




Abstract
Early protocells are commonly assumed to consist of an amphiphilic membrane enclosing an RNA-based self-replicating genetic system and a primitive metabolism without protein enzymes. Thus, protocell evolution must have relied on simple physicochemical self-organization processes within and across such vesicular structures. We investigate freeze-thaw (FT) cycling as a potential environmental driver for the necessary content exchange between vesicles. To this end, we developed a conceptually simple yet statistically powerful high-throughput procedure based on nucleic acid-containing giant unilamellar vesicles (GUVs) as model protocells. GUVs are formed by emulsion transfer in glass bottom microtiter plates and hence can be manipulated and monitored by fluorescence microscopy without additional pipetting and sample handling steps. This new protocol greatly minimizes artefacts, such as unintended GUV rupture or fusion by shear forces. Using DNA-encapsulating phospholipid GUVs fabricated by this method, we quantified the extent of content mixing between GUVs under different FT conditions. We found evidence of nucleic acid exchange in all detected vesicles if fast freezing of GUVs at −80 °C is followed by slow thawing at room temperature. In contrast, slow freezing and fast thawing both adversely affected content mixing. Surprisingly, and in contrast to previous reports for FT-induced content mixing, we found that the content is not exchanged through vesicle fusion and fission, but that vesicles largely maintain their membrane identity and even large molecules are exchanged via diffusion across the membranes. Our approach supports efficient screening of prebiotically plausible molecules and environmental conditions, to yield universal mechanistic insights into how cellular life may have emerged.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Comprehensively defining living matter is difficult [1]. Yet, all living species are capable of decreasing internal entropy and increasing functional complexity at the expense of substances or free energy absorbed from the environment. In order for cellular life to develop this characteristic complexity, even primitive protocells must have compartmentalized their molecular components to separate themselves from each other and the environment, allowing them to maintain and regulate biochemical processes (i.e., develop a metabolism), effectively couple their cellular phenotype to their genotype and prevent takeover by parasitic mutants. This compartment boundary consists of lipid membranes in all known living entities. At the same time, protocells needed to evolve mechanisms to regulate material, energy and information exchange between themselves and their surroundings.
Exchange of material across (sub)cellular compartments is commonly mediated by either proteinaceous membrane channels and transporters, or by compartment fusion and fission events mediated by complex protein machineries. Similarly, growth and division in modern cells is a tightly regulated process that involves genome replication and partitioning, culminating in the separation of new daughter cells through fission of the mother cell compartment. Given the intricate nature of these complex biological processes, it remains a mystery how the first precursors of our modern cells, doubtlessly much more primitive entities before the advent of proteins, could have exchanged material—itself a prerequisite for the emergence of life.
While numerous compartment systems have been proposed as the first autopoietic units [2], the most prominent protocellular models thought to provide a plausible historic route to modern cells are based on membrane vesicles formed by amphiphiles such as lipids [3], which encapsulate a primitive metabolism and an RNA-based self-replicating genetic system [4]. Typical models for the replication of such primitive vesicles accommodate growth either by uptake of micelles or free amphiphiles, or by vesicle-vesicle fusion events followed by division through vesicle fission [5]. Out of these models, self-reproduction based on micelle feeding has been extensively studied. It typically involves formation, growth, and self-reproduction of buffered, submicrometre vesicles by uptake of alkaline micelles formed by simple fatty acids such as oleic acid [6–13]. In a further expansion of this model, reproduction of multilamellar vesicles composed of prebiotically plausible fatty acid and lipid mixtures was shown to occur through a series of alkaline micelle addition, filamentous growth, and gentle agitation with minimal content leakage of encapsulated RNAs [14]. Primitive replication cycles of vesicles can also be induced by increasing lipid concentration through evaporation and shear-forcing or photochemically induced division [15]. It was hypothesized that RNA replication inside vesicles could lead to an increased osmotic pressure and thus growth at the expense of empty vesicles [16]. Such larger, multilamellar vesicles could then undergo division in environments of gentle shear [14]. However, although isolated aspects of this proposed mechanism have been shown, a combined experimental demonstration of intravesicular RNA replication, growth and division remains elusive. In contrast, proliferation via feeding of a protocell with RNA building blocks by content exchange with 'feedstock' vesicles provides a simple but robust alternative scenario for early protocell replication [17–19]. The required driving force for vesicle encapsulation and content exchange can be provided by environmental conditions such as freeze-thaw (FT) cycling [20–23]. Indeed, repeated FT-induced mixing of vesicle contents by vesicle fusion was recently used to enable sustainable proliferation of liposomes with encapsulated protein expression and protein-catalysed RNA replication [24]. These observations suggest that FT-cycling, among other plausible periodic physicochemical processes such as rehydration and dehydration [25], could have promoted primitive cycles of growth and division of protocells.
An attractive experimental system to study such proliferation scenarios are giant unilamellar vesicles (GUVs), i.e. single bilayer liposomes that are larger than 10 μm. Given their size, GUVs can be directly observed by light microscopy. However, preparation of GUVs encapsulating large biomolecules is experimentally challenging. Commonly used experimental procedures, such as controlled swelling [26, 27] and electroformation [28], complicate handling and content encapsulation.
Here, we present a new high-throughput methodology for homogeneous production of nucleic acid-containing GUV populations by water-in-oil emulsion transfer in a microtiter plate [29]. Our protocol allows for direct exposure of GUVs to different environmental conditions inducing content mixing, without any intermediate sample pipetting steps, and for these processes to be directly monitored by imaging. This reduction of handling steps strongly minimizes potential artefacts introduced by sample manipulation, such as unintended GUV rupture or fusion by shear forces. Using GUVs made of 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) as a model system, we have tested different FT-conditions for their potential to drive content exchange across vesicles. We found that fast freezing induced by placing the sample on a metal block at −80 °C, followed by slow thawing at room temperature resulted in efficient nucleic acid exchange in all detected vesicles without any observable vesicle fusion events.
Thus, our results offer an attractive explanation of how intravesicular content, including primitive RNA replicators, could have spread within vesicle populations independently from vesicle fusion and fission events using transient periods of increased membrane permeability.
In the future, our method will allow the rapid exploration of vesicle properties for GUVs made with different amphiphiles and buffer systems, as well as allow the efficient screening of environmental conditions for their potential to induce GUV-based protocell content exchange and propagation.
Methods
GUV preparation
Our method for generating GUVs is based on emulsion transfer in microtiter plates [29, 30]. In contrast to common protocols, we both generated and imaged GUVs in glass bottom microtiter plates, enabling high-throughput parameter optimization. Since the same 96-well plate is used for all experimental steps including GUV generation, freeze-thawing and subsequent imaging, pipetting steps are minimized.

Figure 1. Schematic representation of the GUV generation process. (a) Microtiter plate with wells containing a layer of mineral oil (with POPC lipids) on top of glucose solution. After 30 min of incubation, a lipid monolayer has formed at the water–oil interface. (b) Micro test tubes with the same lipid-in-oil mixture layered over a sucrose solution containing each a differently fluorescently labelled oligonucleotide. (c) Water-in-oil emulsion generated by mechanical agitation. (d) Both emulsions were pipetted in a 1:1 ratio onto the oil phase in microtiter plate wells. (e) GUVS are generated by moving the droplets through the lipid-saturated water–oil interface (emulsion transfer).
Download figure:
Standard image High-resolution imageFirst, POPC (1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids, Inc.) was dissolved in chloroform at a concentration of 8 mM. The solution (75 μl) was then transferred to a 4 ml glass vial and the chloroform was evaporated under argon flow (15 min), followed by removing residual solvent traces by applying a vacuum (1 h). After addition of 1.5 ml mineral oil (Sigma-Aldrich, M5904), the vial was sonicated for 1 h at 40 °C, and incubated overnight at room temperature to dissolve the lipids in the oil.
The glass bottom microtiter plates (Greiner Bio-One, 96-well glass bottom SensoPlate™) were passivated with Pluronic F-127 (Sigma-Aldrich): after plasma-cleaning the microtiter plate, 50 μl of 20 mg ml−1 Pluronic F-127 were added to each well of the plate intended to hold GUVs and incubated for 30 min at room temperature, followed by a wash with 900 mM glucose solution (50 μl). 100 μl of the same glucose solution was then pipetted into each well (outer aqueous phase containing future GUVs) and POPC-mineral oil solution (40 μl) was layered on top, followed by incubation for 30 min at room temperature to form a monolayer at the water–oil interface (figure 1(a)).
To create two distinct fluorescently labelled populations of GUVs, we prepared internal GUV solutions (900 mM sucrose) with DNA oligonucleotides (26 bases (13 repetitions of 5'-CA-3'), IDT) either labelled with a 5' Cy5 fluorophore (4 μM) or a 5' 6-FAM fluorophore (8 μM).
To generate the water-in-oil emulsion for the emulsion transfer, 5 μl of each inner phase solution (each containing one of the two oligonucleotides) was added to 250 μl of the POPC-mineral oil solution in a 0.5 ml micro test tube (figure 1(b)). Droplets were formed by shear forces through mechanical agitation (i.e. repeatedly rubbing each tube for about 1 min across the array of cavities on a micro test tube rack) (figure 1(c)), then 40 μl of both emulsions (80 μl in total) were immediately pipetted into each well of the microtiter plate (figure 1(d)). The microtiter plate was subsequently centrifuged for 3 min at 250 g (Eppendorf Centrifuge 5804 R) in a rotor designed to hold microtiter plates (Eppendorf A-2-DWP) to generate GUVs (figure 1(e)).
All steps that involve handling of the hygroscopic mineral oil, except for the last centrifugation step, are carried out in a glove box under nitrogen flow and a humidity of 3%–8% to minimize the amount of water in the organic phase. While this measure generally increased vesicle yields drastically, it proved to be crucial in warm summer months with indoor humidities of more than 50%, when we sometimes did not see any vesicle formation if samples were handled at ambient conditions.
For the experiments in figures 4, S6 and S7 membrane labels were used in the following mole fractions of membrane label to lipids: 0.013 mol% DOPE-ATTO488 (1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine labelled with ATTO488) (ATTO-TEC GmbH); 0.017 mol% DiD (DiIC18(5)) and DiI (DiIC18(3)) (Thermo Fisher Scientific). The labelled dextrans (3000 MW and 40 000 MW) were both used in a concentration of 0.08 mg ml−1.
Freeze-thawing
Three different FT protocols were used, enabling fast freezing and fast thawing (condition A), slow freezing and slow thawing (condition B) and fast freezing followed by slow thawing (condition C). We only observed very few intact vesicles after slow freezing and fast thawing (condition D; figure S3), and therefore did not include this condition in our further investigations and analysis.
Prior to freezing, the microtiter plates containing GUVs were sealed with a sealing film (Carl Roth Rotilabo®-sealing film for micro test plates) and were positioned at a slight angle for 5–10 min, to allow the GUVs to accumulate on one side of the well.
For condition A, a 10 cm × 6 cm × 6 cm block made of stainless steel was placed in a −80 °C freezer. After the metal block reached the temperature of the freezer, the microtiter plate was placed on top and pressed firmly against it for about 1 min. To allow direct contact with the glass bottom, the metal block must be smaller than the bottom frame of the microtiter plate. After 30 min, the plate and the metal block were taken out of the freezer. The plate was removed from the first metal block and placed onto a second block preheated to 60 °C on a hot plate. After 5 min of thawing at 60 °C, the plate was transferred to the microscope for imaging (see figure 2(b)).
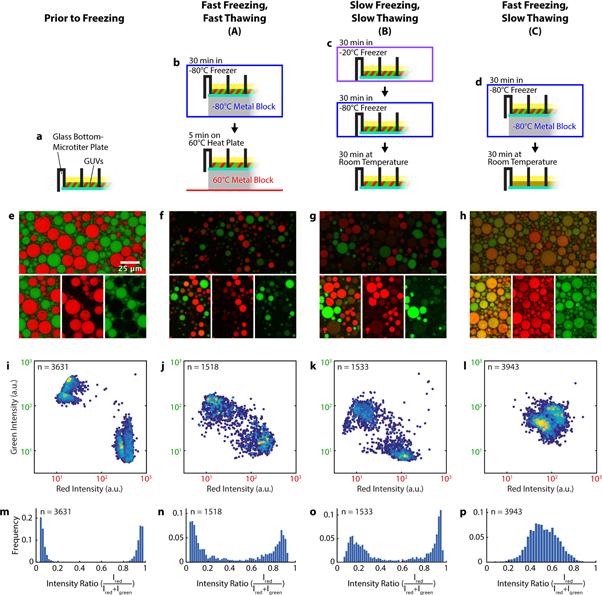
Figure 2. Comparison of different FT-conditions. (a)–(d) Schematic representations of the freeze-thaw protocols for each condition. (e)–(h) Confocal images of the GUVs, fluorescence intensity of Cy5-DNA is shown in red, fluorescence of 6-FAM-DNA in green. The top image corresponds to an unedited image taken with identical microscope settings and contrast settings across all conditions. The contrast of the images underneath (smaller sections of the top image) has been individually adjusted, shown is both a merged image and each individual channel. (i)–(l) Intensity scatter plots, Cy5 fluorescence intensity (red) is plotted against 6-FAM fluorescence intensity (green) on a logarithmic scale. Each data point represents an individual vesicle. The colour of each point reflects the density of points in the region similar to a heat map, from dark blue (low) to yellow (high). Number of experimental repeats was 5 for (i) and 4 for (j)–(l). (m)–(p) Histograms of the ratio of red intensity over total intensity for each of the respective conditions.
Download figure:
Standard image High-resolution imageFor condition B (figure 2(c)), the microtiter plate was placed into a −20 °C freezer at a slight angle for 30 min, and subsequently into a −80 °C freezer for another 30 min. Afterwards the GUVs were thawed at room temperature for 30 min.
Condition C is a combination of the previous two. The GUVs were frozen for 30 min at −80 °C on a metal block and subsequently thawed for 30 min at room temperature (see figure 2(d)).
Imaging
Imaging was performed with an LSM 780/CC3 confocal microscope (Carl Zeiss, Germany) equipped with a C-Apochromat, 40x/1.2 W objective. We used PMT detectors (integration mode) to detect fluorescence emission (excitation at 488 nm for 6-FAM and 633 nm for Cy5) and record confocal images. To capture many GUVs in high resolution, tile scans were performed by recording a defined number of adjoining single images of the sample (the 'tiles'), moving the sample using motorized stages with high precision. Following the scanning process, the mosaic image is assembled using the stitching algorithms provided by the Zeiss software.
Analysis
Fluorescence images were analysed using custom software written in MATLAB (MATLAB R2014b, The MathWorks, Inc.) adapted from Ranasinghe et al [31]. Briefly, vesicles were detected (i.e. regions of interest identified) in each field of view across two images sequentially acquired for each colour channel ('red' and 'green') by converting both images to a binary image (mask) using a threshold intensity set to be the mean single pixel intensity of the bandpass-filtered images plus 0.5–2 times this value. This factor was determined empirically by visual inspection of the binary image obtained and comparison with the original data. In the next step, objects (vesicles) are detected by tracing continuous regions of ones in the binary image. Signals are assumed to correspond to single vesicles if the area of the region is >10.8 μm2 (250 pixel). The mean intensity for the centres of each region/vesicle (i.e. the originally detected region minus a shell of 5 pixel) are then calculated from both original images for vesicles detected in either of the two channels. Finally, the intensities are plotted in scatter plots coloured according to data density (figures 2(i)–(l)) and the ratio of red/green intensity was calculated for each vesicle.
MATLAB code for analysing image data is available online.
Results
Generating heterogeneous GUV populations by emulsion transfer in microtiter plates
To study the effect of FT-cycling on vesicle content mixing, we first developed a method to simultaneously generate GUVs of differing content via emulsion transfer in microtiter plates, thus greatly improving experimental throughput and parallelizing the screening for optimal conditions. Briefly, two distinct populations of GUVs were obtained by (i) preparing water-in-oil droplets from two solutions, each containing a different fluorescently labelled oligonucleotide. These two emulsions were then (ii) mixed and (iii) passed through a lipid-lined oil–water interface upon which GUVs were formed (figure 1).
We found the surface passivation of the microtiter plate chambers to be a crucial factor in our experiments. We tested β-casein (2 and 5 mg ml−1), PLL-g-PEG (0.5 and 2 mg ml−1) and Pluronic F-127 (1–50 mg ml−1) as passivation agents in combination with various subsequent washing protocols. Vesicle generation in β-casein-passivated chambers was successful, but only very few vesicles remained intact after the FT cycles. PLL-g-PEG seemed promising while working with vesicles that did not contain oligonucleotides, but after incorporating labelled oligonucleotides, the experiments failed. Pluronic F-127 at high concentrations (10–50 mg ml−1) was the only passivating agent to work under relevant conditions. In contrast to the other passivation methods, we also observed the highest overall vesicle yield and no aggregation on the glass surface.
GUV content mixing efficiency depends on both the freezing and thawing rate
With our optimized microtiter plate protocol in hand, we investigated how the yield of content mixing upon freeze-thawing vesicles depended on the heating and cooling rates of the FT process. We tested three different FT protocols with variations in heating and cooling rates. Firstly, the sample was rapidly frozen at −80 °C and rapidly thawed at 60 °C, in both cases maximizing heat exchange by placing the plate on a metal surface (condition A: fast freezing/fast thawing). Secondly, the sample was cooled slowly to −20 °C before cooling it further to −80 °C, then slowly thawed at room temperature (condition B: slow freezing/slow thawing). Thirdly, we combined rapid freezing (step 1 from condition A) with slow thawing (step 2 from condition B) such that fast freezing is followed by slow thawing (condition C). The remaining combination (condition D: slow freezing/fast thawing) is not covered in our detailed analysis, since it resulted in almost no vesicles after the FT process (figure S3).
We applied these FT-cycles to microtiter plates containing mixtures of GUVs encapsulating 26 bp DNA oligos either labelled with Cy5 or with 6-FAM, and recorded two-colour images of the GUVs by confocal microscopy, both before (figure 2(e)) and after the FT cycle (figures 2(f)–(h)). We then used an automated, custom image analysis routine to extract the fluorescence intensities for individual GUVs from the image data.
For conditions A–C, we observed that the majority of GUVs contained both DNAs after a FT cycle, whereas before freezing, two distinct populations of vesicles could clearly be distinguished (figures 2(i)–(l)). The efficiency of this mixing of contents varied between the different FT protocols. This content mixing is accompanied by an overall decrease in GUV fluorescence intensity in both colour channels (figures 2(i)–(l), figure S1).
Under condition A (fast/fast), the ratios of green to red intensities were shifted compared to the control for many GUVs, indicating content mixing. However, two populations of GUVs which either contain more Cy5- or 6-FAM-DNA can still be distinguished (figures 2(j) and (n)), indicating that the mixing is incomplete. Only a small population of GUVs shows approximately equal intensities for both dyes, and hence contains approximately equal amounts of oligonucleotides (figures 2(f), (j) and (n)).
Similarly, condition B (slow/slow) resulted in the majority of GUVs containing a highly uneven ratio of fluorescence signals, and thus an uneven ratio of DNA concentrations (figures 2(k) and (o)). Compared to condition A, however, the overall intensity of these vesicles (and hence the concentration of their contents) was lower (figure S1).
Under condition C (fast/slow), content mixing was by far the most efficient. Most GUVs displayed approximately equal intensity levels for both dye-labelled DNAs (figures 2(l) and (p)), in stark contrast to the first two conditions. In addition, the combined fluorescence intensities from both DNAs contained within the GUVs were similar to those measured for GUVs subjected to condition A (figure S1). Protocol C did not seem to change vesicles numbers noticeably when we visually inspected the data, whereas GUV numbers decreased for protocols A, B and D with almost no intact GUVs remaining for condition D. Thus, condition C not only results in the most complete content mixing but also retains the most GUVs and the most content (labelled oligos) within those GUVs.
We note that after a FT cycle, the GUV size distribution is shifted to slightly smaller vesicle sizes for all tested conditions (figure S2), indicating that the FT cycle induced some degree of vesicle fragmentation.
Inner/outer phase exchange
While fast freezing and slow thawing of vesicles encapsulating differently labelled DNA oligos leads to efficient content mixing, the overall concentration of encapsulated DNA decreased after mixing. We thus investigated whether this noticeable content loss is due to exchange with the surrounding phase during the FT step. To quantify the extent of this exchange, we freeze-thawed GUVs containing 6-FAM-labelled oligos, which were surrounded by an outer phase containing Cy5-labelled oligos (figure 3(a)). While only background-level Cy5 signals were detected inside vesicles before freeze-thawing, the level increases notably after freeze-thawing for the tested conditions (figures 3(b) and (c)). Although this suggests some internalization of the outer phase during the FT process, the intensity of Cy5 measured inside GUVs after FT was still much lower than that measured in the outer phase (on average 18% of outer phase), whereas the average intensity of the encapsulated DNA dropped to ∼43% of the initial value for FT condition C (fast freezing, slow thawing) (figure 3(b)). Increased uptake of surrounding fluid is observed for condition B (figure S5(b)), which is consistent with the larger decrease of fluorescence intensity observed for content mixing between GUVs under this condition (figure 2(k)).

Figure 3. Uptake of the surrounding phase into GUVs by freeze-thawing. (a) Vesicles containing 6-FAM-labelled oligos in the inner phase and Cy5-labelled oligos in the outer phase. (b) Vesicles after a FT cycle according to condition C (figure 2). The GUVs show uptake of the outer phase and some content loss, the extent of which depended on local GUV densities (increasing from left to right, see (d)). (c) Scatterplot of red/green intensities for (a) and (b) and for GUVs after freeze-thawing according to condition B (see figure S5(b)). The horizontal axis indicates Cy5 fluorescence intensity, while the vertical axis indicates 6-FAM fluorescence intensity on a logarithmic scale. Each point represents an individual vesicle. (d) Cy5 and 6-FAM intensities (i.e. one green and red data point per GUV) plotted against the horizontal locations of the GUVs. Lines represent linear fits. Out of 1124 data points, 23 outliers are outside of the y axis scale of this plot.
Download figure:
Standard image High-resolution imageIf content exchange between GUVs were to happen via content release and uptake (and diffusion of the content through the GUV-surrounding phase), we would expect that content exchange is more efficient at high GUV densities, as dilution of the content would be minimized. Conveniently, our FT-protocol involves tilting of the microtiter plates to an angle of 45° before freeze-thawing, which results in a GUV density gradient across the well (figure S5(a)). In areas with high GUV density, the immediate surrounding phase constitutes only a small fraction of the total volume. We therefore tested whether GUV content composition after freeze-thawing is dependent on the location of the GUVs within the well, and hence the local GUV density. In line with our hypothesis, figures 3(b) and (d) show that vesicles in high-density areas (left) keep more of their original content (6-FAM, green) and encapsulate less of the surrounding phase (Cy5, red) than vesicles in low-density areas (right). Thus, GUV density determines the degree GUV content loss and encapsulation of the surrounding phase.
Freeze-thawing does not induce membrane exchange between GUVs
To gain further insights into the mechanisms responsible for content exchange between vesicles, and to specifically test whether this exchange could, at least partially, be mediated by fusion and subsequent fission of vesicles, we performed our FT-cycling with two populations of GUVs with different membrane labels. The lumen of one GUV population also contained a fluorescently labelled cargo of various molecular sizes to allow for simultaneous monitoring of size-dependent content mixing. We generated the GUVs according to the protocol shown in figure 1, but used different lipid-in-oil solutions in the different steps to generate two GUV populations: one population with DOPE-ATTO488 in the membrane, but no labelled content; and a second population with DiD-labelled membranes and TMR-labelled 40 kDa dextran cargo (see figure 4(a) for a depiction of the protocol). Importantly, we found that if the time between mixing the two types of emulsions and the centrifugation step is reduced to a minimum, no significant amount of foreign membrane is incorporated into the membrane of the respective other population of GUVs.

{kind=link}
{kind=link}
{kind=link}
Figure 4. Freeze-thawing of GUV populations with differently labelled membranes. (a) We generated GUVs similarly to figure 1, but here the two populations of GUVs are not only distinct in their content, but also differ in their membrane composition. (b) GUVs before and after freeze-thawing. Blue: TMR-labelled dextran; red: DiD-labelled membrane; green: ATTO488-labelled membrane. Images on the left show all three channels, images on the right show the same sections with only the membrane channels. Prior to freeze-thawing, the two populations differ in membrane labelling and content. After fast freezing and slow thawing (protocol C), we observed that content exchange took place, but two populations of GUVs can still easily be distinguished by their membrane labelling, indicating that no exchange of membrane between GUVs occurred. More images and variations of these experiments are shown in figures S6 and S7, respectively. (c) Intensity profiles along the dotted lines in (b).
Download figure:
Standard image High-resolution image{kind=link}
When we freeze-thawed the GUVs according to protocol C, surprisingly we observed content mixing, while maintaining two populations of distinctly membrane-labelled vesicles (figure 4(b) and (c)). These data suggest that content exchange is possible even for large molecules up to 40 kDa without a significant number of membrane fusion and fission events. Figure S6 shows more images of the experiment. To validate these findings further, we performed a series of two-colour experiments in which we labelled the membrane of at least one population of GUVs with various membrane dyes (DOPE-ATTO488, DiD and DiI) and also used a variety of labelled cargo molecules (dextran 40 000 MW, dextran 3000 MW and the 6-FAM labelled Oligo from previous experiments). A comparison of confocal images before and after freeze-thawing for these experiments is shown in figure S7. In all cases, we saw a homogenization of contents, but could still distinguish the two populations of vesicles by their distinct membrane labels, or lack thereof.
We conclude from these experiments that GUVs mostly retain their membrane identity during the FT process, and that the cause for content exchange between vesicles is not fusion (and subsequent fission) of vesicles.
To investigate vesicle fate throughout the FT process in more detail, we attempted to follow the GUVs during the thawing process by time-lapse microscopy. Although imaging frozen samples throughout the entire thawing process proved unattainable with our experimental equipment, we were able to follow vesicle shape changes after thawing the sample for 20 min at room temperature. At this stage, the content of the complete sample well was fully thawed. However, numerous elongated vesicles could be observed, as well as their fission into smaller spherical GUVs over the following 5 min (figure S8(a)). While we assume that these events are not required for the content exchange between vesicles, the observations explain the shift to smaller vesicles after freeze-thawing (figure S2). Dynamic shape changes like these can be induced by vesicle deflation through osmotic water extraction. To explore whether changes in osmolarity during the FT process are a possible cause, we analysed the osmolarity in horizontal sections from wells that were frozen according to protocol C. We found a gradient of osmolarity with higher concentrations at the bottom of the well (supplementary figure S8(b)) which could be a cause for the GUV destabilization and subsequent fission.
Discussion
The purpose of this study was to develop and test a convenient but powerful methodology for the exploration of changing environmental conditions, such as FT-cycles, as drivers of content exchange between vesicle compartments. We found that mixing of contents in our POPC-GUV model system proceeded to varying degrees in conditions A–C. Condition C (fast freezing, slow thawing) resulted in the most uniform final state. Final GUVs for both A and B still predominantly contain only one of both DNA oligos, with only a very small fraction of resulting GUVs encapsulating approximately equal amounts of both oligos after FT.
Previously, Tsuji et al used a similar FT-protocol to induce content mixing of GUVs and concluded from their findings that content exchange between vesicles is caused by vesicle fusion and fission [24]. In contrast to our work, Tsuji et al centrifuged their GUVs at 18 000 g (more than 70× faster than the speed at which we generate our GUVs, 250 g) to 'increase fusion efficiency' and freeze their GUVs using liquid nitrogen in plastic micro reaction tubes with different heat conductivity and surface properties from our glass bottom wells [24]. A well-studied process is the FT-induced collective fusion of small unilamellar vesicles (SUVs) to form large unilamellar vesicles or GUVs [20, 32–34], but the conditions for SUV fusion are unlikely to be identical to those inducing GUV fusion given their different physical properties.
From our findings, we conclude that under our experimental conditions, vesicles exchange their contents independently from fusion and fission. Rather, content exchange occurs via membrane destablization, content leakage and uptake of content from the surrounding phase during the FT process. Three key observations strongly support this conclusion:
- (i)If the microtiter plate is handled carefully after thawing, such that the GUVs do not move within the wells, we observe a very homogeneous mixing of content on an intermediate range as figure S4 shows. Fusion of (few) neighbouring vesicles and their subsequent fission would likely lead to a higher local diversity of vesicle contents. Instead, on a small scale, all vesicles have approximately the same ratio of dyes and on a larger scale, smooth gradients regarding the vesicle content are visible, but rarely sharp contrasts. This indicates a very thorough mixing across tens to hundreds of vesicles.
- (ii)As described in figure 3(d) and the respective paragraph, the loss of content and the degree of exchange between GUVs and their surrounding phase seems to be dependent on the local density of GUVs. A model in which GUVs exchange their content via the outer phase explains this phenomenon, because a larger ratio of surrounding phase to GUV volume would dilute GUV content once GUVs become porous upon FT-cycling. Conversely, in regions of high vesicle densities, the surrounding phase contributes only to a small fraction of total volume, and hence only a correspondingly small fraction of GUV content is replaced by surrounding phase, as also observed in out experiments.
- (iii)The strongest support for our hypothesis that content exchange is not induced by fusion and fission comes from experiments showing that while mixing of GUV content is highly efficient, GUVs of different membrane compositions do not interchange lipids, and thus remain distinct entities during the process (figures 4, S6 and S7).
It is plausible that the GUVs become more permeable at temperatures slightly below the phase transition temperature of the bilayer lipids (Tm = −2 °C), especially if samples are thawed slowly (conditions B and C). At these temperatures, the GUV outer and inner phases have already thawed, due to the high sugar concentrations that lower the melting temperature. We think that this physicochemical state is restricted to the time when the samples start thawing at the glass bottom of the well (where most GUVs are located). When the upper layers of the well continue to thaw, the layer of GUVs at the glass bottom has already reached a temperature above the phase transition temperature of the lipids, rendering the GUVs impermeable to the DNA oligos or dextran again. A transient increase in membrane permeability during thawing, in combination with diffusion being restricted to the GUV-containing layer close to the glass bottom of the well, could explain how GUVs exchange their content via the surrounding phase without losing most of their content by dilution into the surrounding phase. Only when the entire well thawed, the vesicle content released into the surrounding phase between the vesicles is diluted by exchange with the large volume of outer phase above the GUV-containing layer.
For condition C, an exchange with the outer phase during the freezing step of the protocols is minimized, since freezing of the sample occurs within few seconds. More efficient exchange with the outer phase due to slow freezing explains the increased loss of content and encapsulation of outer phase observed in protocol B (see figures 3(c) and S5(b)). In fact, content loss was so high for condition B that many of the vesicles after freeze-thawing were not detected by our script and were only barely visible by eye.
In summary, we have shown that tightly packed GUVs, if subjected to a FT cycle, can transiently exchange content even in the absence of GUV fusion and fission. In simple protocellular systems based on lipid-encapsulated RNA replicators, such transient periods of content exchange could have facilitated the spread of protocells with functional genomes via direct transfer of replicators. Indeed, cycles of transient compartmentalization and mixing of RNA replicators were previously shown to be sufficient to prevent takeover by parasitic mutants [35]. Moreover, transient increases in membrane permeability could have facilitated uptake of RNA building blocks from other vesicles or the surrounding environment.
Our microtiter plate protocol allows high-throughput in situ imaging of many different conditions in parallel, without risking artefacts introduced by vesicle handling. While emulsion transfer allows the encapsulation of a large spectrum of biomolecules, it should be noted that the lipid bilayer of GUVs generated by the emulsion transfer may contain trace amounts of oil, changing their mechanical properties [36]. Therefore, we cannot exclude that results for GUVs generated by a different method might differ from ours.
In the future, our experimental approach will facilitate efficient screening of the vast parameter space of prebiotically plausible molecules and environmental conditions, to eventually derive unifying mechanistic insights into how cellular live may have evolved. It will be interesting to investigate the impact of the lipid phase transition temperature and test if, for example, DOPC (1, 2-dioleoyl-sn-glycero-3-phosphocholine), which has a much lower Tm of −17 °C compared to POPC (Tm = −2 °C), might inhibit content exchange between vesicles. Additionally, our method promises to explore the effect of freeze-thawing on vesicles made from chemically much simpler amphiphiles, such as prebiotically accessible fatty acids and monoacylglycerols [11, 37]. Finally, the microtiter plate layout of our experiments supports convenient future screening experiments e.g. of heterogeneous environments, such as minerals or different buffer conditions, which are compatible with nucleic acid catalysis and formation.
Acknowledgments
We would like to thank Tabea Kirchhofer and Tina Seemann for their help and advice with the microtiter plate emulsion transfer method. This work is part of the MaxSynBio consortium, which is jointly funded by the Federal Ministry of Education and Research of Germany and the Max Planck Society. Kristina Ganzinger has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 703132.