

Abstract
Significant progress has been made over the past 25 years in the development of in vitro-engineered substitutes that mimic human skin, either to be used as grafts for the replacement of lost skin, or for the establishment of in vitro human skin models. In this sense, laboratory-grown skin substitutes containing dermal and epidermal components offer a promising approach to skin engineering. In particular, a human plasma-based bilayered skin generated by our group, has been applied successfully to treat burns as well as traumatic and surgical wounds in a large number of patients in Spain. There are some aspects requiring improvements in the production process of this skin; for example, the relatively long time (three weeks) needed to produce the surface required to cover an extensive burn or a large wound, and the necessity to automatize and standardize a process currently performed manually. 3D bioprinting has emerged as a flexible tool in regenerative medicine and it provides a platform to address these challenges. In the present study, we have used this technique to print a human bilayered skin using bioinks containing human plasma as well as primary human fibroblasts and keratinocytes that were obtained from skin biopsies. We were able to generate 100 cm2, a standard P100 tissue culture plate, of printed skin in less than 35 min (including the 30 min required for fibrin gelation). We have analysed the structure and function of the printed skin using histological and immunohistochemical methods, both in 3D in vitro cultures and after long-term transplantation to immunodeficient mice. In both cases, the generated skin was very similar to human skin and, furthermore, it was indistinguishable from bilayered dermo-epidermal equivalents, handmade in our laboratories. These results demonstrate that 3D bioprinting is a suitable technology to generate bioengineered skin for therapeutical and industrial applications in an automatized manner.
Export citation and abstract BibTeX RIS
1. Introduction
Skin injuries caused by burns, chronic ulcers from different etiology, infections, cancer surgery, and other genetic and somatic diseases require effective treatment to prevent morbidity or mortality. The World Health Organization estimates that nearly 11 million burn injuries per year worldwide require medical attention, with approximately 265 000 leading to death [1]. To restore the function of the skin after damage and to facilitate wound-healing, autologous grafts (autografts) obtained from own-patients donor sites are commonly used to repair the skin, while avoiding immune-rejection. Unfortunately, the availability of autografts for wound coverage is insufficient when dealing with large and/or severe wounds [2–4]. As a result, several approaches have been explored for skin replacement therapy, such as cultured autologous epithelial autografts (for a review see [5]), but their results are far from ideal, since they are limited by their fragility and the difficulty of handling, unpredictable take rate and sensitivity to mechanical shearing forces for at least two months post grafting [6–8]. In response to these limitations, new approaches for skin engineering have been tested and developed in recent years. These advances have led to the development of more sophisticated laboratory-grown skin substitutes which contain dermal and epidermal components that interact dynamically with each other during in vitro maturation and also after transplantation [9–12]. In particular, fibrinogen (and its derivative fibrin) is a blood component that has been used extensively as a stromal substitute to construct human skin since it has the advantages of low price, availability, and good tolerance to cells; in addition, if required, it can be produced as an autologous scaffold [13–15]. In this context, a human plasma-derived bilayered (including dermis and epidermis) skin model was generated by our group and applied successfully to treat burns and traumatic and surgical wounds [16, 17].
The limitations of the current process: high production costs, the need for specialized personnel, and the time required for production of a surface of therapeutically useful skin (3–4 weeks to generate 1 m2), combined with a foreseen higher demand for artificial skin, have all led to an increasing need to develop new methods that offer automation, standardization, and reduction in time and production costs [7, 18, 19]. Three-dimensional (3D) bioprinting, has emerged as a flexible tool in regenerative medicine and provides a platform to address these needs. 3D bioprinting opens up the possibility of constructing artificial tissues or organs, either autologous or allogeneic, by printing cells, soluble factors and biomaterials in a desired pattern with the help of high-precision Cartesian robots [20–23]. A variety of biomaterials have been widely studied as scaffolds for bioprinting in tissue engineering [24, 25] examples include hydrogels [e.g. 26, 27] which are mostly used for the generation of soft tissues such as skin (for a review, see [28]), and polymers [e.g. 29] and ceramics, which are frequently used for the generation of hard tissues such as bone (e.g. [30, 31]). Additionally, many efforts have been made in the field of biomaterials to design multifunctional scaffolds, which could be used in the future for the 3D printing of advanced tissue engineered constructs e.g. [32], see for reviews [33, 34]. A very common strategy involves printing layers of hydrogel matrix precursors and a posteriori crosslinking in order to form a scaffold that provides structural support to the cells and other extracellular components embedded within it [35–37]. After an in vitro culture period of time, which depends on the tissue generated and is required for tissue fusion, remodelling and maturation, the printed tissue or organ construct can potentially be applied to replace the function of the damaged tissue.
Very recently, a comprehensive review about skin 3D bioprinting was published [38]. As discussed in this review, there are two main strategies concerning the use of skin bioprinting for wound treatment. The first strategy is in situ bioprinting. Using this technique, fibrin-collagen layers containing either amniotic fluid-derived stem cells [39] or human fibroblasts (hFBs) and keratinocytes (hKCs) [40] were printed on full-thickness wounds of nude mice. Although the results of these experiments were promising, further optimization is still required before application to human patients [38].
In the second strategy, two different bioprinting approaches have recently been explored for the in vitro production and in vitro and in vivo analysis of skin constructs containing dermal and epidermal components. In the first approach [41, 42], the authors used a free-form fabrication (FFF) technique to deposit a variable number of layers of crosslinked collagen and collagen containing either hFBs or hKCs. In the second approach [43, 44], laser assisted bioprinting (LaBP) was used to deposit alternating layers, composed of 20 sublayers each, containing immortal murine fibroblasts and immortal hKCs (NIH-3T3 and HaCaT cell lines, respectively) embedded in a collagen matrix. These approaches are mainly aimed at demonstrating the feasibility of generating artificial skin by bioprinting. However, in our opinion (see also [38] for additional comments), they present several drawbacks: (1) In general, they do not use human primary fibroblasts and keratinocytes simultaneously. The used cells might be less sensitive to the stresses of the bioprinting process, and their proliferation and differentiation characteristics are far different from those of the cells contained in human native skin. (2) The printed layered structures are not reminiscent of normal skin and the skin constructs produced did not possess the structural quality of the normal human skin. Moreover, the time required in these multi-layered deposition methods is currently far from allowing the effective generation of the relatively large skin surfaces needed in wound treatment.
Based on our previous experience producing skin equivalents for transplantation to human patients, we decided to follow the second strategy. In this study we have used a FFF 3D bioprinting technique to engineer a human plasma-derived bilayered skin using hFBs and hKCs obtained from skin biopsies. The printed human skin was analysed both in 3D in vitro cultures and in vivo upon grafting to immunodeficient athymic mice (skin-humanized mice), using histological and immunohistological methods [45–47]. Our results showed that the printed skin had structural and functional characteristics as well as appearance and consistency similar to those of normal human skin, and skin equivalents produced manually in our group. We also demonstrated the capacity of our process to reproducibly print large areas of human skin, useful for the treatment of diverse cutaneous pathologies such as burns, ulcers and surgical wounds.
2. Materials and methods
2.1. Bioprinter design and set-up
The model used in this study was the open source Printrbot (original), modified to deposit cell-laden hydrogels (figure 1(A)). Most of the structural parts were generated by a normal 3D printer in acrylonitrile-butadiene-styrene (ABS).

Figure 1. (A) Bioprinter setup and components. Human plasma, hFBs, calcium chloride (CaCl2) and hKCs, respectively, are pumped through four sterile tubes from the extrusion module E to the head A. The extruder needles B deposit the components on the printing plastic dish (P100, Corning 100 × 20 mm). The heated bed C maintains the temperature at 37 °C. The system is controlled by the control unit D, which is composed of an Arduino with RAMPS 1.4 and LCD interface. (B) Picture of the head showing the three tubes (a)–(c) carrying the components of the dermal compartment (hFBs, human plasma and CaCl2), converging to the trifurcated connector which is itself connected to needle 1 and the fourth tube, carrying the hKCs, connected directly to needle 2.
Download figure:
Standard image High-resolution imageAn external module (extrusion module) composed of two electric stepper motors (NEMA17) and four sterile disposable plastic syringes (5 or 20 ml) was designed to contain and extrude the hydrogel precursors and the cell suspensions. The plastic syringes and the sterile connecting tubes were replaced between experiments to avoid contamination. The content of each syringe will be described in section 2.3. Three of these tubes ((a)–(c) in figure 1(B)) converged at the head into a trifurcated connector where their contents were mixed; then, the mixture went through a luer 1.2 × 40 mm extrusion needle without a bevel, acting as a'nozzle' (needle 1 in figure 1(B)). The flow of each syringe (3–6 ml min−1) was directly proportional to its volume content in order to obtain a constant homogeneous mixture in the extrusion needle. Typically, the flow in the extrusion needle was 12 ml min−1. The fourth tube ((d) in figure 1(B)) was directly connected to an independent extrusion needle (needle 2 in figure 1(B)) and had a flow of 4 ml min−1.
The x–y plane contained a heated surface in order to maintain the temperature at 37 °C (figure 1(A)). The printer and the extrusion module were placed in a cell culture laminar flow hood; all the parts were sterilized with UV light in the hood.
The printer firmware was installed in the microcontroller (ATmega2560) of a RepRap Arduino Mega Pololu Shield (RAMPS), and it manages the mechanical sensors and actuators as well as the thermal control of the heated bed. The selected firmware was Marlin, also open source, because it is able to control more than one extruder. Deposition trajectories were generated and sent to the RAMPS using Repetier v0.53. This programme transformed the geometric data into paths or spatial coordinates to be followed by the printer head. It also controlled which of the dispensers should be active and the operative procedure time. A script in C++ was developed to establish the volume of liquid to be deposited.
Liquids were pumped by a module composed of four separated dispensers, as previously described. Each of them had its own syringe. As the RAMPS had only two available ports to connect extruders, to control three of the syringes we designed a system similar to a syringe pump but with a reduction system, based on timing belts and pulleys, which allowed to move three different syringes with only one step motor (supplementary figure 1). Each syringe driving screw had its own angular speed (wi) determined by the corresponding reduction coefficient which depended on the relation between the teeth number (ni) of its pulley and that of the main pulley (n). These coefficients were calculated to obtain a constant stoiquiometry of the three components in the final mixture at the nozzle. The fourth syringe was moved independently by the second step motor.
The motors, NEMA17, presented a step angle of 1.8° and a holding torque of 4.8 kg cm, and were controlled by a DRV8825 driver that provides six different step resolutions (full-step, half-step, 1/4-step, 1/8-step, 1/16-step, and 1/32-step). The final resolution of the deposition depended on this parameter and it could be easily tuned with jumpers. All these drivers, sensors and actuators were connected to the RAMPS 1.4 shield, which was mounted over an Arduino Mega 2560 board that contained a microcontroller ATmega2560.
2.2. Primary hKCs and hFBs culture
hFBs and hKCs obtained from skin biopsies of healthy donors were obtained from the collections of biological samples of human origin; these samples are registered in the 'Registro Nacional de Biobancos para Investigación Biomédica del Instituto de Salud Carlos III'. hKCs were cultured following previously described methods [48] as modified by our laboratory [15, 49]. The growing media for hKCs was a 3:1 mixture of Dulbecco's Modified Eagle Medium (DMEM) (GIBCO-BRL) and HAM'S F12 (GIBCO-BRL) (hKC medium) containing 10% of fetal bovine serum (FBS), 0.1 nM choleric toxin, 2 nM T3, 5 mg ml−1 insulin, 0.4 mg ml−1 hydrocortisone and 10 ng ml−1 EGF (Sigma, St Louis, MO). hFBs were cultured in Dulbecco's modified Eagle's medium (DMEM, Biochrom KG) containing 10% FBS.
2.3. Preparation and printing of fibrin-based skin
Skin substitutes formed by two layers, representing the dermis (lower layer) and the epidermis (upper layer), were generated following the method developed in [15, 17]. The lower layer was a plasma-derived fibrin matrix populated with hFBs and the upper layer was formed by hKCs, seeded on the top of the fibrin scaffold. Fresh frozen human plasma was provided by a local blood bank (Banco de Sangre del Centro Comunitario de Transfusión del Principado de Asturias (CCST) Spain) and was obtained according to the standards of the American Association of Blood Banks [50]. The fibrin matrix was prepared as previously described in [15, 17] with some modifications required for the bioprinting process, as described below.
Figure 2 describes the bioprinting process and in vitro and in vivo experiments. To generate a dermal substitute, 7 × 104 cultured hFBs were resuspended in 4 ml of DMEM and loaded in the first syringe. In the second syringe, a volume of human plasma containing 30 mg of fibrinogen (typically 13 ml) was mixed with 200 μl of tranexamic acid (antifibrinolytic agent Amchafibrin, Fides- Ecopharma). Finally, in a third syringe 2.3 ml of CaCl2 (prepared at 1% w/v in saline, (NaCl 0.9% w/v)) was loaded. The function of CaCl2 is to induce the coagulation of the plasma fibrinogen into a fibrin hydrogel. The total volume of the three syringes was adjusted to 25 ml by adding saline to the third syringe. After this, the syringes were put into the extrusion module of the bioprinter and their content was mixed (as described in section 2.1) and deposited on a P100 tissue culture plate (Corning 100 × 20 mm).
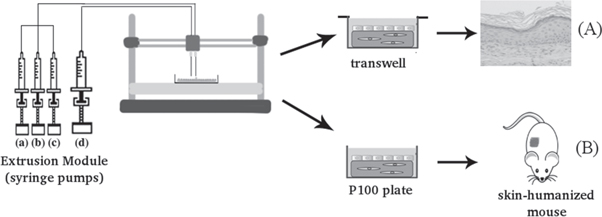
Figure 2. Scheme of the bioprinting process. The extrusion module contained four syringes, loaded with hFBs (a), plasma (b), CaCl2 (c) and hKCs (d), respectively. The contents of the syringes (a)–(c) were continuously pumped out at the appropriate speed, mixed as they arrived at the head, extruded through the needle and deposited on the corresponding plate type (P100 or transwell), following the trajectories dictated by the control unit. This mixture was allowed to polymerize for 30 min at 37 °C to form a fibroblast-containing fibrin hydrogel, which became the dermal compartment of the skin equivalent. Immediately after this polymerization step, the hKCs suspension contained in syringe (d) was similarly deposited on top of this hydrogel to form a confluent monolayer. (A) Equivalents printed on transwell inserts were allowed to differentiate at the air–liquid surface for 17 d and then analysed. (B) Equivalents printed on P100 plates were grafted on to the backs of immunodeficient mice for eight weeks and then analysed.
Download figure:
Standard image High-resolution imagePrinted dermal substitutes were left in a cell culture incubator (at 37 °C in 5% CO2) for thirty minutes to allow them to polymerize, and then 10 ml of hKCs medium, containing 6 × 106 hKCs per P100 plate were loaded into the fourth syringe of the extrusion system and deposited over the dermal equivalent. hKCs were allowed to attach and spread overnight in a cell culture incubator. This number was established in order to generate a confluent hKCs monolayer at this moment. Immediately after the overnight incubation, the printed skin equivalents were transplanted on to the backs of immunodeficient mice (see section 2.5).
Alternatively, skin substitutes were printed on transwell inserts. The components were placed on polycarbonate transwell inserts (1 μm pore) in a 6-well culture plate (Corning Costar Corp., Cambridge, MA, 4.15 cm2) for the 3D in vitro assays (see section 2.5). In this case, the deposition volume and the trajectories were adjusted to the geometry of these plates. After the hKCs attaching and spreading step, hKCs were allowed to differentiate at the air–liquid interface and formed a multilayered skin as explained in section 2.5.
2.4. Immunodeficient mice
Immunodeficient athymic nude mice were purchased from IFFA-Credo-Charles River (St. Aulin-les-Elbeuf, France) and kept and used at the CIEMAT Animal Facility (Spanish registration number 28079-21A) under sterile conditions. The animals were housed in individually ventilated type II cages, a maximum of four mice to each cage with 25 air changes per hour and 10 KGy gamma irradiated soft wood pellets as bedding. Experiments were carried out according to European and Spanish laws and regulations.
2.5. In vivo and in vitro maturation and differentiation of printed skin equivalents
As explained in section 2.3, the printing process was designed to produce a fibroblast-containing fibrin hydrogel covered with a monolayer of hKCs. In vivo and in vitro assays were performed to analyse the viability of these constructs and their capacity to generate a terminally differentiated skin.
For the in vivo assays, once hKCs were attached to the fibrin surface, the cultured equivalents were manually detached from the P100 plate and grafted on to the backs of immunodeficient mice. Four female mice were aseptically cleansed and grafted as previously described [17]. Full thickness circular wounds of 12 mm diameter were produced by means of a punch on the dorsum of each mouse. Then, circular samples of the same diameter were obtained by the same punch from the printed skin substitutes, placed on the generated wounds and covered by the skin, previously removed from these mice, devitalized by three cycles of freezing and thawing. The devitalized skin was kept in place with the help of sutures. The grafts were analysed eight weeks after grafting took place.
For the in vitro assays, skin constructs deposited on transwells were allowed to differentiate at the air–liquid interface for 17 d at 37 °C in a CO2 incubator in differentiating medium (hKCs medium containing 0.5% FBS and 50 μm of ascorbic acid). The medium was changed every three days [51–53].
2.6. Histology and immunostaining
Four-mm biopsies of human skin regenerated either in vitro or in vivo were collected with the help of a punch. For histological analysis, samples were fixed in 3.7% buffered formaldehyde, and embedded in paraffin. 3 μm cross-sections were dewaxed, rehydrated and stained with hematoxylin-eosin (H/E).
For immunohistochemistry experiments, the biopsies were frozen and 5 μm cryosections were analysed using primary specific antibodies against well-known skin markers: anti human-vimentin (V9, BioGenex, San Ramon, CA to distinguish hFBs), antikeratin 5 (polyclonal AF138, BabCO, Berkeley, CA; to label hKCs of the proliferative basal layer), antikeratin 10 (monoclonal AE2, ICN Biomedicals, Cleveland, OH; to label suprabasal hKCs), and antihuman filaggrin (polyclonal AF-62, BabCO) to label the epidermal granular layer. To determine the formation of the dermoepidermal basal membrane, a specific antibody against human-collagen VII (Clone LH7.2, Sigma) was used. To detect blood vessel formation, biopsies from human skin regenerated on nude mice were labelled with anti-SMA (Smooth Muscle Actin, C6198, Sigma, St. Louis, USA). Samples were coverslipped using Mowiol (Hoechst, Somerville, NJ) mounting media containing 46-diamidino-2-phenyl indole (DAPI, ROCHE, Germany, 20 μg ml−1) for nuclei visualization.
2.7. Analysis of epidermal cells viability
To determine if the printing process compromised the viability of the epidermal cells, freshly trypsinized hKCs (as a control) and hKCs, pumped through the extrusion system after trypsinization, were subjected to colony-forming assay as described elsewhere [54, 55].
In brief, approximately 500 hKCs were placed into each well of a six multi-well plate containing a feeder layer of lethally irradiated 3T3 cells. Three wells were seeded with control hKCs and the other three were seeded with extruded hKCs. After nine days, cultures were stained with fluorescent Rhodamine B (R-6626, Sigma St Louis, USA) to estimate the number of epidermal colonies using an inverted fluorescence microscope. The number of colonies was calculated by counting ten fields per well.
3. Results
3.1. Analysis of printed human skin differentiated in vitro
Bioengineered equivalents deposited into transwells were allowed to differentiate at the air–liquid interface for 17 d. As shown in the histological staining (figure 3(B)), printed equivalents generated a tissue with a structure similar to that obtained differentiating handmade skin equivalents (figure 3(A)) and also similar to normal human skin (figure 4(C)). A well-formed, orthokeratotic stratum corneum was present indicating terminal differentiation. The dermal compartment contained well spread hFBs in the fibrin matrix. To analyse the nature of this differentiation more carefully, immunofluorescent analysis was carried out. Expression of keratin K10 was detected in suprabasal cells (figure 3(C)), where this intracellular structural protein is specifically synthesized in normal skin. To visualize the state and persistence of the hFBs, after this relatively prolonged culture time, we stained cryosections with a specific antibody against human vimentin, a cytoskeletal protein characteristic of this kind of cell. This demonstrated the proper growth and spreading of the hFBs comprising the dermal compartment of the bioprinted skin (figure 3(D)). The lack of strateum corneum in figures 3(C) and (D) is due to the cryosectioning method which frequently removes these structures.

Figure 3. In vitro 3D human skin equivalents obtained after 17 d of differentiation at the air–liquid interface. (A) 'Handmade' skin equivalent following our previous protocol. (B)–(D) Printed skin equivalents. (A), (B) Histological analysis of fixed samples, using hematoxilyn-eosin. (C), (D) Immunostaining of frozen samples using an anti-K10 antibody ((C), green immunofluorescence) and an anti-human vimentin antibody ((D), red immunofluorescence). Blue colour in (C) and (D) denotes DAPI staining of the nuclei. Ep and De in (A)–(D) denote the epidermal and the dermal compartments, respectively. The white dotted line indicates the dermo-epidermal junction (basal membrane). Scale bar: 100 μm.
Download figure:
Standard image High-resolution image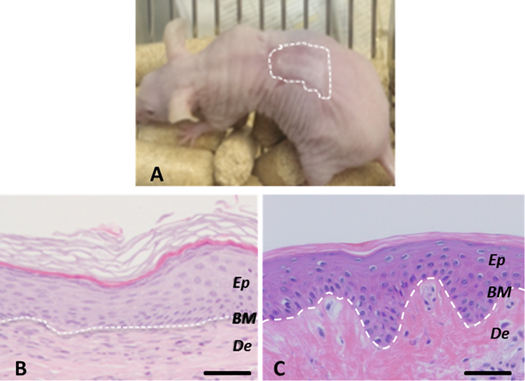
Figure 4. Histological analysis (8 weeks postgrafting) of bioprinted human skin grafted to immunodeficient mice. (A) Visual appearance of the grafted human skin. The dotted line marks the boundary between human and mouse skin. (B) H/E staining of the regenerated human skin. (C) H/E staining of normal human skin. The white dotted line in (B) and (C) indicates the dermo-epidermal junction (basal membrane, BM). Ep and De in (B) and (C) denote the epidermal and the dermal compartments, respectively. Scale bar: 100 μm.
Download figure:
Standard image High-resolution image3.2. Analysis of printed human skin differentiated in vivo
To study if bioprinted skin substitutes had the capacity to differentiate in vivo, we grafted them on to the back of immunodeficient athymic mice. These grafts were performed orthotopically, so that the printed equivalents were placed on the wound beds generated on the back of the immunodeficient mice, as described in [17, 49] (see also section 2.5). Between four and six weeks after grafting, the devitalized mouse skin, used as a biologic bandage, fell off and the grafted human skin became visible (figure 4(A)). It exhibited a characteristic wrinkled, thick and whitish aspect, very similar to the appearance of native human skin and clearly different from the surrounding thin and pinkish mouse skin. Histological analysis demonstrated that the regenerated human skin presented a structure very similar to that of normal human skin (compare figure 4(B) with figure 4(C)). All the strata characteristic of normal skin, stratum basale, stratum spinosum, stratum granulosum and a well-developed stratum corneum are easily identified in the printed skin.
A more detailed analysis of the original printed skin was performed by immunofluorescence using well-established skin markers. Accordinlgy, the basal proliferative stratum was clearly revealed by labelling with an antibody recognizing keratin K5 (figure 5(A), green staining). The correct formation of the dermo-epidermal junction of the skin was confirmed by labelling with an antibody against human collagen VII (figure 4(B) green staining), the protein forming the anchoring fibrils that bind together epidermis and dermis. This structure is very important for the mechanical stability of the skin; its lack leads to severe blistering due to the separation of the two compartments of the tissue, observed in patients suffering from dysthrophic epidermolysis bullosa [56]. We consider the presence of the basal lamina and the stratum corneum as clear indications of proper and complete differentiation of the grafted printed skin. As in normal mature skin, there was a strong suprabasal expression of keratin K10 (figure 5(C), red staining) and of the late differentiation marker filaggrin, characteristic of the granular layer (figure 5(D), green staining). Also rete ridges (a hallmark of mature human skin, not found in mouse skin) were detected in some regions of the grafted printed skin (asterisks in figure 5(D)).

Figure 5. Immunohistochemical analysis (eight weeks postgrafting) of bioprinted human skin grafted to immunodeficient mice using antibodies against skin markers. (A) Keratin K5 detection (green immunofluorescence). (B) Collagen VII (green line between dermis and epidermis) and vimentin (the red colour in the dermal compartment) detection. (C) Human keratin K10 detection (red suprabasal staining: notice that the basal layer is negative). (D) Filaggrin (green staining in the stratum granulosum) and SMA (red staining) detection. Arrows point to some of the capillaries present in the dermal compartment. Asterisks mark rete ridges. Nuclei were stained with DAPI (blue). The white dotted line in panels (A) and (C), indicates the epidermal-dermal boundary. Inside the images: Ep—epidermal compartment, De—dermal compartment. Scale bar: 100 μm.
Download figure:
Standard image High-resolution imageIn addition, immunostaining with a specific antibody against human vimentin, showed the persistence of hFBs exclusively in the dermal compartment of the regenerated skin (figure 5(B), red staining). Another important parameter to assess in the regeneration process is the vascularization of the grafted human skin, which allows oxygenation and nutrition of the new tissue and therefore, its long-term persistence. As shown by the red staining and arrows in figure 5(D), blood vessels (SMA +, red) were detected in the dermis of the printed skin upon in vivo regeneration.
3.3. hKCs survival analysis
hKCs are more delicate and difficult to keep in culture than fibroblasts. In particular, it is well- established that hKCs differentiate terminally when cultured in suspension [57]. Therefore, it is necessary to analyse the viability of these cells after going through the bioprinting process. To do this, we performed colony-forming efficiency assays as described in materials and methods (section 2.7). We found (figure 6) that the number and the size of the colonies were very similar, both before and after the cells underwent the bioprinting process (the same number of cells were seeded in both cases).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Colony-forming efficiency assay. Microscopic appearance (phase contrast) of the hKC colonies grown in the presence of a feeder layer of lethally irradiated 3T3 cells; before (A) and after (B), passing them through the printing system. (C) Number of keratinocyte colonies per microscopic field before (left) and after (right). Scale bar: 200 μm.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
This work shows the automation and in vitro production of printed human skin containing dermal and epidermal components, with structural and functional properties similar to those of skin constructed by conventional manual procedures developed previously in our group, and also to those of in vivo human skin. Due to the relatively simple structure of skin tissue and the strong clinical relevance of methods facilitating the treatment of wounds, production of skin tissue containing dermal and epidermal components by bioprinting is currently an area of active development. To our knowledge, two main in vitro deposition approaches containing in vitro and in vivo analysis have been published [41–44]. The first approach, which was based on a layer by layer deposition technique, demonstrated the feasibility of the multi-layered deposition of fibroblasts and keratinocytes in a collagen scaffold. The dermal compartment contained eight acellular collagen layers, interspersed with three fibroblast-containing collagen layers. On top of this, two keratinocytes-containing collagen layers were deposited. This requires that, after deposition, the fibroblasts migrate to generate a homogeneous distribution throughout the collagen matrix, as can be observed in a normal dermis.
Apart from this, it is difficult to evaluate the quality of the 3D skin obtained, since the authors do not show the localization of any of the several well-known skin differentiation markers. In addition, the histology of the skin tissue does not show proper stratification and terminal differentiation as compared to human skin. As the authors recognize, this can be, at least in part, due to the use of an immortalized keratinocyte cell line (HaCaT cells) instead of primary hKCs. Alternatively, it could be a consequence of the anomalous severe shrinking and compaction of the dermal compartment reported by the authors during the air–liquid interface culture, the step in which keratinocyte stratification and differentiation to form an epidermis takes place. Finally, based on the data provided by the authors, the estimated printing speed allows the deposition of 1 cm2 skin per hour which is a very slow process considering the large surfaces needed for clinical or commercial applications.
The second approach used a LaBP technique to arrange immortal human HaCaT keratinocytes and mouse NIH-3T3 fibroblasts. In this technique, 20 collagen sublayers containing fibroblasts were printed onto a sheet of Matriderm® and subsequently 20 collagen sublayers containing keratinocytes were printed on top of it. A clear problem of this method is again the use of immortalized keratinocyte and fibroblast cell lines; in particular the NIH-3T3 murine fibroblasts that are very different from hFBs. Moreover, the histological and immunohistochemical data presented by the authors indicated relevant differences when comparing their printed skin with normal human skin: (1) From the histological point of view, it is apparent that in the histological sections of the in vivo implants using the dorsal skin full chambers [44], the human epidermis, although apparently well differentiated, as indicated by the presence of a stratum corneum, had an abnormal thickness: it is thinner than the mouse epidermis while it is known that human skin is thicker than mouse skin, in particular in this type of grafting experiment [17, 49]. (2) From the immunohistochemical point of view, K14 is a well-established marker of epidermal basal cells as shown by the own authors in normal mouse skin. However, this marker is found in all the epidermal layers of both in vivo and in vitro differentiated printed skin. Similarly, Ki67, which is a marker of cell proliferation that should be mostly restricted to basal cells, was found evenly distributed through the whole epidermis of in vitro cultures. In agreement with the authors, we think that the time used in these experiments, probably due to the limitations imposed by the dorsal skin full chamber method, is a too short time to obtain a fully differentiated epidermis. It has been reported that HaCaT cells need between three and six weeks to develop a fully differentiated epidermis upon in vivo transplantation to the back of nude mice [58, 59]. The delocalized K14 and Ki67 staining observed in these experiments is also a hallmark of the early stages of HaCaT 3D differentiation [58, 59]. Finally, although it is difficult to make an accurate estimation from the data provided by the authors, a caveat concerning this technology is what would be the time required to produce a skin surface of clinical or commercial interest (50–100 cm2).
From a practical point of view, our method is simpler and quicker than the two described methods since, instead of printing a high number of cellular and acellular layers to form the dermis and epidermis, we deposit simultaneously all the elements (hFBs, human plasma and CaCl2) required to form the dermis and, on top of this, the hKCs required to form a confluent layer of epidermal cells. These constructs give rise to a fully differentiated human skin upon in vitro or in vivo differentiation.
To overcome the limitations present in the two approaches discussed above and, therefore, to fully validate the bioprinting method for the production of a tissue with structural and functional properties, similar to normal human skin, the use of human primary fibroblasts and keratinocytes is necessary. According to this, we used primary human cells together with a fibrin-based dermal matrix previously developed by our group for the production of large skin surfaces, useful in the treatment of severe and extensive burns, wounds with loss of substance and skin fragility diseases [16, 17]. Based on the foregoing, we developed an extrusion bioprinting method that did not harm these biological components, in particular, hKCs, which are known to terminally differentiate when they are kept in suspension [57].
We did not observe any contraction or shrinking of the fibrin hydrogels as reported in [42] with collagen hydrogels. Initially, we analysed the printed skin equivalent upon in vitro differentiation. The results showed that the generated skin had correct architecture (figure 3(B), H/E staining) and differentiation (figures 3(C) and (D), staining with dermal and epidermal markers) as well as persistence, homogeneous distribution and spreading of the hFBs.
Secondly, the structure and functionality of the printed human skin was further analysed on skin-humanized mice. This model recapitulates faithfully the characteristics of the skin from which human cells were obtained (donor's skin) and overcomes the time limitations imposed by the dorsal skin full chamber used in [44]. Our laboratory has extensive experience using it to model diverse cutaneous diseases [45] and processes [60, 61]. To our knowledge, it constitutes the best system to perform long-term experiments with human skin in an in vivo scenario. We used H/E staining (figure 4) and several differentiation markers such as keratin 5 (a marker of proliferative basal keratinocytes), human vimentin (a marker of hFBs), human-collagen type VII (a marker of dermoepidermal basal membrane), keratin 10 (a marker of suprabasal differentiated keratinocytes) and human-filaggrin (a marker of keratinocyte terminal differentiation) (figure 5). These markers labelled the same cells and structures in the bioprinted skin as they do in human normal skin, including the presence of a well-developed stratum corneum and a basal membrane. In addition, it is important to highlight the in vivo formation of rete ridges, a hallmark of mature human skin, not found in mouse skin and so far, not reported with printed human skin.
Neoangiogenesis formation is one of the most crucial events for successful skin grafting to take place. Obviously, this is particularly relevant if one considers the clinical applications of bioengineered skin equivalents. As shown in figure 5(D), and also reported in [44], small blood vessels were found in the grafted bioprinted equivalents beneath the epidermis. They are similar to the capillary network found in the papillary dermis of normal skin, which is critical for nourishing the avascular epidermis. These blood vessels seem to grow in from the depth of the wound bed into the dermal compartment. This is also observed in patients with grafted skin and it is attributed to endothelial growth factors, and other soluble factors, largely produced by keratinocytes and fibroblasts; for review see [62]. This finding represents additional evidence of the functionality of our skin regenerated from human bioprinted equivalents.
We consider that the two approaches discussed above [41–44], are clearly relevant as they describe multi-layered deposition techniques for skin bioprinting. Together with our work, they demonstrate that different bioprinters and bioprinting technologies can be potentially used to produce human skin. Clearly, the design of appropriate bioinks is a critical step for the production of printed skin with structure and functionality increasingly similar to normal human skin. Based on our previous experience of treating patients with skin equivalents, we have developed a simple, flexible and robust method to produce human skin which is useful in the clinic (e.g. for treatment of skin wounds) and in industry (e.g. for drug screening). Our approach allows the deposition of 100 cm2 of human skin in less than 35 min, which is much faster than approaches reported in the literature [41–44].
In addition, other bioprinting approaches use collagen as a 3D matrix, a biomaterial commonly used in the production of dermo-epidermal equivalents that, in our opinion, presents two main disadvantages when compared to the human plasma-based dermal scaffolds used in this work. Firstly, human plasma, unlike animal renatured collagen, provides a more suitable 3D scaffold to promote migration, proliferation and differentiation of the cells in the wound bed [15, 17, 63]. Secondly, human plasma-based scaffolds allow efficient production of collagen by hFBs and the concomitant remodelling of the scaffold to generate a dermal extracellular matrix similar to that found in normal human skin [15, 17].
5. Conclusions
We have developed a simple and robust bioprinting method and bioinks that allow the production of human bilayer skin, using human plasma and primary hFBs and hKCs. Based on careful histological and immunohistochemical in vitro and in vivo analysis, we demonstrated that the printed skin was very similar to normal human skin and indistinguishable from bilayered dermo-epidermal equivalents, previously produced manually in our laboratory and successfully used in the clinic. This method allows the production of human skin in amounts and times appropriate for its clinical and commercial use. The method also opens up the possibility of producing skin equivalents in an automatized and standardized manner, which should lead to a reduction in the cost of the product and an improvement in the production line, thereby overcoming some of the problems presented by the current manual production method.
Acknowledgments
We kindly thank Angélica del Corral, Guillermo Vizcaíno, Miguel Iñigo from UC3M and Blanca Duarte, Almudena Holguin, Nuria Illera and Luisa Retamosa from CIEMAT and IIS-FJD for their excellent technical assistance; and Jesus Martinez (CIEMAT) for animal care. We are also indebted to Dr Alvaro Meana, Eva Garcia (from CCST), Dr Fernando Larcher (from CIEMAT and IIS-FJD) and Dr Marcela Del Rio (from UC3M and IIS-FJD) for helpful discussions and providing materials required for this work. We also thank Lucia Gullon and Alfredo Brisac from BioDan Group for collaboration and support in this project, in particular in the development of the bioprinter. This work was partially supported by grants DPI2014-61887-EXP and DPI2015-68088-P from the Spanish Ministerio de Economía y Competitividad. The authors declare no conflict of interest.