

Abstract
Two-dimensional networks inspired by graphene are of prime importance in nanoscience. We present a computational study of an infinite molecular sheet confined on a water surface to assess its properties and formation mechanism. Terpyridine-based ligand molecules are interlinked by Zn ions to form an extended 2D metal-organic framework. We show that the network is stable on the water surface, and that the substrate affects the dynamic properties of the sheet, exhibiting a confining effect and flattening the sheet by 30%. We use metadynamics to characterize the process of network formation and breaking and determine an intra-network binding energy of 143 kJ mol−1. Based on this mechanistic insight we propose that the 2D network strength can be tuned by varying the rigidity of the ligand through its chemical structure.
Export citation and abstract BibTeX RIS
1. Introduction
Two-dimensional molecular sheets with precise molecular architecture are crucial materials in modern nanoscience [1]. The discovery of graphene has initiated widespread research efforts into 2D monolayers [2, 3]. These materials can be prepared by exfoliation or chemical vapor deposition, but more benign methods to grow 2D systems with atomic-scale control of the sheet are highly desirable [4, 5]. Moreover, the ability to tune the binding strength and stability of the network would be essential to widen the range of possible applications.
A promising alternative technique for the fabrication of infinite coordination polymer sheets was recently introduced [4, 5]. This approach is based on spreading suitable oligofunctional ligand monomers on an air/water interface, increasing lateral pressure to form a dense layer, and interlinking the monomers with metal ions diffused from the liquid phase [5, 6]. The resulting monolayers have large lateral dimensions and can be straightforwardly removed from the liquid substrate by drying. Previous studies have investigated the mechanical strength of these sheets and the transmetalation of the ligand ions [4]. The elastic modulus of the sheet could be computed in good agreement with AFM nanoindentation results [4]. More recently, also other types of networks assembled on water have been reported. In particular, Schlüter et al were able to produce a covalently linked sheet by photochemically polymerizing anthracene-based monomers at an air/water interface [7]. In our previous work, we have examined the properties of a terpyridine-derived trifunctional monomer (TTPB, figure 1(a)) confined on water, as well as the uptake of Zn2+ from the liquid phase [8]. However, the details of the monomer aggregation, the energetics of the linking process as well as the dynamic properties of the extended MOF sheet (figure 1(b)) have so far not been studied. In order to optimize the rational design of these tailored 2D materials and to control their properties, in-depth understanding of these processes is essential.
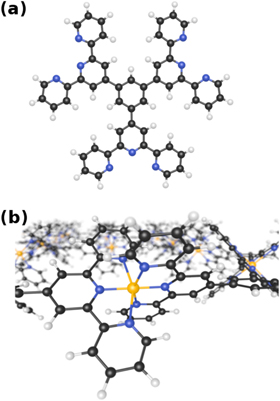
Figure 1. (a) The TTPB molecule. Atom colors: C, black; H, white; N, blue. (b) Network of TTPB molecules formed by interlinking them with Zn ions (orange).
Download figure:
Standard image High-resolution imageIn this contribution we present a large-scale study of water-supported TTPB-Zn sheets using ab initio molecular dynamics based on density functional theory (DFT). As our model is fully quantum mechanical, we are able to study all details of the system, including bond breaking and formation and the solvation of ions. We first examine the properties of the extended sheet, comparing the water-supported network with the free-standing sheet in the gas phase. Subsequently we focus on an isolated TTPB-Zn-TTPB dimer, for which we elucidate the molecular details of the binding mechanism and quantify the free energy of this process using the metadynamics (MTD) method [9, 10].
2. Computational details
We employ the CP2K code [11, 12] and a computational set-up described in previous work [8]. All simulations use DFT at the PBE+D3 [13, 14] level with double-zeta basis sets [15] and GTH pseudopotentials [16]. The time step is fixed at 0.5 fs and the temperature maintained at 300 K using a Nosé–Hoover thermostat chain.
The metadynamics [9, 10] method is used to explore the free energy landscape of the dimerization process. The method employs a history-dependent bias potential to encourage the trajectory to visit previously unexplored regions of the phase space. This potential is constructed by depositing Gaussian 'hills' every 25 fs in a space described by three collective variables (CVs) as defined in section 3.2. By subsequently aggregating all deposited hills, the free energy surface (FES) of the reaction can be reconstructed.
We simulate two systems, a TTPB dimer (TTPB2w-Zn) and an extended Zn-bridged TTPB network ((TTPBw-Zn3)n), supported on liquid water. The former is contained in a 52 × 26 × 75 Å3 box, adsorbed on a 16 Å thick layer of H2O (724 molecules) with solvated Cl− counter ions. A schematic depiction of the simulated system is shown in figure S1 of the supporting information. The latter is modeled as TTPB2 in a hexagonal cell with a = 26.3 Å, bridged by three Zn2+ ions into an infinite network across the cell boundaries, adsorbed on 371 H2O molecules forming a slab of the same thickness of 16 Å. For comparison, we also simulate the free-standing network, i.e. in the gas phase without the supporting water layer, referred to as (TTPBg-Zn3)n. In this case, a uniform background charge is used instead of counter ions.
3. Results and discussion
3.1. Properties of the 2D network
As a first step to understand the 2D network we examine its structure and properties.
The 2D MOF is interlinked with terpyridine-Zn bridges, where six pyridyl groups coordinate each ion as shown in figure 1(b). This gives rise to three inequivalent Zn–N distances: to the axial pyridyls ( ), to the pyridyls in direct contact with the substrate (
), to the pyridyls in direct contact with the substrate ( ) and to those exposed to vacuum (
) and to those exposed to vacuum ( ). The coordination environment is furthermore characterized by the relative orientation of the two terpy groups. The planes of the two ligands (approximated as best-fit planes through the central pyridyl hexagon and the two adjacent carbon atoms on the outer pyridyls) intersect at an angle,
). The coordination environment is furthermore characterized by the relative orientation of the two terpy groups. The planes of the two ligands (approximated as best-fit planes through the central pyridyl hexagon and the two adjacent carbon atoms on the outer pyridyls) intersect at an angle,  , which describes this orientation. Figure 2(a) illustrates these quantities with a schematic representation of the Zn coordination environment.
, which describes this orientation. Figure 2(a) illustrates these quantities with a schematic representation of the Zn coordination environment.
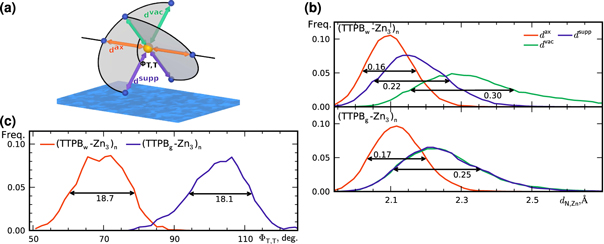
Figure 2. (a) Schematic drawing of a TTPB-Zn-TTPB bridge atop a water surface. The arrows indicate the three inequivalent N–Zn bonds ( ,
,  ,
,  ) and the shaded areas represent the planes of the terpy groups at an angle
) and the shaded areas represent the planes of the terpy groups at an angle  . Atom colors: Zn, orange; N, blue. (b) Distributions of N–Zn distances (Å) in (TTPBw-Zn3)n and (TTPBg-Zn3)n. (c) Distribution of
. Atom colors: Zn, orange; N, blue. (b) Distributions of N–Zn distances (Å) in (TTPBw-Zn3)n and (TTPBg-Zn3)n. (c) Distribution of  in (TTPBw-Zn3)n and (TTPBg-Zn3)n.
in (TTPBw-Zn3)n and (TTPBg-Zn3)n.
Download figure:
Standard image High-resolution imageWe plot time-averaged histograms of  for (TTPBw-Zn3)n and (TTPBg-Zn3)n in figure 2(b). All N–Zn distances are characterized by rather smooth distributions around an average, typically showing a slight skew towards larger values. Estimates of the full width at half maximum (FWHM) of the distributions are indicated in the figures. They vary between 0.16 and 0.30, depending on the local environment.
for (TTPBw-Zn3)n and (TTPBg-Zn3)n in figure 2(b). All N–Zn distances are characterized by rather smooth distributions around an average, typically showing a slight skew towards larger values. Estimates of the full width at half maximum (FWHM) of the distributions are indicated in the figures. They vary between 0.16 and 0.30, depending on the local environment.
In both systems, the axial pyridyl groups form the shortest bonds to Zn (≈ 2.1 Å). They appear largely unaffected by the presence or absence of the water substrate. These distributions are also the narrowest, with an FWHM around 0.16 Å in both cases.  and
and  are equivalent (≈ 2.25 Å) in (TTPBg-Zn3)n, as all participating ligands are exposed to the same environment. The distributions are superimposed and have the same FWHM. In contrast, as seen in the top panel,
are equivalent (≈ 2.25 Å) in (TTPBg-Zn3)n, as all participating ligands are exposed to the same environment. The distributions are superimposed and have the same FWHM. In contrast, as seen in the top panel,  are different on the water-exposed and vacuum-exposed sides of the MOF.
are different on the water-exposed and vacuum-exposed sides of the MOF.  is markedly shortened (compared to (TTPBg-Zn3)n) to an average of 2.17 Å in the presence of the liquid substrate and the FWHM is slightly reduced. On the other hand,
is markedly shortened (compared to (TTPBg-Zn3)n) to an average of 2.17 Å in the presence of the liquid substrate and the FWHM is slightly reduced. On the other hand,  increases to an average value of 2.36 Å, almost 5% longer than in (TTPBg-Zn3)n, and the distribution substantially widens, indicating that the vacuum-exposed groups have an increased flexibility. Remarkably, the overall average
increases to an average value of 2.36 Å, almost 5% longer than in (TTPBg-Zn3)n, and the distribution substantially widens, indicating that the vacuum-exposed groups have an increased flexibility. Remarkably, the overall average  is the same, 2.21 Å, for both (TTPBg-Zn3)n and (TTPBw-Zn3)n. The corresponding
is the same, 2.21 Å, for both (TTPBg-Zn3)n and (TTPBw-Zn3)n. The corresponding  distribution in TTPB2w-Zn is essentially identical to the one of (TTPBw-Zn3)n (figure S2 in the supporting information), indicating that each TTPB-Zn-TTPB environment is decoupled from the others in the network. We can thus conclude that the pyridyl groups in TTPB are independent from one another when forming the network.
distribution in TTPB2w-Zn is essentially identical to the one of (TTPBw-Zn3)n (figure S2 in the supporting information), indicating that each TTPB-Zn-TTPB environment is decoupled from the others in the network. We can thus conclude that the pyridyl groups in TTPB are independent from one another when forming the network.
These data show that the presence of the liquid substrate influences the structural properties of the adsorbed sheet. The water-exposed pyridyl groups are restricted in their fluctuations and pushed closer to the Zn ions. In turn, this appears to destabilize the vacuum-exposed N–Zn bonds, shifting  to larger values compared to the free-standing sheet. We have noted this mild confining effect of the liquid also for the TTPB monomer [8], and confirm it with the observations here.
to larger values compared to the free-standing sheet. We have noted this mild confining effect of the liquid also for the TTPB monomer [8], and confirm it with the observations here.
Beyond intra-sheet bond distances, the 'flatness' of the network as characterized by  is another relevant property of the 2D MOF. Figure 2(c) shows the distribution of
is another relevant property of the 2D MOF. Figure 2(c) shows the distribution of  for (TTPBw-Zn3)n and (TTPBg-Zn3)n. Both curves have approximately the same shape and FWHM, but are centered around average values of 69° and 102°, respectively. The coordination environment around Zn is quite strongly flattened in the adsorbed sheet compared to the ideal octahedral geometry. On the other hand, in (TTPBg-Zn3)n
for (TTPBw-Zn3)n and (TTPBg-Zn3)n. Both curves have approximately the same shape and FWHM, but are centered around average values of 69° and 102°, respectively. The coordination environment around Zn is quite strongly flattened in the adsorbed sheet compared to the ideal octahedral geometry. On the other hand, in (TTPBg-Zn3)n  is somewhat increased, making the pyridyl arms stand out further from that plane. The four equatorial pyridyl groups remain close to the plane of the 2D sheet and consequently (TTPBw-Zn3)n is thinner than (TTPBg-Zn3)n. Specifically, the thickness of (TTPBg-Zn3)n is approximately 13.0 Å, in good agreement with both experimental (15 Å) and computed (12 Å) data for a comparable molecule [4]. The water-confined network has a significantly lower thickness of approximately 9.1 Å. We attribute this to the presence of the water surface, which exerts an attraction towards the adsorbate and flattens it.
is somewhat increased, making the pyridyl arms stand out further from that plane. The four equatorial pyridyl groups remain close to the plane of the 2D sheet and consequently (TTPBw-Zn3)n is thinner than (TTPBg-Zn3)n. Specifically, the thickness of (TTPBg-Zn3)n is approximately 13.0 Å, in good agreement with both experimental (15 Å) and computed (12 Å) data for a comparable molecule [4]. The water-confined network has a significantly lower thickness of approximately 9.1 Å. We attribute this to the presence of the water surface, which exerts an attraction towards the adsorbate and flattens it.
In conclusion, it becomes clear that the dynamic flexibility of the network has a crucial influence on its structural properties. The presence of the liquid layer significantly influences this flexibility on the level of individual ligand groups and their relative orientation. We now explore the implications of this on the binding strength of the 2D network.
3.2. Dimer breaking mechanism
In order to quantify the binding strength of the 2D MOF assembled on water, we rely on the binding free energy of the dimer TTPB2w-Zn as a model. For this purpose, we use the metadynamics method to facilitate the exploration of the potential energy space and reconstruct the FES of the dissociation reaction
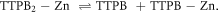
We define the following CVs to map the FES:
-
 , the Zn–N coordination number, expected to change from 6 to 3 in as the dimer dissociates.
, the Zn–N coordination number, expected to change from 6 to 3 in as the dimer dissociates. -
, the distance between the two axial N of the binding site, expected to sharply increase from the average value of 4.15 Å for the bound dimer.
-
, the Zn–O coordination number, which may increase as some pyridyl groups coordinating Zn are replaced by water.
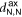
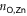
Starting from a bound configuration, the adaptive bias potential allows the system to quickly explore the configurational space, oscillating between bound and unbound states.
The series of images in figure 3 shows snapshots extracted from the MTD trajectory, depicting the breaking and reassembly of TTPB2w-Zn.
- (a)Fully bound TTPB2w-Zn( = 5.84, = 4.21 Å, = 0.09).
- (b)Partial breaking of the binding site, with 1 pyridyl group transiently rotating into a trans conformation ( = 4.63, = 4.68 Å, = 1.94).
- (c)Further bond breaking, leaving the dimer bridged by a single Zn–N bond ( = 4.00, = 5.99 Å, = 1.20).
- (d)Transient rotation of one pyridyl group into the trans conformation, yielding an undercoordinated Zn ion ( = 2.03, = 8.04 Å, = 2.98).
- (e)Re-formation of a Zn–N bond with the second TTPB ( = 3.75, = 4.82 Å, = 1.21).
- (f)Fully re-assembled dimer ( = 5.75, = 3.95 Å, = 0.56).
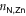

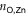
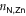
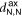
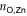
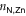
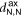
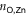
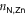
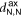
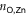
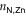
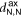
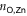
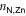
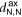
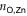
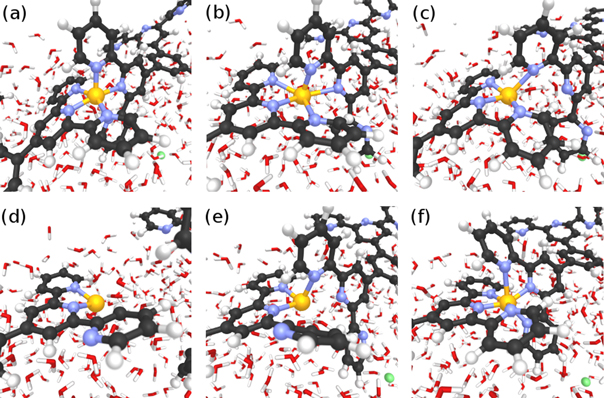
Figure 3. Snapshots from the MTD trajectory simulating the breaking of TTPB2w-Zn. See text for details. Atom colors: H, white; C, black; N, blue; Zn, yellow.
Download figure:
Standard image High-resolution imageAs can be seen from the snapshots, the breaking of the dimer follows a similar stepwise mechanism as the uptake of the Zn2+ from solution [8]. The bias potential causes individual TTPB-Zn bonds to break and the monomers to separate until the dimer has fully broken. This is accompanied by the rotation of individual pyridyl groups, that intermittently reduce the Zn–N coordination (we observed a similar behavior also when TTPB binds metal ions from the liquid phase [8]). The successful re-formation of the full dimer indicates that the available configurational space has been explored sufficiently and the trajectory has visited all relevant minima.
Evidently, the (rotational) flexibility of the ligand arms is a crucial aspect of the binding process. Moreover, the intermittent breaking of Zn-coordination and turning away of the pyridyl ligands competes with a complete and stable binding and thus indirectly weakens the network.
In the process of forming a full 2D network, additional molecules need to be incorporated. The dimer can be considered a first nucleus from which the layer can subsequently grow. As the TTPB monomers are expected to be more mobile on the liquid surface than larger assemblies, we envision that network growth proceeds by stepwise binding of free TTPB. For this to occur in two-dimensions, several terpy-metal-terpy bonds have to be formed at the same time. While our results show that the defect-free network is stable, the kinetics of this growth process make it likely that point defects occur in places where not all three binding sites of the monomer can be occupied. However, the flexiblity of the ligands should make it possible to remove many such defects by mild annealing. Additionally, the formation of 'grain boundaries' can be expected in places where larger network fragments coalesce.
3.3. Energetics of dimer breaking
The reconstructed FES of the TTPB2w-Zn breaking process is shown in figure 4 (left panel) as a function of  and
and  . The system is characterized by two minima, one for the bound state (top left,
. The system is characterized by two minima, one for the bound state (top left, 
 5.8,
5.8, 
 4.0 Å) and one for the unbound state (bottom right,
4.0 Å) and one for the unbound state (bottom right, 
 3.0,
3.0, 
 8.0 Å), where the dimer is dissociated into a TTPB and a TTPB-Zn fragment. The topography of the FES also reveals some intermediate states at higher energy. One corresponds to a bound dimer with undercoordinated Zn2+ at reduced
8.0 Å), where the dimer is dissociated into a TTPB and a TTPB-Zn fragment. The topography of the FES also reveals some intermediate states at higher energy. One corresponds to a bound dimer with undercoordinated Zn2+ at reduced  and small
and small  (see figure 3(b)). Furthermore, Zn can also be undercoordinated in the dissociated state, missing one bond to a pyridyl group (see figure 3(d)).
(see figure 3(b)). Furthermore, Zn can also be undercoordinated in the dissociated state, missing one bond to a pyridyl group (see figure 3(d)).

{kind=link}
{kind=link}
{kind=link}
Figure 4. MTD free energy surface of the TTPB2w-Zn dissociation in the space of the collective variables  and
and  . The initial bound state is indicated in the top left (
. The initial bound state is indicated in the top left (
 6), and the dissociation proceeds towards the bottom right (
6), and the dissociation proceeds towards the bottom right (
 3). The third CV,
3). The third CV,  , has been integrated in this 2D representation. Right panel shows free energy profile along the minimum energy path connecting the bound and unbound states, highlighting the energy barrier to break TTPB2w-Zn.
, has been integrated in this 2D representation. Right panel shows free energy profile along the minimum energy path connecting the bound and unbound states, highlighting the energy barrier to break TTPB2w-Zn.
Download figure:
Standard image High-resolution image{kind=link}
The free energy difference between the bound and unbound dimer amounts to 143.1 kJ mol−1. Taking into account the higher entropy of TTPB + TTPB-Zn relative to TTPB2w-Zn, due to the higher number of particles and larger conformational flexibility, the binding enthalpy of the dimer should be appreciably larger than this. For comparison, the experimental binding enthalpy of Zn(terpy)22+ is −60.7 kJ mol−1 [17], considerably smaller.
The right panel in figure 4 shows the free energy profile along the MEP. The breaking of the dimer is rather steeply uphill in energy, with a shallow local minimum of the undercoordinated dimer. A barrier of 185 kJ mol−1 is crossed before reaching the shallow minimum of TTPB + TTPB-Zn. As this MEP describes the breaking of the dimer, its reverse corresponds to the formation. Thus, only a small barrier of 42.1 kJ mol−1 has to be overcome by the system in order to form a dimer with a binding free energy of −143.1 kJ mol−1.
4. Conclusions
Our results indicate that a 2D MOF based on Zn-bridged TTPB is quite stable in terms of free energy. Combining these findings with the previous work on Zn ion coordination from the liquid phase [8] we can reproduce the full formation of stable terpyridine-based MOFs on water. Moreover, Zn is among the transition metals that bind most weakly to the terpyridine ligands [17]. Our binding energy is thus a lower limit that could significantly be improved by replacing Zn2+ with Fe2+ or Cu2+.
Our study provides insight into the mechanism of the network formation and the intermittent bond breaking which counteracts it. We envision a possible way to address this by modifying the ligand molecule to reduce its flexibility and prevent the rotation of the pyridyl groups. The 3' and 5' carbons of the central pyridyl would have to be covalently linked to the respective 5 and 5'' positions of the outer pyridyls. Such rigidified U-shaped terpyridyls are synthetically accessible [18, 19], and merit evaluation as MOF building blocks in future work. Our data show that there is significant potential to strengthen these 2D MOFs by tuning the ligand rigidity, thus allowing further improvement of these novel membranes.
Acknowledgments
The authors gratefully acknowledge financial support from the Swiss National Science Foundation under grant no. 20021_140441 and generous computing resources from the Swiss National Supercomputing Center (project ID: S415). RK thanks the Japanese High-Performance Computing Initiative for computing time on the K Computer as part of a 'Junior Researcher Promotion' (project ID: hp140077).