



Abstract
We report on the ability of the meteoritic material schreibersite to catalyze the generation of higher sugars from simple carbohydrates in the formose reaction network. Since the analysis of carbonaceous meteorites like the Murchison meteorite it has become generally accepted that a substantial amount of organic material has been delivered to the early earth and, therefore, ought to be considered in scenarios for the origin(s) of life. Also for the open question of accessible phosphorus sources, an extraterrestrial material called schreibersite has been identified that is capable of releasing soluble and reactive phosphorus oxyanions that would react with organics to form for instance nucleotides and membrane associated molecules. We have reinvestigated this material using capillary electrophoresis to monitor its corrosion process in water and probed its ability to phosphorylate a wide range of organics. Although showing a poor reactivity of schreibersite, we have found that the material catalyzes the aldol reaction of small carbohydrates forming larger sugar molecules. This reaction in the formose reaction network is a prebiotically likely route to biologically relevant sugars. The results of our study present one of the first instances of connecting extraterrestrial material to prebiotic chemistry on the early earth.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
1.1. Chemical networks and chemical evolution
Ever since the pioneering work of Oparin [1], Haldane [2] and Miller [3], the scientific field of the origin(s) of life has amplified in its spectrum of theories, experiments and results starting from the 'warm little pond' idea of Charles Darwin [4] to discoveries of extra-solar (super-)earth planets [5, 6] and specific abiotic pathways to relevant building blocks of life [7–18]. Even though the field is now evolving for almost a century, there still is uncertainty about fundamentals like the definition of life and the applicability of the phrase plausible prebiotic condition [19–24]. That is because scenarios for the origin(s) of life are conceivable for a wide range of different parameters, e.g. temperature, pressure, irradiation flux and availability as well as concentration of feedstock molecules. Possible scenarios for the origin(s) of life include various settings such as hydrothermal vents [25], volcanic locations [26, 27], primordial soup [28, 29] and drying lagoons [30–32]. The difficulty with the characterization of life, on the other hand, lies with the many open questions of its evolvement. If one could precisely retrace the steps of the emergence of life on earth, it would become apparent where to draw the line between inanimate and alive. A popular periphrasis, though, was given in 1994 by NASA and says that life is 'a self-sustaining chemical system capable of Darwinian evolution' [23].
A possible sequence of events leading to systems covered by that definition and giving way to the last universal common ancestor (LUCA) is depicted in figure 1. After the formation of the Earth 4.5 billion years ago in which all organic matter was pyrolyzed, small molecules were not only formed through atmo-, hydro- and/or lithospheric chemistry but also delivered by extraterrestrial bombardment. Those small molecules are prone to construct complex reaction networks under changing conditions of versatile energy flux (out-of-equilibrium chemistry) and give rise to more complex molecules of certain features (redox-, photo- and catalytic activity, amphiphilic properties for bilayer assembly). Those reaction networks likely succumb to evolutionary principles such as isolation, competence and adaption although their survival is not measured in reproduction or based on heredity but in self-sustainment and complexity. That phase, thus, can be termed chemical evolution. In such a first self-sustaining system, the emergence of functional polymers becomes probable and levels the transition to Darwinian evolution. Once first protocells are formed Darwinian evolution manifests and allows the generation of LUCA.
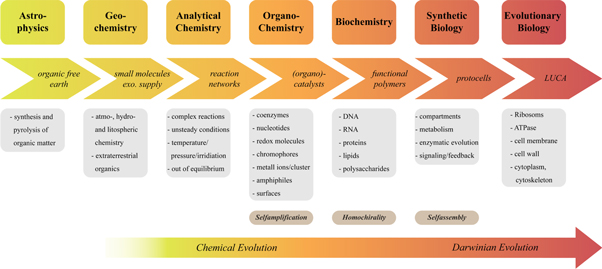
Figure 1. Possible steps of the formation of the Earth towards the last universal common ancestor (LUCA). Scientific fields involved with the studies of the origin(s) of life as well as specific features and molecules of each step are shown, too.
Download figure:
Standard image High-resolution imageAlong that way, however, there is a multitude of open questions that require several scientific disciplines to answer them. The authors' focus lies with the reaction pathways in context with the formation of small molecules and their reactivity towards complex reaction networks.
The present contribution deals with the meteoritic material schreibersite Fe3P as a source for reactive phosphorus oxyanions and its behavior during aqueous corrosion in the presence of organic molecules. It, therefore, focuses on the possible emergence and reactivity of small molecules and their transition into complex reaction networks. Specifically, the ability of the mentioned material to catalyze the formation of carbohydrates from small precursors, aldol/retro-aldol reactions in the formose reaction network, is studied. This way, a connection between extraterrestrial and terrestrial material is drawn and the impact of meteoritic bombardment not only for delivery but also for inducing reactions is highlighted.
1.2. Schreibersite as a possible phosphorylation agent
Phosphorus is one of the key elements of life and amounts up to 1% of the weight of dry cells [33]. It is part of the backbone of DNA/RNA, essential component of the phospholipids in cell membranes, energy storage in form of adenosine triphosphate, constituent of coenzyme such as flavine adenine dinucleotide, nicotinamide adenine dinucleotide and coenzyme A as well as crucial derivatization group in metabolic pathways. Its significance in biological life is contrasted by its natural availability. There is no volatile phase or versatile redox chemistry of phosphorus under terrestrial conditions. The stable and predominantly occurring form is orthophosphate  which exhibits poor solubility in presence of divalent cations in aqueous medium [34]. Phosphorus, therefore, is a limiting ingredient for the origin(s) of life. It is assumed that 95% of the Earth's stock of phosphorus was concentrated in the core of the Earth leaving the continental crust with about 650 ppm phosphorus [35]. An inventory of the possible phosphorus sources in the hadean eon is given in table 1 as posed by Hazen [36] and expanded by Pasek [37].
which exhibits poor solubility in presence of divalent cations in aqueous medium [34]. Phosphorus, therefore, is a limiting ingredient for the origin(s) of life. It is assumed that 95% of the Earth's stock of phosphorus was concentrated in the core of the Earth leaving the continental crust with about 650 ppm phosphorus [35]. An inventory of the possible phosphorus sources in the hadean eon is given in table 1 as posed by Hazen [36] and expanded by Pasek [37].
Table 1. Mineral diversity in the hadean eon on the basis of Hazen [36] and Pasek [37]. Additions by the latter one are given in italics.
| Name | Formula | Classification |
|---|---|---|
| Nickelphosphide | Ni3P | Phosphide |
| Perryite | (Ni,Fe)8(Si,P)3 | |
| Schreibersite | (Fe,Ni,Cr)3P | |
| Beusite | Mn(II)Fe(II)(PO4)2 | Phosphate |
| Bobbierite | Mg3(PO4)2 · 8H2O | |
| Childrenite | Fe(II)AlPO4(OH)2 · H2O | |
| Chlorapatite | Ca5(PO4)3Cl | |
| Farringtonite | Mg3(PO4)2 | |
| Fluorapatite | Ca5(PO4)3F | |
| Galileiite | NaFe(II)(PO4)3 | |
| Graftonite | (Fe(II),Mn(II),Ca)3(PO4)3 | |
| Hydroxylapatite | Ca5(PO4)3OH | |
| Johnsomervilleite | Na10Ca6Mg18Fe(II)(PO4)36 | |
| Luneburgite | Mg3B2(PO4)2(OH)6 · 8H2O | |
| Merrillite | Ca9NaMg(PO4)7 | |
| Monazite-Ce | CePO4 | |
| Newberyite | MgHPO4 · 3H2O | |
| Sarcopside | Fe(II)3(PO4)2 | |
| Stanfieldite | Ca4Mg5(PO4)6 | |
| Struvite | NH4MgPO4 · 6H2O | |
| Triplite | (Mn(II),Fe(II))2PO4(F,OH) | |
| Vivanite | Fe(II)3(PO4)2 | |
| Wagnerite | Mg2PO4F | |
| Wavellite | Al3(PO4)2(OH)3 · 5H2O | |
| Whitelockite | Ca9Mg(PO4)6(PO3OH) | |
| Xenotime | (Y,Yb)(PO4) | |
The majority of the minerals listed in table 1 exhibit only a low solubility in water and, hence, do not have the ability to make phosphorus accessible in aqueous media on the early earth. The low abundance of free phosphorus is accompanied with its generally low reactivity towards organic molecules in its most stable form, orthophosphate. It is still highly discussed by what chemical pathway phosphorus could have been incorporated into organic molecules [37, 38]. Besides reactions in non-aqueous media such as formamide [39–44] and eutectic media [45] as well as by use of condensing agents (urea [46–49] and cyanamide [50–52]) or activated phosphates (pyro-, tri-, poly-, cyclic- or diamidophosphate) [16, 27, 53–55], one scenario is based on the meteoritic material schreibersite (Fe,Ni,Cr)3P [56]. Statistics assume that around 0.1%–10% of the Earths phosphorus inventory was delivered by meteoritic bombardment [57]. This material which contributes about 0.2–0.4 wt% of iron-rich meteorites [58] contains phosphorus in a reduced oxidation state allowing it to be corroded by water and to release reactive phosphorus oxyanions through a radical mechanism [59]. This material has already been examined in literature regarding the composition of the released phosphorus oxyanions (see table 2) and its ability to phosphorylate organic molecules. It was found that schreibersite is capable of phosphorylating glycerol and nucleosides in yields of 2.5% and 5.7%, respectively [60, 61]. The corrosion rate of schreibersite in saline solution is about 0.2% per week and 10% per year [57].
Table 2. Overview of phosphorus oxyanions products of schreibersite corrosion and determined concentrations in various experiments in literature.

|
||||||||
|---|---|---|---|---|---|---|---|---|
| Name | Hypo-phospite | Phosphite | Diphos-phite | Hypo-phosphate | Phosphate | Pyro-phosphate | Triphos-phate | Trimeta-phosphate |
| Formula | [H2O2P]− | [HO3P]− | [H2O5P2]2− | [O6P2]4− | [O4P]3− | [O7P2]4− | [O10P3]5− | [O9P3]3− |
| Oxidation state of P | +I | +III | +III | +IV | +V | +V | +V | +V |
| Charge/P | 1 | 1 | 1 | 2 | 3 | 2 | 1.6 | 1 |
| A (μM) | — | 700 | — | 280 | 1100 | 150 | — | — |
| B (μM) | — | 100 | — | 18 | 130 | 23 | — | — |
| C (%) | 0 | 59 | — | 2 | 31 | 1 | — | — |
| D (%) | 61 | 26 | — | — | 5 | — | — | — |
| E (%) | 87 | 11 | — | — | — | — | — | — |
| F (μM) | — | 1100 | — | 360 | 430 | 450 | — | — |
| G | Yes | Yes | No | No | Yes | No | No | No |
A: 1 g Fe3P, 25 ml DI water, air, 1 d; 293 K; quantification by NMR after addition of NaOH [56].B: 1 g Fe3P, 25 ml DI water, argon, 1 d; 293 K; quantification by NMR after addition of NaOH [56].C: 0.5 g Fe3P, 25 ml 0.1 M H2SO4; 7 d, 298 K; quantification by NMR after addition of Na2S [62].D: 0.5 g Fe3P, 10 ml H2O; UV; 3 h, 77 K; quantification by NMR after addition of Na2S [62].E: 19.1 g meteorite, 20 ml EtOH: H2O 1: 1 v/v; UV, 15 h, 77 K; quantification by NMR after addition of Na2S [62].F: 1 g, 25 ml DI water, air, 1 d; 293 K; quantification by NMR after addition of NaOH [59].G: 0.5 g Fe3P, N2, 1 week; 293 K and 343 K; evaluation by IC-ESI-MS [63].
1.3. Formose reaction
Carbohydrates are essential components of life. They are part of nucleotides in form of deoxyribose or ribose and are, therefore, crucial for DNA, RNA and cofactors. They are also vital constituents in metabolic pathways in all organisms, e.g. glycolysis. A plausibly prebiotic formation of carbohydrates of different length and composition was first discovered by Butlerov in 1861 [10] and interpreted on a molecular level by Breslow in 1959 [64] and is termed the formose reaction. The formose reaction is the oligomerization of formaldehyde in aqueous solution in the presence of a basic catalyst. It results in a complex mixture of straight-chain and branched monosaccharides, polyols and polyhydroxycarbonic acids. The specific outcome and product distribution depends on the concentration of involved feedstock molecules, temperature and reaction time. Its mechanism is not yet fully understood [65–68]. The formose reaction comprises a multitude of reaction paths in which carbohydrates not only self- and cross-add to one another in an aldol reaction type, but also split in retro-aldol reactions, disproportionate or oxidize, especially when performed under oxygen [69–72]. An over-simplified reaction network along with a list of unbranched sugars of up until 6 carbon atoms that all are products of the formose reaction network is given in figure 2. If started from formaldehyde, formally the simplest carbohydrate, the formose reaction can only be catalyzed either in the presence of a co-catalyst capable of enediolization, e.g. higher sugars such as glycolaldehyde and glyceraldehyde (a minimum of 3 ppm [66]), thiazolium salts activating formaldehyde for electrophilic attack [73] or UV-irradiation [74, 75]. As for the basic catalyst, a wide range of compounds are suitable such as Al(OH)3, NaOH, borates, FeO and typically Ca(OH)2 and have already been studied in literature [72]. Most of the inorganic and organic substances known to be efficient catalysts for the formose reaction network, however, are not considered to be present in the hadean eon for the origin of life [36]. A disadvantage of the formose reaction, also, lies with its poor selectivity. Biologically significant carbohydrates such as ribose are only formed in low yield. Besides, the longer the formose reaction proceeds the more carbohydrates are turned into a water insoluble tar discernable from an intensifying yellow–brown color of the solution [70]. It was shown, however, that the presence of borate [69, 76] or phosphorylated carbohydrates [77, 78] overcome that selectivity issue.
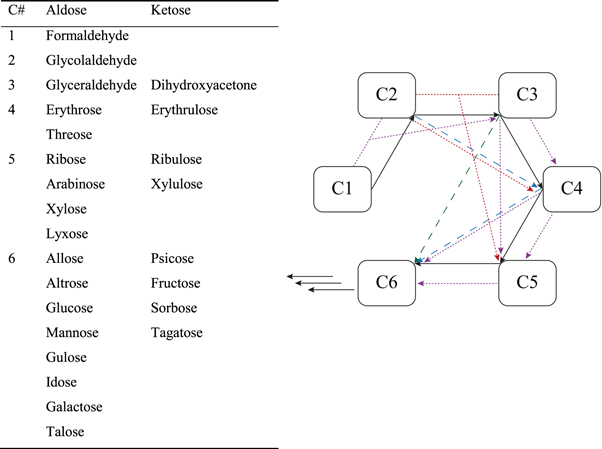
Figure 2. Simplified reaction scheme of the formose reaction and unbranched aldoses and ketoses of up to 6 carbon units. Black arrow: addition of C1; blue arrow: additions of C2; green arrow: additions of C3; violet arrow: addition of either C1 or C2; red arrow: addition of either C2 or C3.
Download figure:
Standard image High-resolution image2. Chemicals and methods
2.1. Chemicals
All chemicals have been used as received except for aqueous sugar standards that were lyophilized to obtain the compounds as solids. Synthetic schreibersite material was purchased from Alfa Aeser (99.5% metal basis) and Sigma-Aldrich (99.5% trace metals basis). Formaldehyde (37% wt in H2O), cyanamide (99%), dicyandiamide (99%), sodium sulfide nonahydrate (99%), thiourea (99), stearic acid (98.5%), ethanolamine (99%), choline (98%), O-ethylhydroxylamine hydrochloride (99%), N-methyl-bis(trifluoroacetamide) (MBTFA) (99%), N, O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (99%), pyridine (99%), glycolaldehyde dimer (mixture of stereoisomers), D-threose (60%), D-erythrose (75%), L-erythrulose (80%), dihydroxyacetone dimer (97%), D-allose (98%), L-arabinose (99%), D-mannose (99%), D-fructose (99%), D-galactose (99%), D-ribose (98%), salicylic acid (99%) BIS-TRIS (98%) and sodium hexafluorophosphate (99%) were supplied by Sigma-Aldrich. Methanol (HPLC grade) and acetonitril (HPLC grade) was bought from Fisher Chemicals and VRW Chemicals, respectively. Urea (99%) and D-xylose (99%) were obtained from Merck. Sugar standards were largely purchased from Carbosynth Limited. Those sugars include: D-xylulose (98%), D-piscose (98%), D-idose (99%), L-ribulose (97%), D-altrose (99%), D-lyxose (99%), L-gulose (98%), D-tallose (99%), D-tagatose (99%), L-sorbose (98%), beta-D-galatoheptose (99%). D-glucose-1-hydrate (99%) was supplied by PanReac AppliChem ITW Reagents. Meso-inositil (99%) was bought from Roth. Dimethyl sulfoxide (DMSO) and ammonia (2M) were obtained from the chemical shop of the department of chemistry and pharmacy of the Ludwig–Maximilian University, Munich. Water was deionized (DI) by a VWR Puranity PU 15 (VWR, Leuven, Belgium).
2.2. Instrumentations
2.2.1. Capillary electrophoresis (CE)
Monitoring of schreibersite corrosion and screening of phosphorylation reactions were performed on an Agilent 3DCE 1600 apparatus (Agilent Technologies, Waldbronn, Germany) controlled by the ChemStation software Rev. B.04.03. Measurements ran on a fused silica capillary (ID 50 μm, OD 360 μm) purchased from MicroQuartz Munich (Munich, Germany).
2.2.2. Gas chromatography (GC)
Separation of silylated sugars from the schreibersite induced formose type reaction were achieved on a Trace GC Ultra system hyphenated to either a PolarisQ MS (quadrupole-ion trap mass spectrometer (MS)) or a ISQ single quadrupole MS (Thermo Scientific, San Jose, California, USA) operated by Xcalibur software 1.4 SR1 or 2.2 SP1, respectively. Injections utilized a split/splitless injector (split mode; at 250 °C). MS detection was, routinely, complemented by flame ionization detection that was operated at 250 °C and carbon-corrected.
2.2.3. Scanning electron microscope with energy dispersive x-ray analysis (SEM-EDX)
Synthetic schreibersite material was analyzed with a FEI Helios G3 UC system equipped with a X-Max-N 80 detector using the AzTec software. Measurements were conducted with 20 kV accelerating voltage at 4 mm working distance.
2.2.4. X-ray photoemission spectroscopy (XPS)
Schreibersite material was loaded into a UHV chamber equipped with a VSW TA10 x-ray source providing non-monochromatic Al Kα radiation and a VSW HA100 hemispherical analyzer. Due to charging of the sample peak shifts were corrected by setting the Na 1s peak to 1072.1 eV. Spectra were acquired before and after cleaning of the sample by Ar+ sputtering (1 kV; 7 μA). The recorded peaks were fitted with a Doniach–Sunjic line shape convoluted with a Gaussian and linear background subtraction for analysis [79].
2.2.5. pH meter
Determination of pH values was conducted with the SCHOTT instrument pH meter Lab 850 by SI Analytics GmbH (Mainz, Germany).
2.3. Methods
2.3.1. CE
Separation conditions were predicted and optimized by the Peakmaster 5.3 Complex software [80]. The background electrolyte (BGE) chosen consisted of 20 mM salicylic acid and 30 mM BIS-TRIS (pH 6.2 and IS 20 mM). Samples were injected with 200 mbar · s, and 0.1 vol% DMSO was used at outlet position before separation as electroosmotic flow (EOF) marker. Separation voltage was set to −30 kV with 40 mbar pressure assistance. The apparent mobility of the EOF was determined to be approximately μEOF = 11 · 10−9 m2V−1 s−1. The capillary was conditioned each day using a sequence of water flush (2 min), 0.1 M sodium hydroxide flush (2 min), water flush (2 min) and BGE flush (5 min). In between measurements, the capillary was rinsed with BGE for 2 min. Indirect-UV electropherograms were recorded with 10 Hz at 210 and 230 nm with a bandwidth of 4 nm and ran at 25 °C. Peak positions were determined by fitting with the Haarhoff–Van der Linde function using OriginPro 2017 SR2 (OriginLab Corporation, Northampton, USA). For identification and calibration, measurements with phosphorus oxyanions standards of different concentration were conducted with sodium hexafluorophosphate as internal standard in triplicates to determine effective mobilities and linearization graphs.
2.3.2. GC–MS
For the separation of sugars, two complementary derivatization strategies were used. Around 4 mg of residue of filtered and lyophilized reaction solution was dissolved in 400 μl pyridine, mixed with 400 μl of a 40 mg ml−1 O-ethylhydroxylamine hydrochloride solution and heated to 70 °C for 30 min on a rocking shaker. One half of that solution was further derivatized with 120 μl BSTFA while the other one was subject to 50 μl MBTFA. Either step was followed by incubation at 70 °C for 30 min on a rocking shaker. BSTFA derivatized sugars were separated on a SE-52 column (14 m length, ID 250 nm, 250 nm film thickness) with 80 kPa helium and a temperature program starting at 50 °C for 2 min and increasing temperature by 10 K min−1 to 140 °C and then 5 K min−1 to 240 °C and keeping that temperature for 2 min MBTFA modified sugars were measured on a OC 225 column (30 m length, ID 250 nm, 250 nm film thickness) with 80 kPa helium using the following temperature gradient: 80 °C for 5 min, 2 K min−1 to 160 °C, 10 K min−1 to 200 °C and keeping that temperature for 12 min. Chromatograms were baseline corrected and peaks were integrated using OriginPro 2017 SR2 (OriginLab Corporation, Northampton, USA). Integration results were corrected by the number of carbon atoms and used for relative quantification of components in mixture. Retention times and fragmentation patterns of standards of all unbranched aldose and ketose sugars up until 6 carbon atoms were used for identification of sugars in reaction mixture.
2.4. Corrosion experiments
2.4.1. Monitoring of schreibersite corrosion
1 g of commercially available Fe3P was placed in glass CE vials and mixed with 500 μl water. Vials were capped, sealed with a crimper and kept at 5 °C, 25 °C, 80 °C or 150 °C for different periods of time. For monitoring of reaction process, vials were shortly removed from the heating block, opened and measured. Right after sample injection, reaction vessels were closed as described and put back into the heating block.
2.4.2. Reaction of organics in the presence of schreibersite
Glass CE vials were charged with 250 mg of synthetic schreibersite material and 0.5 ml of 0.020 M solution of an organic. If solubility of organics prohibited addition at desired concentration, the substance was added as a solid and the mixture was suspended in the given volume of DI water. The reaction mixture was capped, sealed with a crimper and heated to either 80 °C for 7 days or 150 °C for 1 day.
2.4.3. Schreibersite induced formose type reaction
Glass CE vials were filled with 1 g of synthetic schreibersite material and 1 ml of 0.250 M carbohydrate solution. In cross reactions the concentration of each component was 0.125 M. Vials were capped and sealed with a crimper. Reaction mixtures were heated at 80 °C for either 1 or 7 days before being filtered, lyophilized and derivatized for GC separation.
3. Results
3.1. Verification of quality of schreibersite surrogate Fe3P
The synthetic and commercially available Fe3P which was shown to be a reasonable chemical proxy for schreibersite was probed via SEM-EDX and XPS to confirm that the material meets the quality of natural and artificial samples described in literature [81]. Full SEM-EDX and XPS data can be found in the supporting information (figures S1 and S2 is available online at stacks.iop.org/NJP/20/055003/mmedia). Either measurement validates that the material employed in all experiments is Fe3P of high purity that has already undergone aerobic hydrolysis on the surface. It has been shown that this alteration does not corrupt the results of corrosion experiments performed thereafter [62, 81].
3.2. Analysis of schreibersite corrosion
Based on the results of Foster et al (see table 2) that showed that the outcome of the analysis of schreibersite hydrolysis depends on the subsequent work-up of the sample, the author's focus lay with the development of a separation method that would allow the continuous monitoring of the corrosion process under inert conditions so that pH-sensitive phosphorus oxyanions would be detected unambiguously. For that purpose, a CE method with indirect UV-detection was established that separates the common corrosion process products in about 12 min utilizing a BGE of the pH value of 6.2 under which the targeted molecules would be both stable and charged in order to allow separation. It shall be highlighted that many of the phosphorus oxyanions of low oxidation state are instable and hydrolyze under high and low pH. A representative separation is shown in figure 3(A).
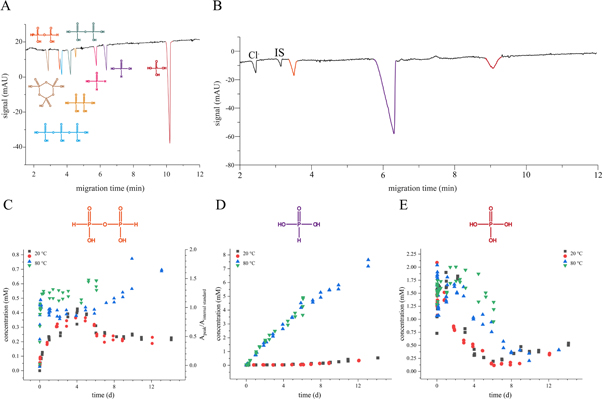
Figure 3. Results on schreibersite corrosion. (A) A representative separation of eight likely schreibersite corrosion products. Separation conditions are given in section 2.3.1. (B) Electropherogram of corrosion process after 14 days at 80 °C. Chloride is an impurity coming from the material. IS is the internal standard. Concentration progress of (C) diphosphite  (D) phosphite
(D) phosphite  and (E) phosphate
and (E) phosphate  over the course of 14 days at 20 °C and 80 °C.
over the course of 14 days at 20 °C and 80 °C.
Download figure:
Standard image High-resolution imageSince sample consumption is in the nL regime and sample work-up is usually not required in CE separation, the schreibersite corrosion process became monitorable in a straightforward fashion. This way, we could screen the process under various conditions of different temperature, pressure, concentration and atmosphere. We observed that the corrosion process proceeds rather slowly at 5 °C and 20 °C but is sufficiently fast at higher temperatures. A typical electropherogram of the material after 14 days at 80 °C is depicted in figure 3(B). On the contrary to literature results summarized in table 2 (entries A–C, F), we did not observe hypo- or pyrophosphate. This, however, is in agreement with Foster et al [63] (see table 2, entry G). Additionally, we detected a signal of the effective mobility of −(54.5 ± 0.5) 10−9 m2 V−1 s−1 which could not be unambiguously attributed to either triphosphate or diphosphite  As triphosphate would be accompanied with its hydrolyzes product pyrophosphate, which has not been detected, and a radical mechanism involving
As triphosphate would be accompanied with its hydrolyzes product pyrophosphate, which has not been detected, and a radical mechanism involving  radicals makes the formation of diphosphite
radicals makes the formation of diphosphite  likely [59], we can assign the peak of the given mobility to be diphosphite
likely [59], we can assign the peak of the given mobility to be diphosphite  The progress of the changes in concentration for 20 °C and 80 °C for the three main products of the corrosion process are shown in figures 2(C)–(E). It is highlighted that phosphite
The progress of the changes in concentration for 20 °C and 80 °C for the three main products of the corrosion process are shown in figures 2(C)–(E). It is highlighted that phosphite  is the major dissolved product accumulating over time.
is the major dissolved product accumulating over time.
Furthermore, studies on the influence of inert atmosphere did not result in observations other than already published [56]. Pressure was found to have no effect on the outcome of the corrosion and the amount of added water solely changed the concentration of the products, not the product distribution. Unlike stated in literature, however, we found that the pH value of corrosion experiments changed drastically and immediately on contact with water. After mixing Fe3P with water, we observed an instant increase of the pH value to around 11 that slowly decreased over time to 9. In order to exclude any impurities of the material to be the cause for that observation, we washed Fe3P five times with water before conducting the pH measurement of the corrosion process. Even then the pH value rose to 9 in about 20 min and stagnated. We attribute the immediate increase of pH when not washing the material to the release of tribasic phosphate  that has formed on the surface due to hydrolysis by the humidity of the air.
that has formed on the surface due to hydrolysis by the humidity of the air.
Besides screening at 5 °C, 20 °C and 80 °C, we conducted corrosion experiments at 150 °C in sealed glass vials. They were in alignment with the previously stated findings but allowed the corrosion process to progress faster. Typically, a corrosion for 14 days at 80 °C equaled one for 1 day at 150 °C in product distribution. Representative electropherograms can be found in the supporting information (figure S4).
3.3. Screening of phosphorylation with organics
As a result of our studies that showed that the corrosion process is sufficiently fast at 80 °C and 150 °C, we screened several organic molecules for their reactivity towards phosphorylation or condensation of phosphates to polyphosphates under the given conditions. However, we could not find detectable amounts of new species in most of the electropherograms. Table 3 gives an overview of the probed molecules. Only for the tested carbohydrates, we observed additional peaks as well as a yellow discoloration of the samples. In combination with the results of Pasek et al [59] that described a tar like residue when reacting glycolaldehyde with schreibersite that they, however, could not analyze, we checked those reaction samples for products associated with the formose reaction.
Table 3. Overview of organic molecules tested in their effect during the schreibersite corrosion.
| O containing molecules | N containing molecules | S containing molecules | Carbohydrates | Membrane relevant molecules |
|---|---|---|---|---|
| Methanol | Ammonia | Sodium sulfide | Formaldehyde | Stearic acid |
| CH3OH | NH3 | Na2S | CH2O | H3C-(CH2)16-COOH |
| Urea | Acetonitrile | Thiourea | Glycolaldehyde | Ethanolamine |
| H2NCONH2 | CH3CN | H2NCSNH2 | HO-CH2CHO | HO-C2H4-NH2 |
| Formamide | Cyanamide | Glyceraldehyde | Choline | |
| HCO-NH2 | NC-NH2 | HO-CH2-CH(OH)-CHO | (H3C)3N+-C2H4-OH | |
| Dicyandiamide | Dihydroxyacetone | Inositol | ||
| (H2N)2C=N-CN | HO-CH2COCH2-OH | C6H12O6 |
3.4. Schreibersite catalyzed formose type reaction
The analysis of the carbohydrate samples from the reactivity studies of Fe3P in the presence of organics confirmed to contain higher sugars such as formed in a formose type reaction. We, therefore, examined the four carbohydrates formaldehyde (C1 sugar), glycolaldehyde (C2 sugar), glyceraldehyde (C3 sugar) and dihydroxyacetone (C3 sugar) in single and cross-reaction setups and analyzed the product distribution. Separation of the reaction products was performed by GC utilizing the derivatization of carbohydrates with BSTFA and MBTFA that allows a relative quantification and identification of detected products. The results of our experiments are shown in figure 4. Typical gas chromatograms can be found in the supporting information (figure S5).
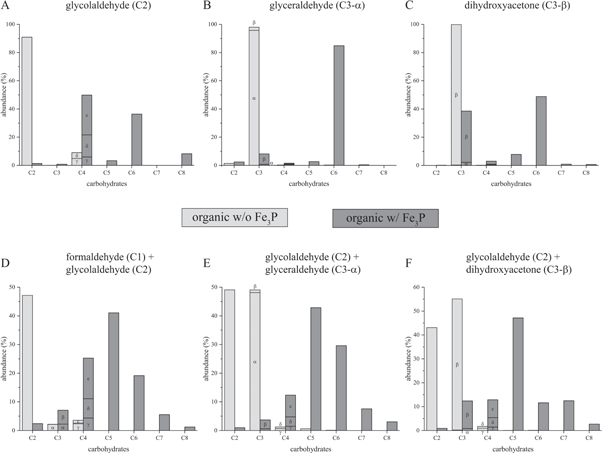
Figure 4. Product distribution of schreibersite catalyzed formose and formose type reaction in the presence of (A) glycolaldehyde, (B) glyceraldehyde, (C) dihydroxyacetone, (D) formaldehyde and glycolaldehyde, (E) glycolaldehyde and glyceraldehyde and (F) glycolaldehyde and dihydroxyacetone. Greek labeling denotes the following compounds: α—glyceraldehyde, β—dihydroxyacetone, γ—erythrose, δ—threose, ε—erythrulose. Higher sugars cannot be quantified individually as a result of overlapping peaks. Therefore, the bulk is quantified. For reaction conditions see section 2.4.3. Reaction time is 1 d. Relative errors of abundance are on average 1% and are omitted for clarity.
Download figure:
Standard image High-resolution imageFormation of sugars was only observed in presence of schreibersite. The comparison of the product distributions of all performed reactions shows the self- and cross-addition of substrates as expected in a typical aldol reaction such as in the formose reaction network. Glycolaldehyde as a C2 carbohydrate predominantly forms C4, C6 and C8 sugars (see figure 4(A)), whereas glyceraldehyde and dihydroxyacetone as C3 sugars generate C6 carbohydrates (see figures 4(B), (C)). The results, further, demonstrate that dihydroxyacetone is less reactive than glyceraldehyde as is indicated by its smaller consumption in the reaction. For dihydroxyacetone, we, also, detected products of C6 constitution that did not match any of the unbranched carbohydrates standards we used for establishing the GC separation method suggesting that branched sugars [70] are formed under these conditions.
We did not observe any conversion to higher carbohydrates within a day when only formaldehyde (C1 sugar) was employed. It can be, hence, concluded that schreibersite is not capable of activating formaldehyde which would require an Umpolung reactions. It is being consumed, though, in the presence of higher sugars (see figure 4(D)). In our cross-reaction experiments (see figures 4(D) to (F)), we could, further, observe a predominance for the formation of C5 sugars with the biologically relevant ribose being a minor constituent. C6 sugars formed were primarily ketoses rather than aldoses.
As it is known that the product distribution of the formose reaction changes with time, we examined the generation of higher carbohydrates for the glycolaldehyde sample after 1 and 7 d. The results are depicted in figure 5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Product distribution of schreibersite induced formose type reaction after 1 and 7 d. Greek labeling denotes the following compounds: γ—erythrose, δ—threose, ε—erythrulose. Higher sugars cannot be quantified individually as a result of overlapping peaks. Therefore, the bulk is quantified. For reaction conditions see section 2.4.3. Relative errors of abundance are on average 1% and are omitted for clarity.
Download figure:
Standard image High-resolution image{kind=link}
The results of this study are in agreement with previous observations of the formose reaction [70, 72]. Under prolonged reaction time the product distribution changes in favor of higher sugars. Also, more retro-aldol products such as C5 sugars become abundant and substrate is transferred into higher molecular structures resulting in a tar like material of low water solubility. Compared to analogous reaction conditions, however, the formation of tar and the associated decomposition of carbohydrates set in significantly later with schreibersite [70, 72]. The conditions introduced by the corrosion process presumably pose a mild environment in which the generation of sugars even over longer period of time is facilitated.
All identified and detected unbranched sugars of the performed experiments can be found in the supporting information (table S3).
4. Discussion
4.1. Monitoring of schreibersite corrosion
We could show that CE is a suitable method for monitoring the corrosion process of artificial schreibersite material as sample consumption is minimal and pretreatment unnecessary. Separations were performed under neutral conditions (pH 6.2) to exclude any bias connected to the pH sensitivity of phosphorus oxyanions. Our results on the composition of the aqueous phase of the Fe3P corrosion are in general agreement with previously published results. Main constituents are phosphite  and phosphate
and phosphate  [56, 59, 62]. In contrast to earlier work, we could not detect pyrophosphate or hypophosphate but diphosphite
[56, 59, 62]. In contrast to earlier work, we could not detect pyrophosphate or hypophosphate but diphosphite  In case of the former, this might be due to a lack of sensitivity. Limits of detection with the indirect UV-detection are around 10–100 μM. In the latter case, it is assumed that diphosphite
In case of the former, this might be due to a lack of sensitivity. Limits of detection with the indirect UV-detection are around 10–100 μM. In the latter case, it is assumed that diphosphite  has not yet been observed as it hydrolyzes with a half-life of 3 min at 20 °C in 0.1 N NaOH solution [82, 83]. The general procedure of detection and quantification of corrosion products in literature, though, involves treatment of the samples with either NaOH or Na2S in order to remove Fe(II) and allow NMR analysis. This practice significantly raises the pH value and would quench any available diphosphite
has not yet been observed as it hydrolyzes with a half-life of 3 min at 20 °C in 0.1 N NaOH solution [82, 83]. The general procedure of detection and quantification of corrosion products in literature, though, involves treatment of the samples with either NaOH or Na2S in order to remove Fe(II) and allow NMR analysis. This practice significantly raises the pH value and would quench any available diphosphite  On that note, it shall be highlighted that also phosphite
On that note, it shall be highlighted that also phosphite  is prone to hydrolysis generating phosphate
is prone to hydrolysis generating phosphate  and molecular hydrogen, but this process only becomes relevant at high temperatures [84]. The differences in observations of NMR based methods and our approach is supported by Foster et al They performed ion chromatography of the corrosion products without pretreatment of the samples and could not detect diphosphate
and molecular hydrogen, but this process only becomes relevant at high temperatures [84]. The differences in observations of NMR based methods and our approach is supported by Foster et al They performed ion chromatography of the corrosion products without pretreatment of the samples and could not detect diphosphate  and hyphosphate
and hyphosphate  either [63].
either [63].
Besides, the results of our CE method provide insight into the progress of the corrosion process for different temperature. Whereas the phosphate  concentration increases sharply in the beginning and then decreases to stagnate at a plateau of around 0.5 mM, concentrations of phosphite
concentration increases sharply in the beginning and then decreases to stagnate at a plateau of around 0.5 mM, concentrations of phosphite  and diphosphite
and diphosphite  increase steadily over time for corrosions performed at 80 °C. At 20 °C diphosphite
increase steadily over time for corrosions performed at 80 °C. At 20 °C diphosphite  reaches a steady state concentration already after around 7 days while phosphite
reaches a steady state concentration already after around 7 days while phosphite  keeps increasing slowly. The immediate rise of the phosphate
keeps increasing slowly. The immediate rise of the phosphate  concentration in the beginning regardless of the temperature is attributed to the already oxidized surface of the material containing phosphate
concentration in the beginning regardless of the temperature is attributed to the already oxidized surface of the material containing phosphate  when stored under air. The advanced decrease of the concentration can be explained by the low solubility of iron(II) phosphate that over the cause of the corrosion is formed and depleting the solution of phosphate
when stored under air. The advanced decrease of the concentration can be explained by the low solubility of iron(II) phosphate that over the cause of the corrosion is formed and depleting the solution of phosphate  The more soluble phosphite
The more soluble phosphite  on the other hand, accumulates.
on the other hand, accumulates.
The immediate release of phosphate  is, also, held responsible for the observed quick rise of the pH value in the beginning of the corrosion. Such an increase can only be explained by phosphate
is, also, held responsible for the observed quick rise of the pH value in the beginning of the corrosion. Such an increase can only be explained by phosphate  in its tribasic state (as in Na3PO4). As the mechanism of the corrosion is still elusive in detail, we argue that any phosphorus oxy compound formed will be in its fully deprotonated form as free protons of any acidic product would be reduced immediately by either iron or phosphorus in Fe3P to form molecular hydrogen. The determined concentrations of phosphate
in its tribasic state (as in Na3PO4). As the mechanism of the corrosion is still elusive in detail, we argue that any phosphorus oxy compound formed will be in its fully deprotonated form as free protons of any acidic product would be reduced immediately by either iron or phosphorus in Fe3P to form molecular hydrogen. The determined concentrations of phosphate  in the corrosion process match the observed pH values in this context. A concentration of 2 mM tribasic phosphate
in the corrosion process match the observed pH values in this context. A concentration of 2 mM tribasic phosphate  as in the beginning of the corrosion experiments when other possibly buffering species are absent equals a pH value of around 11 as was observed. We believe that the basifying conditions of schreibersite have yet not been described in literature since experiments were performed with little material in 25–50 times the water volume we employed in our experiments. Such a dilution corresponds to a decrease in pH by 1.5 units. As the pH declines over time to a value of around 9 due to buffering reactions by iron (formation of insoluble Fe(OH)2), published experiments likely observed a neutral pH of around 7.5.
as in the beginning of the corrosion experiments when other possibly buffering species are absent equals a pH value of around 11 as was observed. We believe that the basifying conditions of schreibersite have yet not been described in literature since experiments were performed with little material in 25–50 times the water volume we employed in our experiments. Such a dilution corresponds to a decrease in pH by 1.5 units. As the pH declines over time to a value of around 9 due to buffering reactions by iron (formation of insoluble Fe(OH)2), published experiments likely observed a neutral pH of around 7.5.
4.2. Screening for phosphorylation and condensation to polyphosphates
The conducted reactions of organics in presence of schreibersite corrosion have shown no detectable amounts of both phosphorylated products or condensed phosphates. This demonstrates the low reactivity of the meteoritic material as is also reflected in the low yields of published but successful phosphorylation reactions with nucleosides [61] and glycerol [60]. It is very likely that the efficiency of those reactions could be enhanced by reducing the amount of water present. Water serves as a quenching agent for reactive phosphorus oxyradical intermediates [59] and, thus, hinders phosphorylation. As phosphorylation reactions in water are disfavored thermodynamically, such an approach might be beneficial in many regards.
4.3. Schreibersite catalyzed formose reaction
As discussed in section 4.1., corroded schreibersite material basifies the aqueous phase in which it is immersed by creating a buffer composed of phosphite  phosphate
phosphate  and iron(II) ions (Fe2+). In such a medium, we observed that simple sugars react in an aldol reaction scheme to generate higher carbohydrates. This formose reaction network is a known abiotic pathway to biologically relevant sugars. We, therefore, tested the reaction with formaldehyde, glycolaldehyde, glyceraldehyde and dihydroxyacetone all of which have been linked to terrestrial [8, 85] and extraterrestrial [11, 86–88] origins. In all cases except for pure formaldehyde, we found typical formose network product distributions after one day. The fact that formaldehyde did not show any reactivity under the given conditions is in full agreement with previous observations in literature [70]. In order to activate formaldehyde, it has to undergo an Umpolung reaction which cannot be achieved by base catalysis. Formaldehyde, however, is being consumed in the presence of an organic co-catalyst. Any organic molecule with the ability to enediolize like glycolaldehyde can serve as such. Therefore, in cross-experiments that would constitute formaldehyde and glycolaldehyde we observed a full consumption of the carbon material enriching specifically C5 sugars. As typical of formose reaction networks the conversion itself shows little selectivity, and biologically important carbohydrates such as ribose are only minor components. It has been shown, though, that phosphorylation of carbohydrates drastically influences the stability of reaction products and, hence, changes the formose network outcome [77, 78]. In our future experiments, we will, therefore, examine possibilities to realize the generation and phosphorylation of sugars simultaneously by employing Fe3P since the corrosion of this meteorite material creates a phosphor oxyanion enriched milieu and, thus, supplies all the necessary ingredients for such a reactivity. Unlike any other catalyst in the context of the formose reaction network studied so far in literature this is a unique feature of Fe3P. Schreibersite, also, belongs to the few likely members of a hadean mineral inventory capable of triggering this network besides examples such as Al(OH)3, Ca(OH)2 and borates [36, 72]. Furthermore, we showed that Fe3P creates a reaction environment in which the generation of carbohydrates as well as the formation of tar occur more slowly in comparison to published experiments [72]. Stabilizing effects of the phosphate
and iron(II) ions (Fe2+). In such a medium, we observed that simple sugars react in an aldol reaction scheme to generate higher carbohydrates. This formose reaction network is a known abiotic pathway to biologically relevant sugars. We, therefore, tested the reaction with formaldehyde, glycolaldehyde, glyceraldehyde and dihydroxyacetone all of which have been linked to terrestrial [8, 85] and extraterrestrial [11, 86–88] origins. In all cases except for pure formaldehyde, we found typical formose network product distributions after one day. The fact that formaldehyde did not show any reactivity under the given conditions is in full agreement with previous observations in literature [70]. In order to activate formaldehyde, it has to undergo an Umpolung reaction which cannot be achieved by base catalysis. Formaldehyde, however, is being consumed in the presence of an organic co-catalyst. Any organic molecule with the ability to enediolize like glycolaldehyde can serve as such. Therefore, in cross-experiments that would constitute formaldehyde and glycolaldehyde we observed a full consumption of the carbon material enriching specifically C5 sugars. As typical of formose reaction networks the conversion itself shows little selectivity, and biologically important carbohydrates such as ribose are only minor components. It has been shown, though, that phosphorylation of carbohydrates drastically influences the stability of reaction products and, hence, changes the formose network outcome [77, 78]. In our future experiments, we will, therefore, examine possibilities to realize the generation and phosphorylation of sugars simultaneously by employing Fe3P since the corrosion of this meteorite material creates a phosphor oxyanion enriched milieu and, thus, supplies all the necessary ingredients for such a reactivity. Unlike any other catalyst in the context of the formose reaction network studied so far in literature this is a unique feature of Fe3P. Schreibersite, also, belongs to the few likely members of a hadean mineral inventory capable of triggering this network besides examples such as Al(OH)3, Ca(OH)2 and borates [36, 72]. Furthermore, we showed that Fe3P creates a reaction environment in which the generation of carbohydrates as well as the formation of tar occur more slowly in comparison to published experiments [72]. Stabilizing effects of the phosphate  phosphite
phosphite  and iron(II) enriched environment will be the focus of future studies. In any event, our studies present indication that schreibersite could be involved in a prebiotic scenario that gives rise to enriched aqueous phases of accessible dissolved phosphorus and carbohydrates simultaneously. As those are significant ingredients for metabolism, schreibersite could be conceived as a trigger for chemical reaction networks to form and chemically evolve to complex self-sustaining systems.
and iron(II) enriched environment will be the focus of future studies. In any event, our studies present indication that schreibersite could be involved in a prebiotic scenario that gives rise to enriched aqueous phases of accessible dissolved phosphorus and carbohydrates simultaneously. As those are significant ingredients for metabolism, schreibersite could be conceived as a trigger for chemical reaction networks to form and chemically evolve to complex self-sustaining systems.
5. Conclusion
In the present contribution, we established a separation method based on CE to monitor unbiasedly the corrosion process of schreibersite and report on the ability of the material to catalyze the formation of carbohydrates from prebiotic precursors in a formose type reaction. The method we present distinguishes itself from previously reported approaches as it does not require any pretreatment steps that are believed to adulterate the product distribution. This is due to the severe pH sensitivity of phosphorus oxyanions. The introduced method utilizes pH neutral conditions (pH 6.2) and is performed within less than 12 min. With it we could detect phosphite  phosphate
phosphate  and presumably diphosphate
and presumably diphosphate  We, further, recorded their concentration profiles for corrosions performed at 20 °C and 80 °C and found that phosphite
We, further, recorded their concentration profiles for corrosions performed at 20 °C and 80 °C and found that phosphite  is the major component accumulating over time. Based on those findings, we tested the corrosion process in the presence of different organics aiming at triggering the formation of either polyphosphates or phosphorylated organic molecules. We, however, could not find any hints for the success in those reactions but observed in carbohydrate samples the formation of higher sugars. Further studies revealed that schreibersite basifies the water in which it is immersed which together with released Fe(II) cations catalyzes reactions in the formose reaction network. This reaction is a prebiotically route to biologically relevant sugars. We could show that all simple carbohydrates like formaldehyde (only in presence of organic co-catalyst), glycolaldehyde, glyceraldehyde and dihydroxyacetone are susceptive to the reaction conditions introduced by the corrosion of schreibersite. In the reaction samples that contained mixtures of the given feedstock molecules preferably C5 sugars such as the biologically relevant ribose were formed, though ribose was only a minor constituent. In conclusion, our results demonstrate that schreibersite efficaciously catalyzes the aldol reactions in the formose reaction network while creating a phosphorus enriched milieu at the same time. It is the only known member of the hadean eon mineral inventory to show that ability. Our findings, thus, implicate a prebiotic scenario in which phosphorus and sugar chemistry are connected. It poses one of the few instances in literature so far in which interplanetary material is directly involved in prebiotic chemistry on the early earth.
is the major component accumulating over time. Based on those findings, we tested the corrosion process in the presence of different organics aiming at triggering the formation of either polyphosphates or phosphorylated organic molecules. We, however, could not find any hints for the success in those reactions but observed in carbohydrate samples the formation of higher sugars. Further studies revealed that schreibersite basifies the water in which it is immersed which together with released Fe(II) cations catalyzes reactions in the formose reaction network. This reaction is a prebiotically route to biologically relevant sugars. We could show that all simple carbohydrates like formaldehyde (only in presence of organic co-catalyst), glycolaldehyde, glyceraldehyde and dihydroxyacetone are susceptive to the reaction conditions introduced by the corrosion of schreibersite. In the reaction samples that contained mixtures of the given feedstock molecules preferably C5 sugars such as the biologically relevant ribose were formed, though ribose was only a minor constituent. In conclusion, our results demonstrate that schreibersite efficaciously catalyzes the aldol reactions in the formose reaction network while creating a phosphorus enriched milieu at the same time. It is the only known member of the hadean eon mineral inventory to show that ability. Our findings, thus, implicate a prebiotic scenario in which phosphorus and sugar chemistry are connected. It poses one of the few instances in literature so far in which interplanetary material is directly involved in prebiotic chemistry on the early earth.
Acknowledgments
Generous financial support by the Max-Planck-Society is appreciatively acknowledged. SP thanks the Fonds der Chemischen Industrie for a PhD fellowship. JS thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. Jacob Lucian Gorenflos López is acknowledged for support in developing the GC separation. The authors, further, thank Dr Steffen Schmidt (Professor Dr Bein, LMU) for conducting the SEM-EDX measurements.
Author contributions
SP and OT jointly designed the research project and experiments. SP performed, analyzed and evaluated all corrosion experiments. JS developed CE separation method and MH as well as SL developed GC separation method. JS and SP examined and evaluated the kinetics of the schreibersite corrosion in water and performed reactivity studies with organics. AH measured and evaluated XPS of Fe3P. SP wrote the paper. OT initiated and oversaw the project.