




Abstract
We review the recent progress in the field of helically assembled π-conjugated polymers, focusing on aromatic conjugated polymers with interchain helical π-stacking that exhibit circularly polarized luminescence (CPL). In Part 1, we discuss optically active polymers with white-colored CPL and the amplification of the circular polarization through liquid crystallinity. In Part 2, we focus on the stimuli-responsive CPL that results from changes in the conformation and aggregation state of π-conjugated molecules and polymers. In Part 3, we discuss the self-assembly of achiral cationic π-conjugated polymers into circularly polarized luminescent supramolecular nanostructures with the aid of other chiral molecules.
Export citation and abstract BibTeX RIS
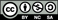
Content from this work may be used under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Because their π-electrons are delocalized over the main chain, conjugated polymers (CPs) possess profound semiconducting properties, such as electrical conductivity, luminescence, and photo-harvesting behavior, and are therefore regarded as candidate materials for organic optoelectronic devices [1–10]. The electronic and optoelectronic properties of CPs can be controlled by modulating the conformation, arrangement, and alignment of the main chains for which the self-organization [11–14], macroscopic alignment [15–24], intra- and inter-molecular interactions [25–32], and synthesis on templates [33–36] are intentionally incorporated. This means that both the primary structure and the higher-ordered structures of CPs are essential factors for achieving advanced optoelectronic properties and functions [9, 10, 37–39]. The formation of CPs via self-assembly is one of the most useful ways to construct hierarchical higher-ordered structures in CPs. The self-assembly forces are classified as π−π interactions, van der Waals interactions, solvophobic interactions, electrostatic interactions, and so on. In particular, the interchain π−π interaction is the most fundamental and indispensable force for constructing ordered structures of CPs. In fact, recent studies have revealed that π−π interactions efficiently yield assemblies with higher-ordered and even hierarchical structures in conjunction with acid−base, electrostatic, and hydrogen-bonding interactions [30, 40–44].
Furthermore, helical CPs have attracted considerable interest because they provide unique optical functions, such as circular polarization in absorption and luminescence [32, 45–47], and novel physicochemical properties [48]. In addition, helical CPs can also be used as chiral templates and chiral sensors [1]. Various types of helical CPs, represented by helical polyacetylenes [49], amino acid-containing substituted polyacetylene derivatives [50, 51], poly(phenylacetylene) derivatives [13, 31, 52–54], poly(meta-phenylene arylene) derivatives [43, 44, 55, 56], and polythiophene (PT) derivatives [40, 57], have been proposed. These helical CPs are constructed with intrachain-twisted, intrachain-spiral, or interchain-helical π-stacked structures [14, 58, 59]. Among these structures, the interchain-helical π-stacking interaction is intriguing because it enables aromatic CPs (ACPs) to self-assemble into an interchain helix, thereby generating amplified chiroptical properties such as circularly polarized luminescence (CPL). CPL is a fascinating property of ACPs that arises from their molecular chirality or helicity, which eliminates the requirement for macroscopic alignment of the polymers that is required for linearly polarized luminescence. The strength of the circular polarization is evaluated with the dissymmetry factor. The luminescence dissymmetry factor (glum) is defined as follows: glum = ΔI/I = (IL − IR)/I = 2(IL − IR)/(IL + IR), where IL and IR are the left- and right-handed CPL intensities, respectively.
In this review, we focus on the recent progress in interchain helically assembled ACPs and discuss their chiroptical properties with respect to CPL, namely (i) white-colored CPL and amplification of the helicity via liquid crystallinity, (ii) reversible switching of CPL by external stimuli, and (iii) supramolecular structures of ionic ACPs with CPL.
2. Recent studies of π-conjugated molecules and polymers with circularly polarized luminescence
2.1. Part 1: White-colored circularly polarized luminescence and amplification of helicity via liquid crystallinity
2.1.1. Optically active polymers with white-colored CPL
To date, numerous colored CPL varieties have been obtained by changing the structures of the conjugated backbones. Among these CPL varieties, white-colored CPL is especially noteworthy because the luminescence band extends over the visible region. Optically active polymers with a broad luminescence band and their CPL properties have been reported. One example is the vinyl polymer P1, which contains 2,7-bis(4-tert-butylphenyl)fluorene units in the side chains and a chiral menthol unit at the terminal of the main chain (scheme

Scheme 1. Optically active polymers with a broad luminescence band.
Download figure:
Standard image High-resolution image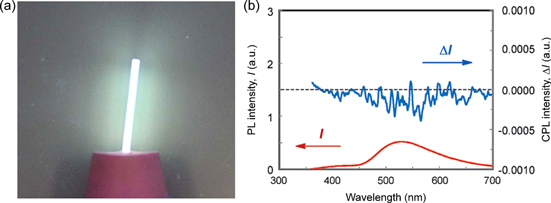
Figure 1. (a) A photograph of a quartz plate coated with P1 film and illuminated by UV light (λ = 365 nm). (b) Photoluminescence (PL) and circularly polarized luminescence (CPL) spectra of P1 film annealed at 120 °C for 6 h. Adapted with permission from ref. [60]. Copyright 2011 The Royal Society of Chemistry.
Download figure:
Standard image High-resolution image2.1.2. Amplification of helicity via liquid crystallinity
Furthermore, it is well known that liquid crystallinity is effective in reinforcing the supramolecular helicity and chirality of polymers. The substituted polyacetylene derivative P3 (scheme

Scheme 2. Lyotropic liquid crystalline poly(phenylacetylene) derivatives with hydrophilic (P3) or lipophilic side chains (P4).
Download figure:
Standard image High-resolution image
Figure 2. (a) Circular dichroism (CD) spectra of P3 with (R)- or (S)-NaPLA in water and UV−vis absorption spectrum of P3 with (R)-NaPLA in water. The concentration of P3 is 4.0 × 10−3 m. The mixing ratio is [NaPLA]/[P3] = 0.5. (b) Schematic illustration of chiral amplification in macromolecular helicity in dilute solution and liquid crystal (LC) state. P3 has interconvertible, right- (green chain) and left-handed (blue chain) helical conformations. In dilute solution, a small amount of chiral acid (NaPLA) induces excess of one-handed helical sense on the P3 helix. In lyotropic LC state, the induced chirality is significantly amplified, resulting in a one-handed helical supramolecular assembly. Adapted with permission from ref. [63]. Copyright 2006 American Chemical Society.
Download figure:
Standard image High-resolution image
Figure 3. (a) Polarized optical microscopy (POM) image of the lyotropic chiral nematic LC (N*-LC) phase of (S)-P4 in toluene (c = 10 wt%) showing a double-spiraled texture. Inset shows a fingerprint texture with a half helical pitch (p/2) of 1.5 μm. (b) Schematic representation of the N*-LC phase of (S)-P4. The polymers self-assemble into the interchain helical structure. The polymer interchain distance in an N*-LC domain is described in the top view. Adapted with permission from ref. [64]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution image2.1.3. White-colored CPL amplified through liquid crystallinity
It was recently reported that a mixture of thienylene- and phenylene-based ACPs with interchain helicity generates white-colored CPL [65]. Generally, the fluorescence color of ACPs depends on the structure of the repeating unit and on the conjugated length of the polymer. Thus, thienylene- and phenylene-based ACPs, P5−7, with chiral substituents were designed and synthesized to exhibit fluorescence colors of blue (B), green (G), and red (R), respectively (scheme
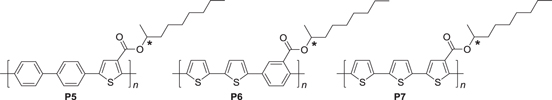
Scheme 3. Thienylene- and phenylene-based aromatic conjugated polymers (ACPs) with chiral nonyl substituents.
Download figure:
Standard image High-resolution image2.1.3.1. Liquid crystalline properties
Polymers P5−7 exhibited enantiotropic main-chain liquid crystallinity at high temperatures and across a wide range of elevated temperatures. Polarized optical microscopy (POM) revealed the presence of an enantiotropic LC phase in all of the polymers. Figure 4 shows the optical textures of the LC phases of (R)-P6 and (R)-P7. (R)-P6 exhibited an oily streak texture (figure 4(a)) and a fingerprint texture (figure 4(b)) at approximately 200 and 150 °C, respectively, during the cooling process; both of these textures can be assigned to the N* phase. Meanwhile, (R)-P7 (figure 4(c)) exhibited a focal conic texture with a unique red color at approximately 156 °C during the cooling process, which is also attributed to the N* phase. (S)-P6 and (S)-P7 exhibited optical textures similar to those of the corresponding (R)-type polymers. P5s showed no distinct optical textures attributable to the N* phase. P5s were found to possess high clearing points (greater than 300 °C), which may be due to the higher molecular weight of the polymers.

Figure 4. POM images of (R)-P6 at (a) 200 °C and (b) 150 °C and of (c) (R)-P7 at 156 °C during the cooling process. Reprinted with permission from ref. [65]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution image2.1.3.2. Circular dichroism
CD spectra of the P5s, P6s, and P7s in chloroform (c = 1.0 × 10−4 m) are shown in figures 5(a)–(c). Cotton effects are observed in the π−π* transition bands of the conjugated main chains. (R)-P5 exhibited a positive bisignate Cotton effect1 that appeared in the π−π* transition band of the conjugated main chain that consisted of biphenylene-thienylene moieties, first with positive and then negative components at longer (420 nm) and shorter (370 nm) wavelengths, respectively. At the same time, (S)-P5 exhibited a completely reversed CD behavior, which was first negative and then positive (figure 5(a)). This behavior implies that a strong exciton coupling occurs in the main chains, leading to the formation of an interchain π-stacked structure [66]. The P6s and P7s also exhibited bisignate Cotton effects, in which the signals for the (R)- and (S)-configurations were mirror images (figures 5(b) and (c)). The observation of the bisignate Cotton effect for the polymers with three aromatic rings in the repeating unit suggests that the polymers are stacked with twisting through π−π interactions, resulting in a helical π-stacked assembly. Figure 6 presents a schematic representation of the helical π-stacked structure of the polymer assembly.
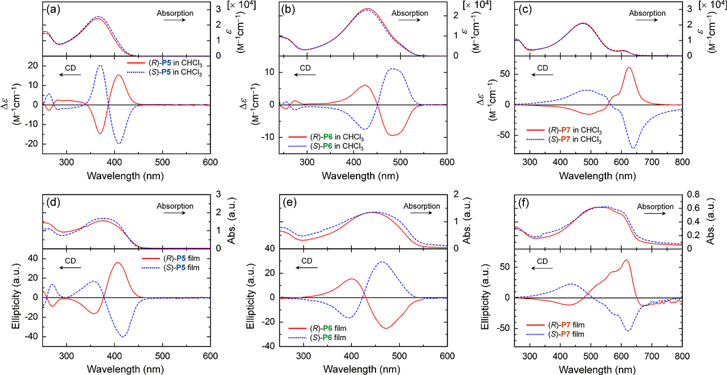
Figure 5. UV−vis absorption (upper) and CD (lower) spectra of polymers in chloroform (c = 1.0 × 10−4 m) for (a) P5s, (b) P6s, and (c) P7s and the as-cast films of (d) P5s, (e) P6s, and (f) P7s. The solid and dotted lines indicate the spectra of polymers with (R)- and (S)-configurations, respectively. Reprinted with permission from ref. [65]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution image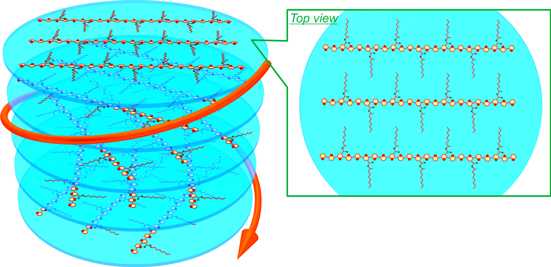
Figure 6. Schematic of the interchain helically π-stacked structure of P7 assembly. Reprinted with permission from ref. [65]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution imageThe Cotton effects in the CD spectra decreased in intensity with increasing temperature and finally disappeared at 70 °C for the P5s and P6s and at 80 °C for the P7s. Additionally, the absorption bands of the polymers shifted toward shorter wavelengths with increasing temperature. These results also imply that the polymer assembly with a helical π-stacked structure exists even in dilute solution at room temperature.
The (R)- and (S)-polymer thin films also exhibited mirror-opposite Cotton effects in the region of the π−π* transition (figures 5(d)−(f)). At the same time, the CD intensity of the film increased by approximately 2−10 times that of the solution. These results are attributed to strengthening of the helical π-stacking between the polymer main chains in solid states, such as films.
2.1.3.3. CPL property
Figure 7(a) presents the photoluminescence (PL) and CPL spectra of the P5s, P6s, and P7s in chloroform (1.0 × 10−4 m), and these polymers exhibit circular polarization in their fluorescence bands. The CPL signals for the polymers with (R)- and (S)-configurations appear in opposite directions, resulting in almost mirror images in solution and cast films; for example, the (R)-P6 exhibits a positive signal, whereas the (S)-P6 exhibits a negative signal.
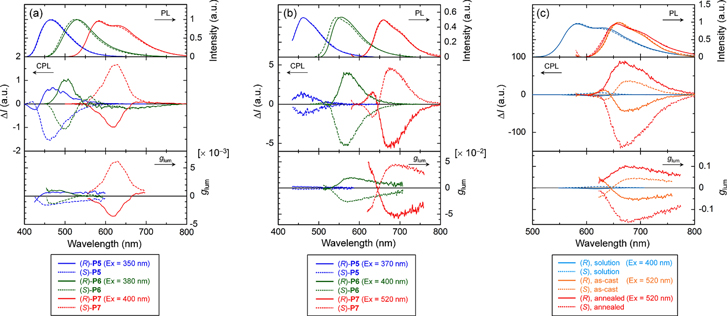
Figure 7. PL (upper), CPL (middle), and luminescence dissymmetrty factor glum (lower) spectra of the polymers in (a) chloroform (c = 1.0 × 10−4 m) and (b) as-cast films. (c) PL (upper), CPL (middle), and glum (lower) spectra of P7 in chloroform (c = 1.0 × 10−4 m), the as-cast films, and the annealed films. Reprinted with permission from ref. [65]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution imageFigure 7(b) presents the CPL spectra of the polymer films. The glum values of the polymer films for the P6s and P7s increased relative to the corresponding solution by approximately 1 order of magnitude, whereas the glum values for the P5s only increased by approximately 2−3 times relative to the corresponding solution. This increase may be attributed to the strong π−π interactions between polymer backbones in the solid state, as mentioned above. The P6s and P7s, which have a bithienylene moiety in the repeating unit, possessed larger glum values than the P5s, which have biphenylene moieties rather than bithienylene moieties. This difference may be explained by the difference in coplanarity between bithienylene and bithienylene. Incorporating bithiophene into the polymer backbone leads to a higher degree of coplanarity and stronger π−π interactions, which result in larger glum values relative to the P5s.
2.1.3.4. Amplification of helicity
The polymer films were annealed for 30 min at 150 °C, which falls in the LC temperature range, and were then subjected to CPL measurements. Figure 7(c) presents the CPL and glum spectra of the solutions, as-cast films, and annealed films for the P7s. After annealing the as-cast films, the absolute glum values increased by at least 1 order of magnitude and produced glum values as high as 10−1, which is similar to those of chiral polyfluorenes [67, 68]. These glum values are very high despite the regiorandom structure of the polymers. This value may be due to the helical π-stacking during annealing at the LC temperature. Interestingly, the CPL signals inverted after annealing the P7s; that is, the glum of the as-cast (S)-P7 film showed a positive signal of +4.1 × 10−2 at 680 nm that became a negative signal of −1.5 × 10−1 at 666 nm after annealing. Annealing in the LC phase encourages the self-assembly of the polymer backbone and the formation of macroscopic π−π stacks, which lead to high absolute glum values [67, 68]. Annealing may also lead to helical packing of the polymer backbones [69] in the N* phase, which may be responsible for the inversion of the glum signal for the P5s and P7s. However, the P6s did not exhibit an inversion of the glum signal. In the P6s, the chiral substituent is linked to phenylene, whereas it is linked to thiophene in the P5s and P7s, as indicated above. Although the structural changes in the polymer thin films caused by annealing are not yet understood, any differences in the chemical structure may be related to the inversion of the glum signals caused by annealing. The hierarchical arrangement of higher-ordered structures by chiral liquid crystallinity, such as the self-ordering of the polymer aggregates in the N* phase by annealing at the LC temperature, is a promising route for highly enhancing the dissymmetry factor up to 10−1 in the CPL of the chiral CPs.
2.1.3.5. White-colored CPL
Finally, the authors mixed solutions of the P5s, P6s, and P7s, which exhibit blue, green, and orange fluorescence, respectively. The actual ratio of P5, P6, and P7 employed in the mixture was 1:1:5. Figure 8(a) presents the CPL spectra of the polymer mixtures. System 1 is the mixture of ( R )-P5, ( R )-P6, and ( S )-P7, which displays a positive glum, and system 2 is the mixture of ( S )-P5, ( S )-P6, and ( R )-P7, which displays a negative glum. These mixtures presented mirrored CPL spectra from 400−700 nm and a glum value on the order of 10−3, indicating white CPL. Figure 8(b) presents a photograph of the polymer mixture in chloroform, which emits white light.
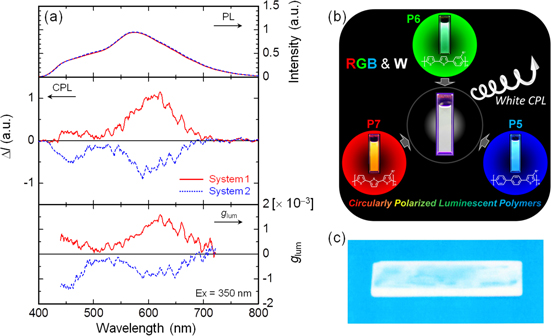
Figure 8. (a) PL (upper), CPL (middle), and glum (lower) spectra of the polymer mixtures in chloroform [P5, 5.0 × 10−5 m; P6, 5.0 × 10−5 m; P7, 2.5 × 10−4 m], where system 1 is the mixture of (R)-P5, (R)-P6, and (S)-P7, and system 2 is a mixture of (S)-P5, (S)-P6, and (R)-P7. (b) RGB and white fluorescence of system 1, the mixture of (R)-P5, (R)-P6, and (S)-P7, in chloroform and (c) white fluorescence of the cast film of system 1 on a quartz substrate. The film was prepared by dissolving the mixture and an excess of polystyrene in chloroform followed by casting onto the quartz substrate. UV light at 365 nm was used for excitation. Reprinted with permission from ref. [65]. Copyright 2012 American Chemical Society.
Download figure:
Standard image High-resolution imageHowever, the cast films prepared using the above mixtures exhibited red, not white, fluorescence due to polymer aggregation, which allows either an energy transfer from the excited states of P5 and P6 to that of P7 or the reabsorption of the fluorescence of P5 and P6 by P7. To suppress both energy transfer and reabsorption, the interchain distances in the polymers need to be increased. Therefore, the polymer mixture in system 1 was dispersed in an excess of polystyrene by dissolving both solutions in chloroform and then casting them onto a quartz substrate. This cast film sufficiently separated the polymers from each other in terms of their interchain distances to create a so-called 'pseudodilute solution' state.
Figure 8(c) depicts the white fluorescence of the film on a quartz substrate. The white luminescence is circularly polarized emission. The glum values increased by a factor of 1.5−5.8 compared with the corresponding values in solution but remained on the order of 10−3 because the polymers were dispersed in an excess of polystyrene to avoid aggregation by decreasing the interchain interactions, which results in conditions similar to those observed in the solutions.
2.2. Part 2: Reversible switching of circularly polarized luminescence by external stimuli
For the purpose of functionalizing CPL, it is very interesting to control chiroptical properties, especially CPL, through the use of external stimuli [70].
2.2.1. Solvent
It has been demonstrated that the interchain helicity of chiral ACPs is largely affected by the surrounding environment, such as solvent, resulting in opposite CPL signs [71]. P8 is a poly(para-phenylene vinylene) derivative with bulky chiral groups in both side chains (scheme
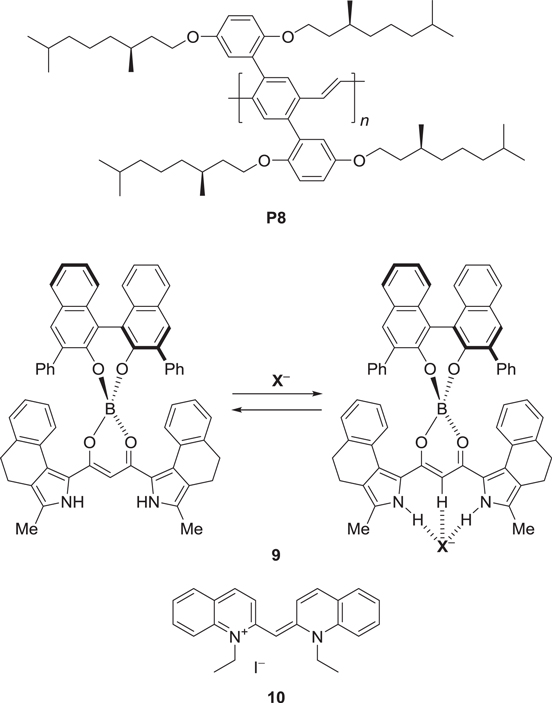
Scheme 4. Conjugated polymer (P8) and molecules (9 and 10) exhibiting external stimuli-responsive CPL.
Download figure:
Standard image High-resolution image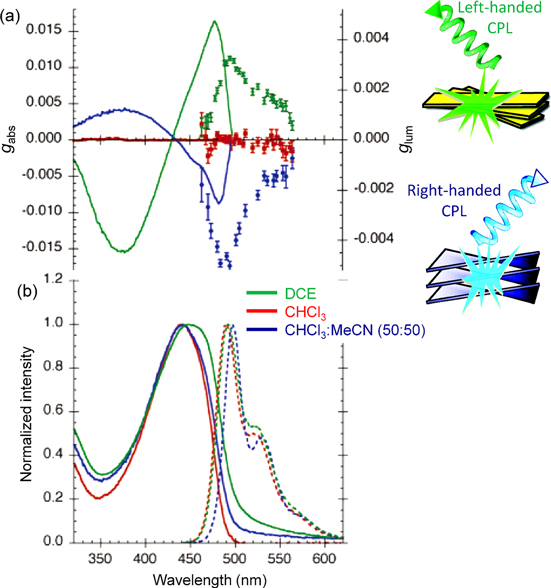
Figure 9. (a) The dissymmetry factors in absorption (lines) and luminescence (markers), and (b) normalized absorption (solid lines) and luminescence (dashed lines) spectra of P8 solutions in DCE, CHCl3, and CHCl3−MeCN (50:50). Adapted with permission from ref. [71]. Copyright 2006 American Chemical Society.
Download figure:
Standard image High-resolution image2.2.2. Anion addition
Chemical-stimuli-responsive CPL has been realized using the luminescent anion receptor 9 (scheme

Figure 10. Spectral changes for 9 (1.0 × 10−5 m in CH2Cl2) in (a) UV−vis absorption (bottom) and CD (top) and (b) fluorescence (bottom) and CPL (top) measurements upon the addition of Cl− as the TBA salt (50 equiv for UV−vis absorption and 200 equiv for the other measurements; 9: black line; 9·Cl−: red line), and photographs of the corresponding solutions (insets). Reprinted with permission from ref. [73]. Copyright 2011 American Chemical Society.
Download figure:
Standard image High-resolution image2.2.3. Thermal treatment
The thermal switching of CPL between emission and quenching has also been reported through the use of the association-dissociation transition of a π-conjugated dye, 10 (scheme
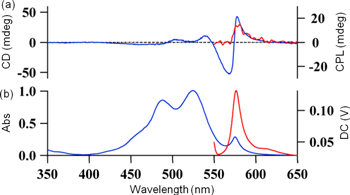
Figure 11. (a) CD (blue) and CPL (red) spectra, and (b) UV−vis absorption (blue) and PL (red) spectra of the induced J-aggregates of 10 (c = 3 × 10−5 m) with (+)-Rochelle salt (c = 1.8 m) in aqueous solution at 5 °C. Excitation wavelength: 470 nm. Reprinted with permission from ref. [73]. Copyright 2012 The Chemical Society of Japan.
Download figure:
Standard image High-resolution image2.2.4. Vortex flow
Switching circular polarization in absorption and luminescence by vortex flows has also been reported. When stirring a benzene solution of zinc porphyrin dendrimer 11 (scheme
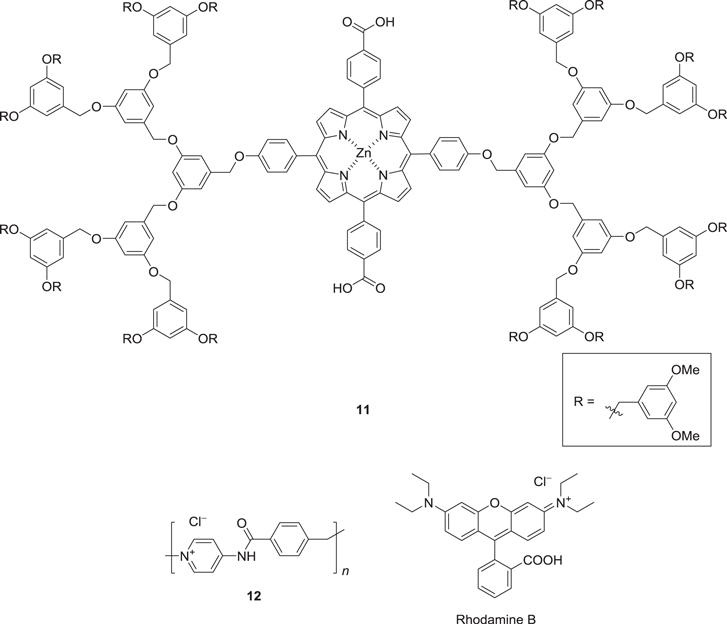
Scheme 5. Structures of (i) a zinc porphyrin dendrimer 11 and (ii) an oligomeric host gelator 12 and a guest luminophore, rhodamine B.
Download figure:
Standard image High-resolution image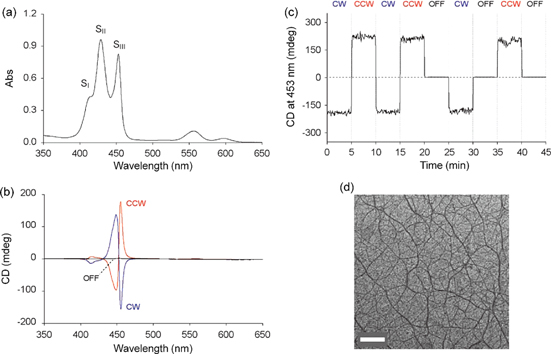
Figure 12. (a) UV−vis absorption and (b) CD spectra of a benzene solution of 11 (c = 6.0 × 10−6 m) in a 10 × 10 × 40 mm3 quartz optical cell. The CD spectra were measured upon bottom rotary stirring at 1350 rpm in clockwise (CW; blue) and counterclockwise (CCW; red), and without stirring (OFF; black). (c) Change in CD intensity at 453 nm in response to a stepwise variation of the stirring conditions at 20 °C. (d) A transmission electron micrograph of air-dried sample of 11 benzene solution on a specimen grid. Scale bar: 500 nm. Adapted with permission from ref. [74]. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image2.2.5. Photoirradiation
It has been reported that reversible photoisomerization-driven switching occurs between the emission and quenching of CPL [47]. Novel conjugated polymers P13 and P14 were synthesized by introducing both a chiral moiety and a photoresponsive dithienylethene (DE) moiety into the side chains of ACPs (scheme
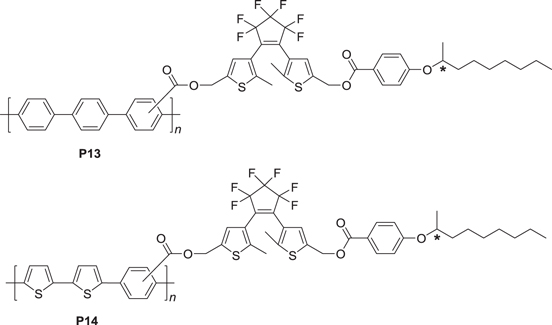
Scheme 6. Structures of photoresponsive chiral ACPs.
Download figure:
Standard image High-resolution image2.2.5.1. Photoisomerization
The conversion ratio from the open to closed forms of DE moiety was confirmed by 1H NMR analysis. The ratios in a photostationary state (PSS) were determined to be 10% and 7−9% for the cast films of P13 and P14, respectively. PSS is an equilibrium state between the open and closed forms of DE moiety under UV irradiation.
2.2.5.2. Absorption and CD properties
The cast films of the polymers exhibited Cotton effects in the corresponding absorption region. The CD spectra of the P13 and P14 films are shown in figures 13(c) and (f), respectively. The (R)- and (S)-P13 films exhibited bisignate CD couplets that were mirror images of each other. This result implies that the polymers in the film are assembled to form a one-handed helical interchain π-stacked structure. On the other hand, the (R)-P14 and (S)-P14 films showed CD bands that were mirror images of each other. However, the CD bands exhibited no splitting shape and relatively weak signals compared with those of the P13 films. This result suggests that the P14 polymers form loosely π-stacked helical structures. The non-straight and rather bent backbone of P14, which consists of bithienylene-monophenylene repeating unit, might be less suitable for the formation of interchain π-stacking, thereby resulting in a weak CD signal. Interestingly, there are no differences in the CD spectra between the open state and PSS, irrespective of the P13 and P14 films and of (R)- and (S)-configuration. This result indicates that the helical π-stacked structure of the P13 film and the chiral structure of the P14 film remain unchanged even after the reversible photoisomerization between the open form and the closed one in the side chain.
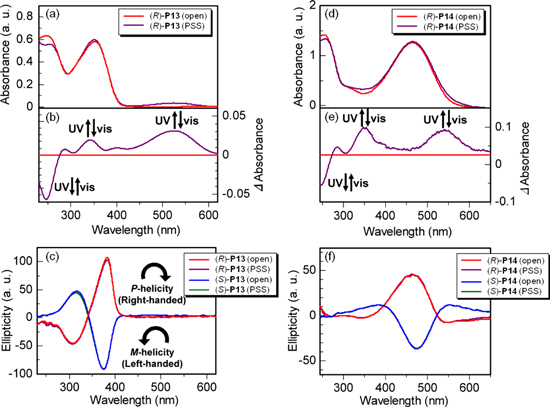
Figure 13. UV−vis absorption spectra and difference spectra between the PSS and the open state of (a, b) (R)-P13 and (d, e) (R)-P14 cast films, respectively, and CD spectra of (c) P13 and (f) P14 cast films in the open state and the PSS of the dithienylethene moiety. Reprinted with permission from ref. [47]. Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image2.2.5.3. Photoluminescence
A fluorescence quenching behavior was observed in the cast polymer films, as shown in figures 14(a) and (c). The fluorescence band at 413 nm for (R)-P13 and that at 570 nm for (R)-P14 in the open state drastically decreased in intensity and then quenched upon UV irradiation. However, the quenched fluorescence was regenerated by irradiation with visible light, leading to a reversible photoswitching in fluorescence, as shown in figures 14(a) and (c). The ratios of the fluorescence intensity between the open state and PSS (Iopen/IPSS) were evaluated as follows: Iopen/IPSS (at 413 nm) = 240 for the (R)-P13 film and Iopen/IPSS (at 570 nm) = 80 for the (R)-P14 film. The fluorescence quenching might be due to an efficient energy transfer from the excited polymer main chain to the closed form of the DE moiety. However, the excited DE of the closed form changes to its ground state through a nonradiative transition.
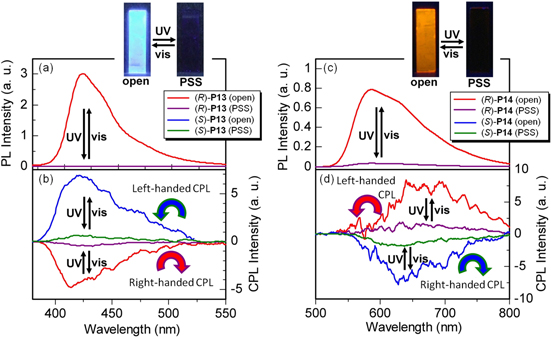
Figure 14. PL spectra of (a) (R)-P13 and (c) (R)-P14 cast films and CPL spectra of (b) P13 and (d) P14 cast films. The wavelength of excitation light was 350 nm for P13 and 380 nm for P14. Insets show photographs of (R)-P13 and (R)-P14 films in the open state and PSS under UV irradiation (excitation wavelength = 370 nm, 4 W, handheld lamp). Reprinted with permission from ref. [47]. Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image2.2.5.4. CPL property
Notably, the cast films of the polymers show CPL and CD in the wavelength region corresponding to the transitions of the polymer main chains. The formation of helical π-stacked structures of P13 and P14 is supported by the observation of CPL in the cast films, as shown in figures 14(b) and (d). The open forms of P13 and P14 exhibit Cotton effects in CPL spectra. Figure 14(b) and (d) also show that the CPL intensities of the P13 and P14 films drastically decrease, irrespective of the (R)- and (S)-configuration, upon photoisomerization of DE moiety from the open form to the closed one. Namely, the quenching of CPL occurs in the PSS, which is associated with the quenching of fluorescence itself. The reverse photoisomerization from the closed form to the open one causes the CPL band to reappear. This photoswitching behavior was observed over 10 cycles.
The ratio of the CPL intensities (ΔIopen/ΔIPSS) between the open state and the PSS of the (R)-P13 film at 414 nm and those of the (R)-P14 film at 650 nm were 13 and 7, respectively, which are smaller than the corresponding ratios of the fluorescence intensities (240 and 80 in Iopen/IPSS for (R)-P13 and (R)-P14, respectively). The degree of circular polarization of luminescence was evaluated using glum. As shown in figure 15, the glum values for the P13 and P14 films were estimated to be on the order of 10−2 at 414 nm and 650 nm, respectively. Finally, note that the helically π-stacked structure formed in the films of the present conjugated polymers is so rigid that it is not affected by the photochemical opening and closing isomerization reaction of the DE moiety in the solid state. Thus, it was possible to control the reversible switching of CPL between emission and quenching through photochemical irradiations by retaining the chirality of the polymers.
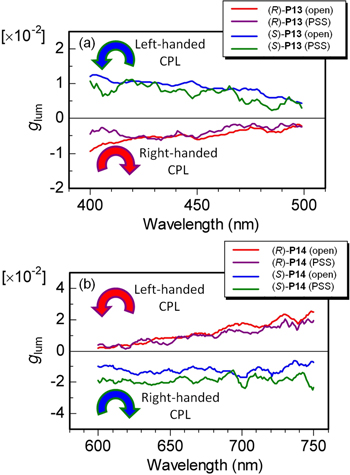
Figure 15. Luminescence dissymmetry factors (glum) of (a) P13 and (b) P14 films in open state and PSS. Reprinted with permission from ref. [47]. Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image2.3. Part 3. Supramolecular structures of ionic ACPs with circularly polarized luminescence
Ionic ACPs are amphiphilic polymers that contain hydrophilic ionic groups in the side chains and a hydrophobic aromatic conjugated backbone. In addition, the conjugated backbones easily aggregate with each other due to their high π-stacking ability, and therefore, ionic ACPs can form a hierarchical nanophase-separated structure. Thus, ionic ACPs are considered to be promising candidates as blocks to construct highly ordered supramolecular nanostructures.
2.3.1. Supramolecular helical nanowire with CPL
A molecular wire is a representative supramolecular nanostructure. An achiral cationic PT derivative, P15 (scheme
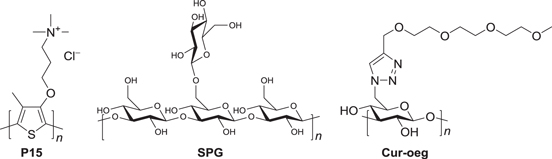
Scheme 7. An achiral cationic polythiophene derivative (P15) and chiral polysaccharides (SPG and Cur-oeg).
Download figure:
Standard image High-resolution image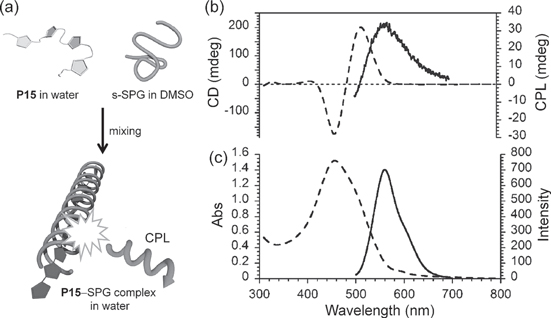
Figure 16. (a) Schematic illustrations of the supramolecular helical nanowire formation in solution. (b) CD (dashed line) and CPL (solid line), and (c) UV−vis absorption (dashed line) and fluorescence (solid line) spectra of P15−SPG complex in water−DMSO. Excitation wavelength: 400 nm. Adapted with permission from ref. [77]. Copyright 2009 The Chemical Society of Japan.
Download figure:
Standard image High-resolution image2.3.2. Stimuli-responsive supramolecular helical nanowire
On the other hand, an analogous P15 complex with a polysaccharide, Curdlan derivative (Cur-oeg, see scheme
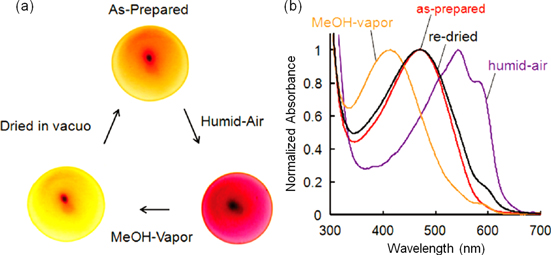
Figure 17. (a) Photographs and (b) UV−vis absorption spectra of a P15−Cur-oeg complex film. The as-prepared film was exposed to humid air and methanol vapor, and then dried in vacuo again. Reprinted with permission from ref. [79]. Copyright 2010 American Chemical Society.
Download figure:
Standard image High-resolution image2.3.3. Polymer spherulite consisting of hierarchical helical assemblies
Recent progress has indicated that an achiral π-conjugated polymer, P16, bearing cationic side chains form an interchain helically π-stacked assembly with anionic chiral compounds (BNPs). Notably, the polymer assembly is stabilized by both electrostatic and π−π interactions, and they hierarchically self-organize into a spherulite with circularly polarized blue luminescence (blue CPL) [30]. This is the first example of a chiroptical spherulite that is hierarchically constructed from π-conjugated polymers.
A cationic water-soluble poly(para-phenylene) derivative (P16) was synthesized by introducing tetraalkylammonium cations at the terminal sites of the side chains. An anionic water-soluble and axially chiral binaphthyl derivative (BNP) was synthesized by introducing two sulfonate anions at the terminal sites of the substituents, which were linked via propoxyl chains to the 2 and 2' positions of the 1,1'-binaphthalene, as shown in scheme
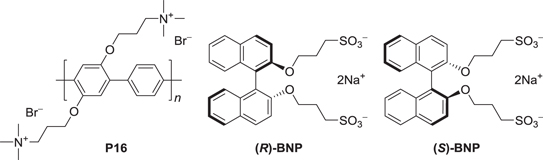
Scheme 8. An achiral cationic poly(para-phenylene) derivative (P16) and axially-chiral anionic binaphthyl derivatives (BNPs) with (R)- and (S)-configurations.
Download figure:
Standard image High-resolution image2.3.3.1. Chiroptical properties of P16, BNP, and their mixture
Figure 18(a) presents the UV−vis absorption and CD spectra of P16, BNP, and a mixture of P16 and BNP (abbreviated as P16−BNP) in methanol−water (50:50). P16 exhibited an absorption band at 340 nm, which corresponds to the π−π* transition of the π-conjugated backbone. BNP showed an absorption band at 228 nm, which corresponds to the π−π* transition of the binaphthalene. BNP exhibited a bisignate Cotton effect in the absorption band region due to the axial chirality of the binaphthalene. P16−BNP showed an additional absorption band at a wavelength (384 nm) that was 44 nm longer than that of P16. This result implies that the effective conjugation length of the P16 backbone increases, accompanied by an increase in the coplanarity of the polymer backbone, which could be due to the formation of an interchain π-stacked structure. Furthermore, P16−BNP exhibited an induced CD band in the π−π* transition region of the π-conjugated backbone (figure 18(a)). The strong bisignate Cotton effects observed at 376 and 341 nm imply the presence of an exciton-coupling phenomenon between the main chains [66]. The induced CD band, as well as the corresponding absorption band, increased in intensity with increasing concentrations of BNP (figure 18(c)). These results imply that P16 forms an assembly with BNP in methanol−water and that the assembly has induced chirality of the polymer moiety, which is caused by chirality transfer from the axially chiral compound (BNP) to the achiral polymer (P16). That is, the cationic P16 polymers are able to come sufficiently close to each other with the aid of the anionic BNP to yield an interchain helical π-stacked assembly [46, 80–86]. Notably, the absorption band at 384 nm and the CD band with the bisignate Cotton effect decreased in intensity with increasing temperature, and both absorption features finally disappeared at 75 °C (figure 18(d)). This result indicates that the helically π-stacked assembly is gradually destroyed upon heating to afford free P16s and BNPs. Because the aliphatic chiral compound, which bears two anionic sulfonate groups, gives no ICD even when it is mixed with P16, the π−π interaction between the conjugated polymer and the aromatic chiral compound, in addition to the presence of electrostatic interactions between the compounds, is indispensable for the formation of the π-stacked assembly.
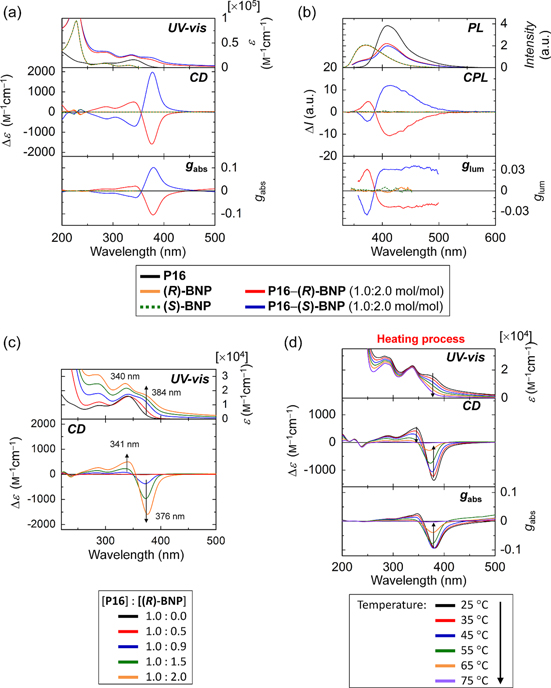
Figure 18. (a) The UV−vis absorption, CD, and absorption dissymmetry factor (gabs) spectra and (b) PL, CPL, and glum spectra of P16, BNP, and a mixture (P16−BNP) (1.0:2.0 mol/ mol) in methanol−water (50:50). The concentrations are as follows: P16, 5.8 × 10−5 m; BNP, 2.0 × 10−5 m; P16 in the mixture, 2.0 × 10−5 m. (c) The UV−vis absorption (upper) and CD (lower) spectra of P16−( R )-BNP in methanol−water (50:50) at various molar ratios. The concentration of P16 is held constant at 2.0 × 10−5 m. (d) The UV−vis absorption, CD, and gabs spectra of P16−( R )-BNP (1.0:2.0 mol/ mol) in methanol−water (50:50 v v−1) at various temperatures during the heating process. The concentration of P16 is 2.0 × 10−5 m. Adapted with permission from ref. [30]. Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageFigure 18(b) presents the PL and CPL analyses of P16, BNP, and P16−BNP in methanol−water (50:50). Not only did P16 exhibit blue fluorescence, but P16−BNP did as well, although the fluorescence intensity of P16−BNP was weaker than that of P16. This may be due to the presence of the so-called 'concentration quenching in fluorescence' phenomenon resulting from the formation of a polymer assembly with an interchain π-stacked structure [46, 84, 85]. It is intriguing that both P16−( R )-BNP and P16−( S )-BNP exhibited negative and positive monosignate Cotton effects at 414 nm in the CPL spectra. This result indicates that the former and the latter have right- and left-handed CPL, respectively. Note that the PL and CPL bands at approximately 370 nm arise from the chiral inducer, BNP.
Both Cotton effects in the CD and CPL spectra of P16−( R )-BNP and P16−( S )-BNP show mirror images in sign, implying that the helical sense of the polymer assembly strictly depends on the chirality of the chiral inducer, BNP. According to exciton coupling theory [66], positive and negative bisignate Cotton effects in CD spectra indicate right- (P-helicity) and left-handed (M-helicity) screw structures, respectively. Therefore, P16−( R )-BNP, which shows a negative bisignate Cotton effect in the CD spectrum, has a left-handed π-stacked structure with M-helicity. This is consistent with the negative monosignate Cotton effect observed in the CPL spectrum, which indicates the emission of circularly polarized light with M-helicity. On the other hand, P16−( S )-BNP, which shows a positive bisignate Cotton effect in the CD spectrum and a positive monosignate Cotton effect in the CPL spectrum, has a right-handed π-stacked structure with P-helicity. The absorption dissymmetry factor (gabs) values were −9.8 × 10−2 (at 376 nm) and +2.1 × 10−2 (at 341 nm) for P16−( R )-BNP and +9.9 × 10−2 (at 378 nm) and −2.8 × 10−2 (at 342 nm) for P16−( S )-BNP. The glum values were −2.4 × 10−2 (at 414 nm) for P16−( R )-BNP and +3.1 × 10−2 (at 415 nm) for P16−( S )-BNP. The values of gabs and glum increased with increasing amounts of BNP until the molar ratio of [BNP]/[P16] reached 2.0. The observation of dissymmetry factors on the order of 10−2−10−1 in the CD and CPL spectra indicate that the polymer assembly has a considerably strong helicity due to the interchain π-stacked structure.
2.3.3.2. Polymer spheres
Figures 19(a) and (b) present scanning electron microscopy (SEM) images of the P16−BNP (1.0:2.0 mol/ mol) assembly. The SEM images reveal a spherical morphology formed by the polymer assemblies (figures 19(a) and (b)). The sphere sizes range from 140 to 255 nm, with an average size of approximately 200 nm. This result is in good agreement with the results of dynamic light scattering measurements, which indicated an average sphere size of 220 ± 57 nm. Figure 19(c) displays a fluorescence microscopy image of polymer spheres prepared by spin-coating at 1000 rpm on a glass plate. A mercury-vapor lamp with a wavelength of 365 nm was used as the excitation light, and a low-pass filter was employed to remove wavelengths shorter than 420 nm. As shown in figure 19(c), each sphere exhibits blue fluorescence.

Figure 19. Scanning electron microscopy images of P16−BNP (1.0:2.0 mol/ mol) prepared by (a) drying the mixed solution of P16 and BNP at room temperature and (b) freeze-drying the mixed solution. (c) A fluorescence microscopy image of P16−BNP (1.0:2.0 mol/ mol) spin-coated on a glass plate. Fluorescence was excited by UV light (λ = 365 nm) and a low-pass filter was employed to remove wavelengths shorter than 420 nm. (d) POM image of P16−BNP (1.0:2.0 mol/ mol) in methanol−water (50:50) at room temperature. The crossed arrows represent the directions of the polarizer and the analyzer of polarized optical microscope. Reprinted with permission from ref. [30]. Copyright 2012 WILEY-VCH Verlag GmbH & co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image2.3.3.3. Spherulites consisting of polymer assemblies
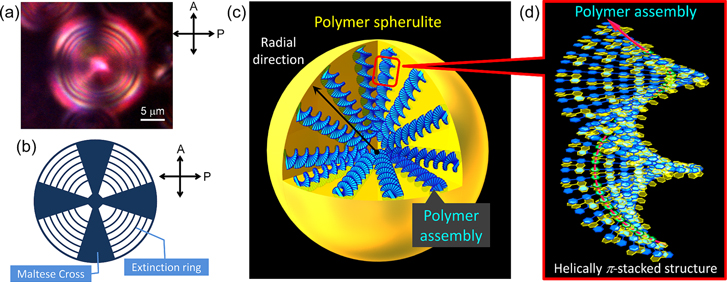
To elucidate the inner structure, the polymer spheres were observed using POM at room temperature. To obtain finer POM images of the polymer spheres, it is desirable to use a sample of large particles for the POM measurement. To increase the particle size, the polymer spheres were prepared by adding the BNP solution to the P16 solution very slowly and then leaving the mixture at room temperature for 7 days without stirring. Note that Maltese Cross patterns with extinction rings are observed in all spheres, as shown in figure 19(d). This result indicates that each sphere is constructed from a polymer spherulite [87–89]. The polymer spherulite is a spherical aggregate that consists of crystalline lamellae and interspaced noncrystalline sites, where the former (crystalline lamellae) radiate from the center of the spherulite. The appearance of a Maltese Cross pattern implies that the axis of the interchain π-stacked structure coincides with the polarization directions of the analyzer or the polarizer of a polarized optical microscope (figures 20(a)–(c)) [89]. Furthermore, the extinction rings indicate the formation of twisted lamellar structures along the radial axis of the spherulite. In this study, the twisted lamellae consist of helical π-stacked conjugated polymers. Plausible models of the polymer assemblies and spherulite are depicted in figures 20(c) and (d). The spherulites observed in POM are spherical aggregates of the polymer assemblies bearing the interchain helically π-stacked structure, whose helical axes are parallel to the radial direction of the spherulite, as shown in figure 20(c). The present system is the first example of a chiroptical spherulite that is hierarchically constructed from π-conjugated polymers. Note that the Maltese Cross pattern disappeared without a change in the pitch of the extinction rings upon thermal heating. At the same time, the absorption and CD bands at approximately 384 nm decreased in intensity with a slight blue-shift and disappeared (figure 18(d)). These results suggest that the decomposition of the polymer spherulite might occur simultaneously with that of the polymer assembly, although the spherulite is a hierarchically higher-ordered structure than the polymer assembly.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 20. (a) A Maltese Cross image with the extinction rings observed in POM for a spherulite of P16−( R )-BNP (1.0:2.0 mol/ mol) in methanol−water (50:50) at room temperature. (b) The schematic pattern of the Maltese Cross image with extinction rings. (c) A plausible model of a spherulite consisting of polymer assemblies. (d) The plausible model of polymer assembly with the helically π-stacked structure. P16 and BNP are represented in blue and yellow, respectively. Reprinted with permission from ref. [30]. Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution image{kind=link}
3. Conclusions
We have discussed ACPs with interchain helical structures from the perspectives of the self-assembled structures and chiroptical properties. The ACPs self-organize to form helical π-stacked assemblies due to chiral side chains or intermolecular interactions with other chiral compounds.
In Part 1, thienylene- and phenylene-based LC conjugated copolymers containing a chiral nony group in the side chain were discussed. The polymers with three aromatic rings in the repeating unit exhibited bisignate Cotton effects in the CD spectra of solutions and films and formed a polymer assembly with an interchain helical π-stacked structure. The temperature dependence of the absorption and CD spectra indicated that the polymer assembly exists even at room temperature but dissociates into single polymers at high temperatures. These polymers exhibited RGB-colored fluorescence and also showed CPL both in solution and as a film. The films exhibited enhanced CPL signals because of the strengthening of the helical π-stacking in solid states. The annealed films exhibited very high CPL with glum values up to 10−1, which were due to the large interchain interactions achieved during self-ordering in the N*-LC phase. It was found that the hierarchical arrangement of the higher-order structure by chiral liquid crystallinity is promising for obtaining CPL with a very high dissymmetry factor in the chiral conjugated polymers. Furthermore, when some of the polymers were mixed in the appropriate ratio, CPL was observed across most of the visible region, indicating white CPL.
In Part 2, novel photoresponsive chiral π-conjugated polymers were discussed by focusing on the DE moieties linked with chiral alkyl groups into polymer side chains. It is implied that the cast films of P13 polymers, consisting of a straight backbone of a terphenylene moiety as a repeating unit, have helically π-stacked structures of main chains. In contrast, the cast films of P14, consisting of a bent backbone of a bithienylene-monophenylene moiety as a repeating unit, are suggested to possess weakly π-stacked structure. The cast films of the two types of conjugated polymers exhibited right- or left-handed CPL with relatively large dissymmetry factors on the order of 10−2. The emission and quenching of CPL of the cast films are reversibly switched through the photoisomerization of the DE moiety introduced in the side chain.
In Part 3, the mixture of a cationic poly(para-phenylene) derivative (P16) and an anionic chiral binaphthyl derivative (BNP) was discussed by focusing on the hierarchically self-organized assembly with an interchain helically π-stacked structure stabilized by the presence of both electrostatic and π−π interactions. The electrostatic and π−π interactions are essential for the stability of the π-stacked structure, resulting in large dissymmetry factors of |10−2| − |10−1| in absorption and luminescence. It was demonstrated that the polymer assemblies further gathered to form spherulites, which can be regarded as semi-crystalline nanospheres, and the spherulites exhibited blue CPL.
These results suggest that polymers with interchain helicity could be useful as advanced chiroptical materials for next-generation plastic optoelectronics.
Acknowledgements
This work was supported by Grants-in-Aid for Science Research (A) (No. 25246002) and (No. 25620098) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
- 1
Positive bisignate Cotton effect means that positive and negative Cotton effects of CD couplet are observed at longer and shorter wavelengths, respectively. On the other hand, negative bisignate Cotton effect means that negative and positive Cotton effects of CD couplet are observed at longer and shorter wavelengths, respectively.