in PBS), unfixed samples in a stainless steel well chuck. A total of 36 indentations were carried out on each sample, 50 nm apart. Indent sites were selected using the high precision X-Y stage within the testing system (Agilent). Poissons ratio was assumed to be 0.5 for each sample [42, 43]. Allowable drift rate was 0.1 nm s−1. A NanoSuite (Keysight Technologies) test method 'G-Series DCM CSM Flat Punch Complex Modulus' was used for all testing [44, 45].
Gene Expression analysis
RNA was extracted from constructs using standard Trizol (Fisher Scientific) methods and purified using Qiagen's RNeasy spin column system. cDNA was synthesised using the Promega's ImProm-II™ Reverse Transcription System.
Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the LightCycler® 480 Instrument II (Roche Life Science) and Sensifast™ SYBR® High-ROX (Bioline) system. Results were normalized to HepG2s of the same passage number grown on tissue culture plastic and compared to the housekeeping gene Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH). Analysis was performed using the 2–[delta][delta] Ct method [46, 47], n = 5. Albumin (Alb), Cytochrome P450 Family 1 Subfamily A Polypeptide 1 (Cyp1A1), Cytochrome P450 Family 1 Subfamily A Polypeptide 2 (Cyp1A2), Cytochrome P450 Family 3 Subfamily A Polypeptide 4 (Cyp3A4), Collagen Type I alpha 1 (Col1A1), Collagen Type 4 alpha 1 (Col4A1) and Fibronectin Type 1 (FN1) were investigated, forward and reverse primers (Sigma) are detailed in supplementary table 1 is available online at stacks.iop.org/BPEX/4/065015/mmedia.
Statistical analysis
One-way ANOVAs with Fishers, Games-Howell and Tukey [48–50] post-hoc testing were performed using Minitab 17 Statistical Software and graphs generated using Origin software (OriginLab, Northampton, MA). Error bars indicate standard deviation. A minimum of n = 3 and max of n = 6 was used for all analysis.
Results
Cell attachment and survival on scaffolds
When compared to normal ECM derived from untransfected cells (N-ECM) and the scaffold alone (SO), a lower number of HepG2s adhered to the synthetically derived vector driven (SD-ECM) scaffold-ECM constructs (figure 2). According to the CellTiter-Blue® results (figure 2(A)), the SD ECM layer maintained the growth of the HepG2s between days 3 and 5. However, this result is not concurrent with the Picogreen® DNA quantitation (figure 2(B)). This is most likely due to different data extraction methods and validation methods in the Picogreen and CellTiter-Blue assays. Both assays possess depth dependencies with regards to their efficiency and effectiveness in extracting data from fibrous scaffold constructs. Additionally, the assays were validated using 2D monolayer cell cultures. This would explain high standard deviations in the Picogreen® DNA quantitation dataset and slight different trends observed between the assays. These finding are corroborated by our recent published work of cells on electrospun scaffolds [14]. Live/Dead® Viability/Cytotoxicity images (figure 3) demonstrate the metabolic viability of the functional HepG2 cell layer (FL), and that at day 5 the cells appear to be confluent and living, with low levels of cell death in each condition.
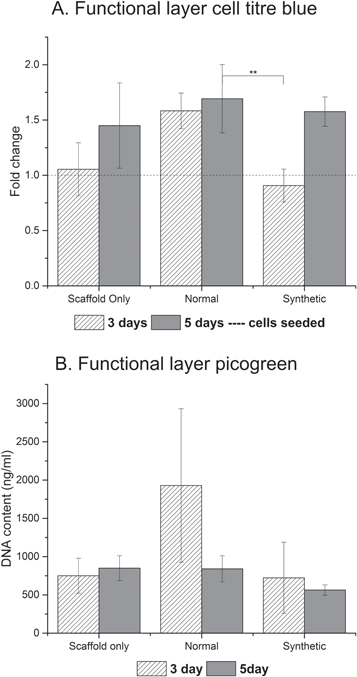
Figure 2. Cell titre blue assay indicating metabolic activity (A) and DNA quantitation of the functional layer (B). One way ANOVA with Games Howell post hoc testing, ** = p value <0.01.
Download figure:
Standard image High-resolution image
Figure 3. Live Dead imaging demonstrating the living functional layer present at each time point, with minimal cell death.
Download figure:
Standard image High-resolution imageMechanical characterisation of scaffolds
Reassuringly, both storage (G') and loss (G'') modulus demonstrate significant differences between the three conditions (figure 4). Fibronectin is known to influence both the mechanical profile of the ECM [51, 52] and influence the maintenance and structure of other vital ECM proteins, such as collagen [53]. Equally, cells are known to respond to the mechanical and topographical influence a scaffold exerts [54].
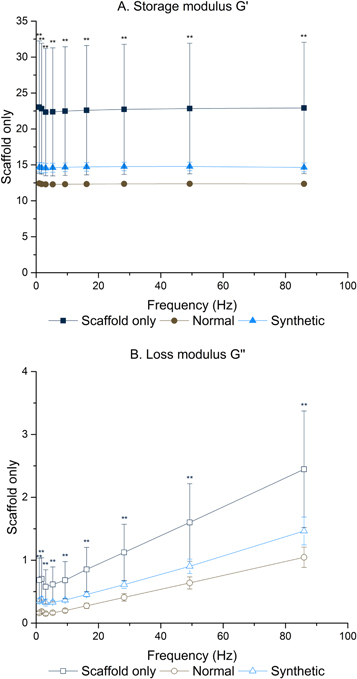
Figure 4. Significant mechanical differences were observed between all scaffold conditions. One way ANOVA with Games Howell post hoc testing, ** = p value <0.01.
Download figure:
Standard image High-resolution imageTesting was performed at frequencies experienced by the human liver in vivo [55]. Storage modulus (G') ranged from 22.92 ± 9.14 to 12.29 ± 0.14 MPa and loss modulus (G'') from 2.45 ± 0.93 to 0.15 ± 0.02 MPa at the frequencies detailed in supplementary tables 2 and 3.
Biochemical characterisation of the hybrid polymer-ECM scaffolds
Differences in the biochemical profile of the different ECMs were demonstrated by immunohistochemistry performed on decellularized hybrid scaffold sections (figure 5). Hepatic cells are very responsive to extracellular matrix proteins; particularly Collagen I, Laminin and Fibronectin [56–58]. Fibronectin is ubiquitous in healthy liver [59, 60], and antibody staining reveals altered levels of fibronectin in the synthetically derived ECM, as expected due to the introduction of the fibronectin vector (figure 5(H)). ECM proteins do not exist in isolation, and fibronectin is known to influence the generation and laydown of other ECM proteins including collagen I and laminin [53, 61], as evidenced in figures 5(G) and (I) when compared to N-ECM. The N-ECM is collagen I rich (figure 5(D)) with some fibronectin and laminin also present (figures 5(E) & (F)). Of note is that the SD-ECM appears to be concentrated on the outer layers of the electrospun scaffold. This could be due to the transfected cells being in fewer number than those which were not transfected (N-ECM), so they did not penetrate the scaffold to the same extent.
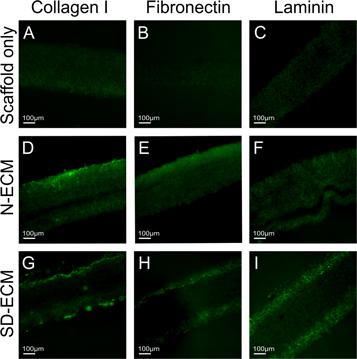
Figure 5. IHC staining showing the retention of major liver ECM proteins Collagen I (D), (G), Fibronectin (E), (H) and Laminin (F), (I) following decellularization. Additionally, SO condition shows no positive staining as expected (A), (B), (C).
Download figure:
Standard image High-resolution imageGene expression of HepG2s in response to hybrid polymer-ECM scaffolds
Genes associated with both liver function and ECM production were assayed for gene expression (figure 6). Albumin expression, a marker of appropriate liver cell differentiation and function, appears upregulated between day 3 and day 5 in each condition, confirming appropriate development of the cells. At day 5, expression is significantly upregulated in comparison to HepG2s grown on tissue culture plastic (TCP) on the SD-ECM constructs; with the highest levels of expression observed in SO and SD-ECM conditions. Additionally, albumin mRNA expression is downregulated in comparison to TCP at day 3 in all conditions (figure 6(A)). Cytochrome P450s (Cyp) are a family of enzymes involved in metabolism of xenobiotics and toxic compounds in the liver [62–64]. Cyp1A1 mRNA expression is significantly altered in comparison to TCP (figure 6(B)); upregulated at day 3 and downregulated at day 5. Cyp1A2 mRNA expression is consistently significantly downregulated in all but one condition; day 3 N-ECM (figure 6(C)). Cyp3A4 mRNA expression is upregulated in every condition, with a significant upregulation observed in response to SD-ECM (figure 6(D)).

Figure 6. Q-PCR results showing significant changes in expression of major liver genes when compared to TCP, Albumin (A), CYP1A1 (B), CYP1A2 (C), CYP3A4 (D) an ECM genes Col 1A1 (E), Col 4A1 (F) and Fibronectin (G) between the hybrid scaffolds and tissue culture plastic. (A, C, D, E, F, G) = One way ANOVA with Games Howell post hoc testing, * = p value <0.05. (B) = One way ANOVA with Tukey post hoc testing, * = p value <0.05.
Download figure:
Standard image High-resolution imageIn addition, we assayed for three ECM genes important for normal liver composition [59, 65]; Fibronectin (figure 6(E)), Collagen I (figure 6(F)) and Collagen IV (figure 6(G)). Considering the plastic nature of ECM, these genes are of interest with regards to ongoing modification of the tissue microenvironment despite hepatocytes not being the main producers of ECM proteins in the liver [59, 66].
Collagen I is consistently upregulated, with significant upregulation observed at day 5 on SD-ECM. Equally, Fibronectin mRNA expression is significantly upregulated on day 5 SD-ECM constructs, though downregulated at day 3 on SO and N-ECM constructs. Collagen IV mRNA expression is consistently upregulated in each condition, with significant changes observed in all but day 3 N-ECM and SD-ECM. While such alterations in gene expression are promising, we refrain from further assumption regarding cell response without further proteomic and functional analyses.
Albumin production
Albumin levels are indicative of hepatocyte health and response to the microenvironment [57]. Each condition results in differing levels of albumin production, with significant differences in protein levels observed between SO and SD-ECM at both 3 days and 5 days (figure 7), indicating that the N-ECM encouraged albumin production more than SD-ECM.

Figure 7. Albumin production between conditions, showing significant changes between SO and SD-ECM conditions at both time points. One way ANOVA with Games Howell post hoc testing, * = p value <0.05. ** = p value <0.01.
Download figure:
Standard image High-resolution imageConfirmation of decellularization
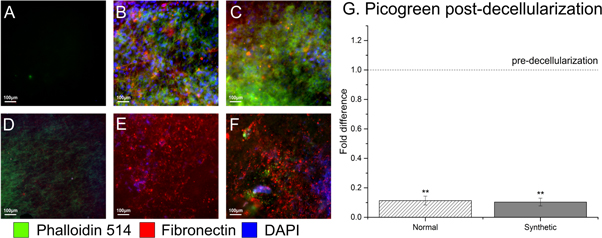
Decellularization was confirmed using Picogreen® DNA quantitation (figure 8(G)) and histological staining (figures 8(A)–(F)). A tenfold reduction in DNA combined with visual confirmation of an absence of DAPI nuclear staining and Phalloidin-514 actin staining on decellularized constructs, plus the presence of fibronectin antibody staining on both the N-ECM and SD-ECM decellularized constructs (figures 8(E) & (F)) provides evidence of efficient decellularization methods, and that synthetically derived fibronectin is present on the hybrid scaffolds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Effectively decellularized constructs, with minimum remnant DNA detected by IHC (E), (F) or picogreen (G). Scaffolds pre-decellularization shown in (A), (B) and (C). One way ANOVA with Games Howell post hoc testing, ** = p value <0.05. Scaffold only (A), (D), N-ECM (B), (E) and SD-ECM (C), (F).
Download figure:
Standard image High-resolution image{kind=link}
Discussion
The extracellular matrix provides a microenvironment for cells which is not yet fully understood, nor replicable by existing manufacturing methods [16, 67–69]. By manipulating human cells to produce a customizable blend of ECM components, and combining this with replicable electrospun scaffolds and decellularization methods we overcome the shortage of donor derived ECM bioscaffolds and the issues regarding animal-sourced biomaterials [16, 70]. Additionally, this platform has the potential to be used to model not only 'healthy' ECM microenvironments, but also those of disease and developing states.
By using fibrous electrospun scaffolds we mimic the morphology of healthy fibrillary collagen [10, 11, 71, 72]. PLA was used to fabricate the scaffolds, chosen for its compatibility with hepatocytes [56] and its use in multiple types of medical device due to its predictable biodegradation rate and mechanical properties [73, 74].
Fibronectin was chosen as our protein to synthetically overexpress due to its vital role in the liver [51, 75] and its interaction with other ECM components such as collagen and laminin [52, 53]. Fibronectin is a large dimeric adhesive glycoprotein which exists in both cellular and plasma forms, with roles in regenerating tissues, embryonic development and regulation of cell behaviours such as adhesion and migration [76]. The role of fibronectin in the liver is still unclear, with inhibition of fibronectin production or deposition improving fibrosis outcomes [77, 78]. However its vital role in the hepatic ECM is demonstrated by mutant mice who were specifically null in only the liver for both plasma and cellular fibronectin. The fibronectin-null livers not only develop highly disorganized/diffuse collagenous ECM networks, but when fibrogenesis was induced the null livers experienced more extensive fibrosis; thought to be due to fibronectins role in regulating TGF-β1 bioavailability [75]. Additionally, homozygous fibronectin null mutants display early embryonic lethality, while heterozygotes (with 50% of the normal plasma fibronectin levels) appear normal; suggesting a dose dependent role for fibronectin in development [79].
As an initial assessment of these novel hybrid scaffolds we investigated the attachment and function of a commonly used liver cell line, HepG2s, when cultured on the synthetically derived hybrid scaffolds versus a wild type 'normal' hybrid scaffold and the scaffold alone. The HepG2 cell line was derived from the hepatocarcinoma of a 15 year old Caucasian male. They are often used because they are virus free, possess liver specific functions such as ammonia metabolism and albumin synthesis and secrete some growth factors [80]. We analysed cell attachment and viability, albumin production and gene expression of both liver function genes and ECM genes at 3 and 5 day time points. Additionally, we validated our decellularization method and performed both immunohistochemical and raman spectrum analyses (data not shown) of the hybrid scaffold-ECM constructs upon which the HepG2s were seeded.
Our results indicate not only that synthetically derived ECMs provide a viable method of biofunctionalising electrospun polymer scaffolds, but that the composition of the synthetically derived ECM-polymer hybrid scaffolds influences liver cells. That albumin production is significantly altered between SO and SD-ECM conditions, but not N-ECM conditions supports this assertion. Gene expression of key hepatic genes was altered on day 5 of SD-ECM conditions in every gene tested, whereas SO and N-ECM conditions only influence CYP1A2 and COL4A1 expression; demonstrating that the composition of the ECM is highly influential in tissue engineering. This in turn leads to questions regarding donor-donor variability of current decellularization work and its influence upon hepatic behaviour and a need for more reproducible hepatic microenvironments that this platform provides.
The researchers responsible recognise that, while this study forms a robust initial proof of principle regarding the exploitation of synthetic biology for scaffold manufacture, and has produced a novel hybrid synthetically derived ECM- polymer scaffold with great potential for liver tissue engineering, further work is required to analyse results and increase translatability. Vector technologies and synthetic biology present obvious concerns with regards to patient safety [30, 81], and care should be taken to ensure all bacterial/viral constructs are removed from the ECM layer. Detergent based methods used to strip the ECM of cells can be detrimental to the bioscaffold [70, 82], disrupting native tissue ultrastructure, decreasing glycosaminoglycan (GAG) content and reducing collagen integrity [83, 84] as well as disrupting lipid-lipid, lipid-protein and protein-protein interactions [85]. Care should be taken to optimise this procedure in the future. HepG2s provide a convenient method of initial viability testing of the scaffolds, but they are derived from a carcinoma and as such criticism of their clinical relevance is well placed. Further studies will utilise primary or stem cell derived hepatocytes to combat such criticism. Additionally, recognition of the value of further proteomic and functional assays (such as ELISAs and Alkaline Phosphatase quantitation) in analysing the function of hepatocytes will be vital for expanding this work, however at this time these were deemed unnecessary considering the obvious critiques of the use of the HepG2 cell line. Further, the importance of ensuring decellularization agents are removed from the constructs should not be underestimated, due to their influence upon cells and ECM [21, 70]. While such criticisms are important to consider, this work clearly demonstrates the potential of synthetic biology for the design of bespoke ECMs and provides a robust initial platform upon which further, improved studies can be built.
Conclusion
This study demonstrates a novel method of creating a biologically bespoke hybrid ECM-polymer scaffold; utilising clinically translatable electrospun scaffold technologies and synthetic biology methods both easily modified to fulfil Good Manufacturing Practice (GMP) guidelines. In order to achieve this, a sacrificial ECM-producing cell layer was transfected using a protein producing fibronectin vector on an electrospun scaffold, biofuctionalizing the scaffold with a biologically bespoke ECM. Scaffolds with wild type untransfected cells and no initial cell layer at all were used as controls. This sacrificial cell layer was successfully removed with a detergent based method and the hybrid synthetically derived ECM-polymer scaffolds seeded with HepG2 liver cells for validation. Results were validated using multiple well characterized methods, including mechanical quantitation, Q-PCR and immunohistochemistry. The synthetically derived PLA-ECM scaffolds exert biological influence upon liver cells, manipulating their microenvironment and resulting in alterations in gene expression profile, protein synthesis and cell attachment and survival. Such data demonstrates promise as a unique method of creating biologically bespoke ECMs and exerting influence upon cell populations both in vivo and in vitro.
These novel scaffolds exhibit great promise both as an implantable patient treatment for liver tissue engineering, for adaptation to other tissues and as a useful tool for development of 3D liver cell culture platforms with potential for both in vivo cell analysis and novel pharmaceutical research.
Acknowledgments
The authors would like to thank Prof Alistair Elfick for use of lab facilities (IBioE, University of Edinburgh). We would also like to thank Steve Mitchell (BioSEM) andDr David Kelly (COIL) (University of Edinburgh). This work is funded by an Engineering & Physical Sciences Research Council [EPSRC] doctoral training partnership studentship, UK Regenerative Medicine Platform II [RMPII] grant MR/L022974/1 and MRC computational and chemical biology of the stem cell niche grant (CCBN) MR/L012766/1.
Author disclosure
The authors declare no competing interests.