
Abstract
For decades, protein folding and functional dynamics have been described in terms of diffusive motion across an underlying energy landscape. With continued advances in structural biology and high-performance computing, the field is positioned to extend these approaches to large biomolecular assemblies. Through the application of energy landscape techniques to the ribosome, one may work towards establishing a comprehensive description of the dynamics, which will bridge theoretical concepts and experimental observations. In this perspective, we discuss a few of the challenges that will need to be addressed as we extend the application of landscape principles to the ribosome.
Export citation and abstract BibTeX RIS
1. Introduction
The energy landscape perspective of biomolecular dynamics provides a quantitative framework with which to consistently integrate theoretical concepts and experimental observations. Recognizing the potential of this approach, there has been decades of investigation into the character of protein folding landscapes. The many insights enabled by the landscape perspective have led to a growing interest to understand the energetics of large biomolecular assemblies. Here, we provide a brief overview of issues that must be addressed in order to extend energy landscape techniques to the ribosome (figure 1(A)).
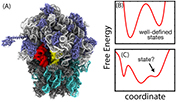
Figure 1. Energetics of biomolecular assemblies. (A) The ribosome is a large molecular assembly that interconverts between many conformational states. Traditionally, ribosome dynamics has been described in terms of discrete states, which is appropriate when the one-dimensional free-energy function has well-defined, deep minima (panel (B)). However, since shallow/broad minima can also be present (panel (C)), the proper definition of a state is not always clear. For these cases, it is sometimes possible to describe the kinetics in terms of one-dimensional motion along a free-energy surface. In order to implement this approach for the ribosome, a major challenge will be to identify the kinetically relevant coordinates for each conformational process.
Download figure:
Standard image High-resolution imageThere are many experimental and theoretical approaches for probing the energy landscapes of biopolymers. From the theoretical side, the objective is to construct appropriate potential energy functions, in order to explore the thermodynamic and kinetic behavior of the system. This may be accomplished by numerically evaluating the partition function [1], or performing simulations to obtain a Boltzmann-distributed sampling of the phase space. Regardless of the technical details, the principal scientific challenge is to select a suitable model, which may posses any level of energetic and structural resolution. From an experimental perspective, single-molecule experiments can provide one-dimensional projections of the dynamics, thereby enabling the direct measurement of free-energy barriers. However, it is often unknown which coordinates/probes are most suitable for describing the barrier of interest. While an appropriately chosen coordinate will allow for characterization of the dominant features of the free energy [2, 3], the use of sub-optimal coordinates can lead to artifacts.
In recent years, there has been an interest in understanding the factors that shape the energy landscape of the ribosome (figure 1(A)). It has been generally thought that by moving away from state-like descriptions, and adopting the energy landscape framework, a more comprehensive understanding of the ribosome may be obtained. Here, we present our view of the pressing challenges facing the scientific community as it attempts to probe the ribosome's energy landscape. In particular, we will describe examples of how concepts previously applied to protein folding can guide the study of free-energy barriers and kinetics associated with ribosome function.
2. Probing folding landscapes
One aim of landscape investigations has been to identify the overall characteristics of protein energetics. In the works of Bryngelson and Wolynes [4], it was argued that in order for proteins to fold on physiological timescales, the energy landscape associated with folding should possess only small traps, relative to the energetic gap between the folded and unfolded ensembles. That is, these landscapes should be minimally frustrated. This suggested that protein folding energetics may be approximated by models that are completely unfrustrated (i.e. structure-based, or Gō-like, models. For review, see [5]). In these models, only interactions found in the native configuration are stabilizing. This approximation to the energetics has provided a basis for exploring many factors that can impact folding dynamics. As a few examples, within the context of a smooth landscape, it has been possible to explore the influence of chain length [6], non-native interactions [7] and native-fold complexity [8] on folding rates and mechanisms. While these are notable successes, when studying more detailed mechanistic properties (e.g. the timescale of hydrogen-bond formation within the native ensemble), one will typically employ explicit-solvent models, which are frequently analyzed using kinetics-based techniques [9, 10].
The energy landscape framework provides strategies for the systematic analysis of free-energy barriers. While folding occurs in a high-dimensional space, it is often possible to accurately describe the dynamics in terms of a few key variables. For example, the lowest free-energy path may sometimes be described by one-dimensional projections of the dynamics [11, 12]. However, in order to rigorously compare experiments and simulations, one must identify which coordinates are most relevant. An essential property of an appropriate coordinate is that it be correlated with the committor probability, thereby allowing it to identify the transition state. For this, the end-to-end distance [2, 3] and the number of native contacts formed [13, 14] have been shown to satisfy these criteria for the folding of small proteins. Building on such insights, single-molecule methods are now able to precisely probe the barriers associated with biomolecular folding [15].
3. Energy landscapes of the ribosome
The ribosome is a massive assembly composed of over 50 polymer chains (figure 1(A)), and its function is associated with energy-releasing chemical steps (GTP hydrolysis), large-scale conformational rearrangements, as well as chemical synthesis (peptide bond formation). Ribosome function also involves protein-RNA interactions, biomolecular recognition events and order-disorder transitions. With this variety of physical attributes, there are many opportunities for energy landscape techniques to advance our quantitative understanding of the ribosome.
Ribosome dynamics has traditionally been described in biochemical terms, where decades of analysis has identified dominant states and rates associated with function [16, 17] (cf. figure 1(B)). Consistent with this foundation, it is standard for observations in single-molecule [18, 19], crystallographic [20] and cryo-EM [21] experiments to be interpreted through a state-centric lens. This view has proven to be very effective when describing long-lived intermediates and chemical reactions. However, since ribosome dynamics may be associated with disorder and shallow free-energy minima figure 1(C)), a more versatile conceptual and quantitative framework is needed.
There is currently a growing number of experimental and computational studies that are aiming to characterize the ribosome's energy landscape. From the experimental side, the most quantitative efforts have used cryo-EM projections to plot the probability as a function of a reaction coordinate [21, 22]. These represent exciting developments, though systematic application of landscape techniques demands a more complete theoretical treatment. For example, as is the case for folding, the analysis of landscapes requires knowledge of appropriate reaction coordinates and the system's diffusive properties. While a few studies have quantified diffusion in the ribosome [23, 24], there is a limited understanding of how to describe the numerous barriers encountered during function.
To illustrate some current challenges in understanding ribosome energetics, we will describe examples of how our group is applying landscape concepts to this large assembly. First, we describe how simplified models are implicating a critical role of molecular sterics (i.e. excluded volume) during functional dynamics. We then discuss the use of energy landscape techniques to identify appropriate metrics for describing conformational transitions in the ribosome. It is our goal to highlight the trajectory of landscape applications to the ribosome, in order to demonstrate how next-generation experimental and theoretical techniques may provide deeper insights into the intricate dynamics of this massive assembly.
3.1. The influence of sterics on dynamics
Simplified energetic models, such as structure-based models (reviewed in [5]), may be employed to identify how molecular structure contributes to the free-energy barriers associated with functional dynamics. However, since the principle of minimal frustration does not necessarily hold for the ribosome, it is important to consider the rationale for applying these models to study this system. In the case of aa-tRNA accommodation (figure 2(A)), the endpoint configuration is stable and the initial configuration is energetically disfavored. Based on this, it was suggested that aa-tRNA may behave as a loaded 'molecular spring' that relaxes during the accommodation process [25]. This notion may be effectively described by a structure-based model, where the endpoint is explicitly defined as the global potential energy minimum. While this is a simplified description of the energetics, the application of this type of model has predicted multiple long-lived intermediates during accommodation [26]. Since the model is devoid of energetic roughness, the presence of intermediates indicates that detailed structural features give rise to complex multi-step dynamics.

Figure 2. Theoretical models can reveal the contribution of structure to the free-energy landscape. (A) During accommodation, the aa-tRNA molecule (yellow) makes close interactions with Helix 89 (H89). (B) The free-energy barrier encountered during accommodation is nearly absent when H89-tRNA steric interactions are not included [27]. (C) During mRNA-tRNA translocation, tRNA molecules (red and yellow) must pass through the dense steric environment imposed by the ribosome. (D) Through simulations, 'virtual mutations' may be employed to probe how structural features contribute to the dynamics. As an example, this type of analysis has shown that tilting of the ribosomal 30S head (described by θ in panel (C)) is attenuated when specific steric interactions are removed [28]. Together, simulations of accommodation and translocation illustrate the critical influence of molecular structure on the free-energy of the ribosome. The directions of tRNA displacements are indicated by arrows.
Download figure:
Standard image High-resolution imageTo characterize the contribution of structure to the free-energy landscape of the ribosome, one may employ models in which specific structural perturbations are introduced. For example, we recently calculated the free-energy barrier associated with aa-tRNA accommodation [27], and showed that it is nearly eliminated upon removal of Helix 89 (H89, figure 2(B)). It was further demonstrated that EF-Tu can reduce this barrier by confining the mobility of the tRNA molecule. Interestingly, this mode of function is analogous to the influence of molecular crowders on protein dynamics, where the folded ensemble can be stabilized through a reduction in configurational entropy of the unfolded ensemble [29]. While the observed change in barrier height demonstrates a physical relationship between molecular structure and dynamics, the lack of energetic roughness and explicit water molecules can lead to an overestimate of the rates in these models [6]. One way that we have aimed to circumvent this limitation is to use diffusion coefficients obtained from explicit-solvent models [23, 24], and then calculate rates directly from the free-energy profiles predicted by simplified models [27].
In the context of mRNA-tRNA translocation (figure 2(C)), simulations with a structure-based model suggested that steric interactions between the so-called PE loop and tRNA molecule can lead to tilting motion of the ribosomal 30S-head domain [28]. By repeating these calculations after removing the PE loop, it was shown that the dominant path of interconversion, as well as the degree of tilting (figure 2(D)), are dependent on these purely repulsive interactions. Interestingly, a subsequent independent single-molecule study [30] provided qualitative support for the predicted sterically-induced tilting motion. The similarity between this experimental observation and the results obtained with a 'sterics-only' model suggests that molecular shape is largely responsible for determining the dominant pathways.
3.2. Quantitatively describing free-energy barriers
The power of the energy landscape perspective can only be realized after the kinetically-relevant degrees of freedom are identified. As with protein folding, to describe a given conformational transition, it is necessary to find reaction coordinates that most closely follow the lowest free-energy pathway. The size of the ribosome makes systematic identification of coordinates extremely challenging. This is exacerbated by the large number of functional substeps in the ribosome and the presence of transient order-disorder transitions [5].
In our group's initial efforts to identify appropriate reaction coordinates for the ribosome, we have performed long simulations of tRNA accommodation [31] and hybrid-state formation [32] using simplified models. For each rearrangement, over 100 spontaneous barrier-crossing events were observed. We then projected the dynamics along different interatomic distances, each representing possible labeling sites in single-molecule experiments. In total, 169 and 1369 interatomic distances were considered for accommodation and hybrid-state formation. This analysis revealed that the distance probed in many single-molecule experiments (R8,47) will underestimate the free-energy barriers (figure 3). However, alternative coordinates were found to accurately describe both processes. Accordingly, these results suggest how future single-molecule measurements may be used to quantify the free-energy barriers associated with ribosome function.

{kind=link}
{kind=link}
Figure 3. The view depends on the perspective. (A) aa-tRNA accommodation on the ribosome may be monitored by interatomic distances. (B) The measured free-energy (or, potential of mean force, PMF) barrier height can vary with the choice of coordinate [31]. These calculations show how experimental observations may depend on which coordinate is employed.
Download figure:
Standard image High-resolution image{kind=link}
4. Outlook
With continued technological advances, it is now time for a rigorous physical-chemical understanding of ribosome dynamics to be established. Using the theoretical infrastructure developed in the context of protein folding, the field is positioned to extend the application of energy landscape techniques to the study of this large assembly. Through close integration of computation and experimentation, it will be possible to dissect the complex interplay of structure and energetics during ribosome function. We anticipate that such endeavors may ultimately reveal physical principles that govern biological dynamics.
Acknowledgments
This work was supported by a National Science Foundation CAREER Award (MCB-1350312).