Abstract
The two-dimensional (2D) material-based thermal switch is attracting attention due to its novel applications, such as energy conversion and thermal management, in nanoscale devices. In this paper, we observed that the reversible 2H–1T' phase transition in MoTe2 is associated with about a fourfold/tenfold change in thermal conductivity along the X/Y direction by using first-principles calculations. This phenomenon can be profoundly understood by comparing the Mo–Te bonding strength between the two phases. The 2H-MoTe2 has one stronger bonding type, while 1T'-MoTe2 has three weaker types of bonds, suggesting bonding inhomogeneity in 1T'-MoTe2. Meanwhile, the bonding inhomogeneity can induce more scattering of vibration modes. The weaker bonding indicates a softer structure, resulting in lower phonon group velocity, a shorter phonon relaxation lifetime and larger Grüneisen constants. The impact caused by the 2H to 1T' phase transition in MoTe2 hinders the propagation of phonons, thereby reducing thermal conductivity. Our study describes the possibility for the provision of the MoTe2-based controllable and reversible thermal switch device.
Export citation and abstract BibTeX RIS
1. Introduction
Two-dimensional (2D) transition-metal dichalcogenides (TMDs) have attracted research attention due to their outstanding mechanical, electrical, magnetic and phonon properties, which makes them promising for use in applications in transistors, sensors and energy conversion devices.[1–3] In recent years, the controllable phase transition of TMDs has intrigued researchers,[4,5] which can undoubtedly promote the development of these high-performance devices.[6,7] The phase-transition-induced changes in the structural, electronic and catalytic properties of TMDs were widely explored.[8,9] The 2H-TMDs can be used in energy storage devices as well as photodetectors, since they are semiconductors with high mobility.[10] In contrast, the 1T-phase exhibits an enhanced hydrogen evolution reaction performance due to its metallic characteristics.[11–13] In practice, the phase transition from the 1T to the 2H phase can be realized through thermal treatment and infrared radiation,[12,13] while the inverse transition is induced by strain and ion intercalation.[14]
Recently, a lithium intercalation method was reported to convert 2H-MoS2 to 1T-MoS2, resulting in a small thermal conductivity switching ratio (0.8).[15] The thermal conductivity switching ratio is defined as the ratio between the high (2H-MoS2) and the low (1T-MoS2) thermal conductivity values.[16] In addition to gate voltage and strain,[8,17] the 2H–1T–1T' phase transition of MoTe2 is able to be induced via laser radiation.[18] During this phase transition, the phonon plays a vital role and couples with the laser-activated electrons, showing different phonon behavior in 2H-MoTe2 and 1T'-MoTe2. Compared with the extensive studies on the effect of phase transitions on the electronic and optoelectronic properties of TMDs, the impacts on thermal conductivity are much less explored. Obviously, a better knowledge of the thermal conductivity of different phases can promote our understanding into the physical insights of monolayer TMDs and facilitate its applications over a wide range, including thermal management and thermoelectrics.[19]
Here, based on density functional theory and the Boltzmann transport equation, we studied the thermal conductivities of MoTe2 of the 2H and 1T' phases, and found that the thermal conductivity of the latter is much lower than that of the former. The underlying mechanism is explained by the weaker bonding, bonding inhomogeneity, more scattering of phonons and overall softening of vibrational modes in 1T'-MoTe2. Meanwhile, we demonstrate that 1T'-MoTe2 possesses lower phonon group velocity and stronger anharmonicity than 2H-MoTe2. These results suggest that monolayer MoTe2 can be used as high-performance thermal switches.
2. Simulation methods
The Vienna ab initio simulation package (VASP)[20] was used in our calculations. The generalized gradient approximation within the Perdew–Burke–Ernzerhof function is the exchange–correlation function.[21] The convergence criteria for the electronic (ionic) relaxations are 10−7 eV (10−4 eV/Å).[22] The cutoff energy for the plane wave is 600 eV, the K-mesh is 25 × 25 × 1 and the vacuum length is 30 Å.[23]
We obtained the harmonic interatomic force constants (IFCs) using Phononpy[24] and the anharmonic IFCs using thirdorder.py.[25] A 4 × 4 × 1 supercell was adopted in the calculations of the harmonic and anharmonic IFCs. Then, the convergence of thermal conductivity was tested from the 4th to 11th-nearest neighboring atoms, as shown in Fig. S1. The results are converged when the cutoff radius reaches 7th-nearest-neighboring atoms. To determine the scale-broad setting and the Q-grid in BTE, their convergences were tested. We found that the results are converged when the scale-broad setting is above 1.00 and the N1 × N1 × 1 is above 60 × 60 × 1, as exhibited in Fig. S2. Thus, based on both IFCs, the thermal conductivity was computed using the 7th-nearest neighbors and 60 × 60 × 1 Q-grid and 1.0 scale-broad setting, as implemented in ShengBTE.[25] The thickness of the monolayer (H) is the height of the bulk cell in the Z direction divided by the layer number,[26] which are 14.63 Å and 13.59 Å for 2H-MoTe2 and 1T'-MoTe2, respectively, as listed in Table 1.
Table 1. The calculated lattice parameters (a and b), Mo–Te bonding lengths (LMo-Te) and monolayer thickness (H) of 2H and 1T' phases, and the corresponding lattice parameters (a and b) in the previous experimental report.[6]
| a (Å) | b (Å) | LMo-Te (Å) | H (Å) | a (Å) and b (Å) in the experiment[6] | |
|---|---|---|---|---|---|
| 2H | 3.54 | 3.54 | 2.73 | 14.63 | a = b = 3.551 |
| 1T' | 3.45 | 6.37 | 2.75, 2.79, 2.83 | 13.59 | a = 3.452, b = 6.368 |
The relationship of the thermal conductivity (κL), specific heat (C), group velocities (v) and relaxation time of the phonon (τ) in ShengBTE is as follows:
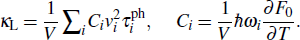
Here, V is the volume of the structure, Ci
is the specific heat for the phonon mode (i), vi
stands for phonon group velocity of mode i,  stands for the relaxation time of mode i, ℏ is the reduced Planck constant, ωi
is the frequency of mode i, F0 is the Bose–Einstein statistics, and T is the temperature.
stands for the relaxation time of mode i, ℏ is the reduced Planck constant, ωi
is the frequency of mode i, F0 is the Bose–Einstein statistics, and T is the temperature.
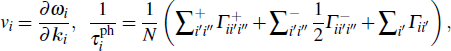
where
k
is the wave vector. Here,  (
( ) are the three-phonon absorption (emission) scattering rates.
) are the three-phonon absorption (emission) scattering rates.
3. Results and discussion
3.1. Structures of 2H-MoTe2 and 1T'-MoTe2
The structures of 2H-MoTe2 and 1T'-MoTe2 exhibit ABA and ABC stackings, respectively. The Mo atom locates in a hexagonal and distorted octahedral coordinate in 2H-MoTe2 and 1T'-MoTe2, respectively, as shown in Figs. 1(a) and 1(b). Interestingly, recent research reported that the two phases could be converted into each other within a short time (∼ ps) via laser radiation.[18] The lattice constants are a1 = b1 = 3.54 Å for 2H-MoTe2, and a2 = 3.45 Å, b2 = 6.37 Å for 1T'-MoTe2, agreeing with previous experimental results,[8,17,27] as listed in Table 1. Then, the bond length was measured in the optimized unit cell. The 2H-MoTe2 has a Mo–Te bonding length of 2.73 Å, while 1T'-MoTe2 shows three Mo–Te bonding lengths of 2.75 Å, 2.79 Å, and 2.84 Å.

Fig. 1. Top and side views of monolayer (a) 2H-MoTe2 and (b) 1T'-MoTe2. The unit cells are marked by the red dotted line.
Download figure:
Standard imageTo distinguish the differences in Mo–Te bonding between the two phases, we analyzed the overlap between Mo and Te atomic orbitals. It is known that the larger the atomic orbital overlap, the stronger the bonding.[28] The overlap can be quantified by the covalency (Ccovalency) between the atomic orbitals based on the electronic density of states (DOS).[29] As shown in Figs. 2(a) and 2(b), the total electronic DOS of MoTe2 is dominated by Mo-4d and Te-5p orbitals, suggesting that these two orbitals form the Mo–Te bonding. The covalency between Mo-4d and Te-5p is calculated using the following expression:[30]
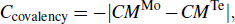
in which CMMo is the band center of Mo-4d and CMTe is the band center of Te-5p
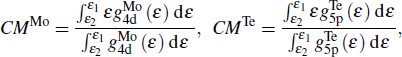
where  and
and  are the orbital projected DOS from Mo-4d and Te-5p atomic orbits, respectively.
are the orbital projected DOS from Mo-4d and Te-5p atomic orbits, respectively.

Fig. 2. The total and projected electronic density of states (DOS) of (a) 2H and (b) 1T' phases, and (c) their electron localization functions (ELFs). The color indicates the relative amplitude of the ELF. The bonding types of 1T'-MoTe2 are identified using Arabic numerals.
Download figure:
Standard imageThe covalencies of the 2H and 1T' phases are –2.15 and –3.36, respectively, indicating that the Mo–Te bonding in 2H-MoTe2 is stiffer than that in 1T'-MoTe2. This result is further verified by the electron localization function (ELF) as shown in Fig. 2(c). The 2H-MoTe2 has stronger bonding, while 1T'-MoTe2 shows three weaker types of bonds (bonding inhomogeneity). Consequently, the weaker bonding and bonding inhomogeneity in 1T'-MoTe2 results in lower thermal conductivity, as shown in the following section.
3.2. Thermal conductivity of 2H-MoTe2 and 1T'-MoTe2
The thermal conductivity is calculated based on rectangular cells, which are plotted in the gray wireframe in Fig. 1. The 1T'-MoTe2 displays more significant anisotropy in thermal conductivity than 2H-MoTe2 (see Fig. 3(a)). Impressively, 2H-MoTe2 has a larger thermal conductivity along the Y direction than that along the X direction, whereas 1T'-MoTe2 exhibits higher thermal conductivity along the X direction than that along the Y direction. As shown in Fig. 3(b), the thermal conductivities of the 2H (1T') phase along the X and Y directions are 43.26 (11.98) W/(mK) and 45.84 (4.95) W/(mK) at 300 K, respectively. For the 2H phase, these calculated values are consistent with previous reports.[31] The thermal conductivity switching ratios along the X and Y directions are 3.61 and 9.26 in MoTe2, respectively. Such switching ratios are comparable to, or even higher than, those of the previously reported thermal modulation ratios of MoS2 (0.8),[15] graphene (2.7),[32] 2D ferromagnets (1.31–5.73),[33] and polymers (8–10),[34] etc.[35,36] Recent research reported that the two phases could be converted into each other within a short time (∼ ps) via laser radiation,[18] gate voltage,[8] and strain.[17] Moreover, the thermal conductivity of 2D materials was measured in many ways, such as via the time-domain heat reflectance method and the laser flash method.[37] Monolayer MoTe2 therefore can be used for high-performance thermal switching devices in practice.

Fig. 3. (a) The thermal conductivities versus the temperature and (b) the thermal conductivities of 2H-MoTe2 and 1T'-MoTe2 at 300 K. The yellow percentages indicate the contributions of out-of-plane (ZA), transverse (TA) and longitudinal (LA) acoustic and optical phonons to the total thermal conductivity.
Download figure:
Standard imageMeanwhile, the contributions of acoustic and optical phonon branches to thermal conductivity are presented in Fig. 3(b). More than 75% of the contribution to thermal conductivity originates from acoustic branches. More convincing evidence can be seen from the cumulative thermal conductivity with frequency, in which phonons at frequencies below 3 THz, i.e., acoustic phonons, mainly contribute to the accumulation of thermal conductivity (see Fig. S3). To further explore the origin of different thermal conductivity between the 2H and 1T' phases, we next explore the effect of phase transition on the harmonic and anharmonic properties of phonons.
3.3. Physical insights into the tunable thermal conductivity
Figures 4(a) and 4(b) display phonon dispersions of the 2H and 1T' phases, respectively. The unit cell of the 2H (1T') phase has 3 (6) atoms and, accordingly, 9 (18) phonon branches. There are no imaginary frequencies, suggesting dynamical stability. Compared with 2H-MoTe2, 1T'-MoTe2 has a negligible phononic gap between acoustic and optical branches, which enhances the phonon scattering process. Based on the phonon eigenvectors, the phonon vibrational modes of the 2H and 1T' phases at the G-point are illustrated in Figs. S4(a) and S4(b), respectively. The phonon vibrational modes of 1T'-MoTe2 exhibit more directions and weaker strength than those of 2H-MoTe2, showing that the 2H to 1T' phase transition induces more scattering and overall softening of vibration modes. Consequently, 1T'-MoTe2 has reduced phonon propagation speed and lower thermal conductivity compared with 2H-MoTe2.

Fig. 4. Phonon dispersions of the (a) 2H-MoTe2 and (b) 1T'-MoTe2 unit cells, and (c), (d) the corresponding Brillouin zone. Phonon group velocity of the (e) 2H and (f) 1T' phases. LA: in-plane longitudinal acoustic phonons, TA: transverse acoustic phonons, and ZA: out-of-plane flexural phonons.
Download figure:
Standard imageFigures 4(e) and 4(f) shows that compared with 1T'-MoTe2, 2H-MoTe2 has higher group velocities of ZA and LA phonons, and lower group velocities of TA phonons. The optical phonon group velocity of 1T'-MoTe2 is larger than that of 2H-MoTe2. Overall, the acoustic phonons of 2H-MoTe2 bear larger group velocities compared with those of 1T'-MoTe2. Therefore, the reduced group velocity with the 2H to 1T' phase transition is responsible for the lower thermal conductivity in MoTe2.
Phonon relaxation time is an important factor which governs thermal conductivity.[5,38] Figure 5(a) shows that 1T'-MoTe2 shares smaller phonon relaxation time than 2H-MoTe2 over the whole frequency range, which implies that the phonon scattering rate can be reduced by the phase transition from the 2H to the 1T' phase. This result agrees with our aforementioned analysis of the phonon dispersion. Phonon anharmonicity is important to phonon propagation,[39,40] which is evaluated using the Grüneisen parameter (γ) in this study. The γ is calculated using  .[41] As presented in Fig. 5(b), the values of γ for ZA phonons are negative, and those for TA, LA and optical phonons are positive. The main difference in γ between the two phases is the γ of ZA phonons, and the 1T' phase has a larger γ of ZA than the 2H phase. Therefore, there is a stronger anharmonicity in 1T'-MoTe2, further confirming the lower thermal conductivity.
.[41] As presented in Fig. 5(b), the values of γ for ZA phonons are negative, and those for TA, LA and optical phonons are positive. The main difference in γ between the two phases is the γ of ZA phonons, and the 1T' phase has a larger γ of ZA than the 2H phase. Therefore, there is a stronger anharmonicity in 1T'-MoTe2, further confirming the lower thermal conductivity.

Fig. 5. (a) The phonon relaxation lifetime and (b) the Grüneisen constants of 2H-MoTe2 and 1T'-MoTe2.
Download figure:
Standard imageFinally, we found that the anisotropy in thermal conductivity is attributed to unevenly distributed Mo and Te atoms in the geometric configurations of the rectangular cell. To clearly describe the difference in anisotropic behavior between 2H-MoTe2 and 1T'-MoTe2, we calculated the Grüneisen parameter γq,s
for the s th mode using  along the G–Y (X) and G–X (Y) directions (a is the lattice parameter). As shown in Fig. S5, the absolute values of γq,s
for the ZA, TA, and LA phonon modes of 1T'-MoTe2 along the Y direction are larger than that of the X direction, as that of 2H-MoTe2 along the X direction is slightly greater than that of the Y direction, which supports our predicted thermal conductivity anisotropy.
along the G–Y (X) and G–X (Y) directions (a is the lattice parameter). As shown in Fig. S5, the absolute values of γq,s
for the ZA, TA, and LA phonon modes of 1T'-MoTe2 along the Y direction are larger than that of the X direction, as that of 2H-MoTe2 along the X direction is slightly greater than that of the Y direction, which supports our predicted thermal conductivity anisotropy.
3.4. Mechanical properties of 2H-MoTe2 and 1T'-MoTe2
The phonon vibrational modes of a material are closely related to its mechanical properties, and the strength of a mechanical property is measured by Young's modulus.[43] For example, compared with Y2Zr2O7, Y3NbO7 has a lower Young's modulus, which indicates a softer structure and softening of the vibration modes.[44] Therefore, to provide more insights into the deferent phonon vibrational modes between the two phases, we calculated the Young's modulus based on the elastic modulus tensor.
The stiffness tensor (Cij , i,j = 1, 2, 6) is the second partial derivative of the strain energy (Es) as a function of strain (ε):[45,46]
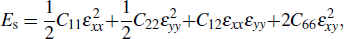
with ε = (a – a0)/a0, where a (a0) is the strained (unstrained) lattice constant. As listed in Table 2, the elastic constants of 2H-MoTe2 are C11 = 83.99 N/m, C22 = 84.02 N/m, C12 = 21.43 N/m, and C66 = 31.15 N/m, and those of 1T'-MoTe2 are C11 = 61.57 N/m, C22 = 87.77 N/m, C12 = 39.66 N/m, and C66 = 31.42 N/m, satisfying the Born criteria  and C66 > 0.[47,48] The structures of both phases therefore display mechanical stability.
and C66 > 0.[47,48] The structures of both phases therefore display mechanical stability.
Table 2. Mechanical properties of 2H-MoTe2, 1T'-MoTe2 and graphene.[42] Cij (in N/m): elastic constants, Y (in N/m): Young's modulus, and v: Poisson's ratio.
| C11 | C22 | C12 | C66 | Yx | Yy | vx | vy | |
|---|---|---|---|---|---|---|---|---|
| 2H-MoTe2 | 83.99 | 84.02 | 21.43 | 31.15 | 88.52 | 88.56 | 0.26 | 0.26 |
| 1T'-MoTe2 | 61.57 | 87.77 | 39.66 | 31.42 | 62.22 | 43.65 | 0.45 | 0.64 |
| Graphene | 364.6 | 364.6 | 67.3 | 148.7 | 352.2 | 352.2 | 0.19 | 0.19 |
Then, the Young's modulus (E) and orientation-dependent Young's modulus (Y) are derived as[49]
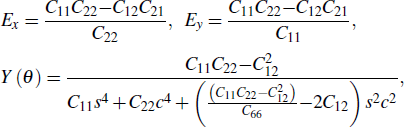
where s = sin θ and c = cos θ. The Young's modulus value of 2H-MoTe2 in the X (Y) direction is 88.52 N/m (88.56 N/m), higher than that of 1T'-MoTe2 (62.22 N/m for the X direction and 43.65 N/m for the Y direction). We find that the stiffness of 1T'-MoTe2 is smaller, confirming the softening of the vibration modes. As shown in Fig. 6, 1T'-MoTe2 exhibits an obvious anisotropic Young's modulus, in which the value in the X direction is much larger than that in the Y direction. On the other hand, 2H-MoTe2 shows little anisotropic Young's modulus, where the value of the Young's modulus in the Y direction is slightly smaller than the value in the X direction. The anisotropy in the Young's modulus complies with the anisotropy of thermal conductivity calculated above.

Fig. 6. The orientational-dependent Young's modulus and Poisson's ratio of 2H-MoTe2 and 1T'-MoTe2.
Download figure:
Standard imageAccording to the stiffness tensor, the Poisson's ratio (v) and orientation-dependent Poisson's ratio are computed as follows:[49,50]
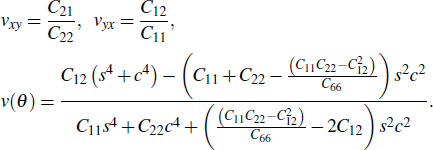
Both the Poisson's ratios of 2H-MoTe2 along the X and Y directions are 0.26, less than those of 1T'-MoTe2 (0.45/0.64 for the X/Y direction). These values are greater than that of graphene (0.18), implying that 2H-MoTe2 and 1T'-MoTe2 are more flexible than graphene, and ensuring that the two phases can be transformed into each other. It is obvious from Fig. 6 that the values of v vary together with the change of direction, which also demonstrates the anisotropic properties.
4. Conclusion
The thermal conductivities' switching ratio between the 2H and 1T' phases of MoTe2 reaches 9.26. Such large thermal switching originates from the softening of the structure, smaller group velocity and stronger anharmonicity of the monolayer MoTe2 structure after 2H–1T' phase transition. Our study qualifies monolayer MoTe2 as an active and reversible thermal switching material with the characteristics of large switching ratios and fast modulation.
Acknowledgments
We gratefully acknowledge the use of the computing resources at the Agency for Science, Technology and Research (A*STAR) and NSCC, Singapore. Project supported by the China Scholarship Council (Grant No. 202107000030), RIE2020 Advanced Manufacturing and Engineering (AME) Programmatic (Grant No. A1898b0043), and A*STAR Aerospace Programme (Grant No. M2115a0092).