


Abstract
We have explored surface processes on Ti0.5Al0.5N(001) interacting with residual and environmental gases, namely O2, H2O and CO2, using ab initio molecular dynamics. Dissociative adsorption of O2 occurs on Ti sites, which are unusual sites, as Al2O3 is more stable than TiO2. This may be understood based on the electronic structure. We suggest that an increased Ti–O bond strength relative to Al–O surface bond strength is the electronic origin for the early stages of TiO2 formation on Ti0.5Al0.5N(001). Another unexpected atomic mechanism, identified as O covers the surface: Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, generating vacancies, and hence enabling mobility at the interface. In the case of H2O and CO2, the dominating physical mechanism is dissociative adsorption, where O–H and N–H as well as C–O and Ti–O dipoles are formed, respectively. These fundamental surface processes are relevant for initial stages of oxidation, surface diffusion and nucleation of reaction layers upon exposure to residual and environmental gases.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
GENERAL SCIENTIFIC SUMMARY Introduction and background. Predictive modelling in solid state physics requires detailed knowledge of all underlying fundamental mechanisms on the nanoscale. For instance, surfaces have to be considered when solids interact with another medium. However, very often these surface models are idealized, as surface structure and energetics are usually described in vacuum.
Main results. In this work, we consider a technologically important benchmark system utilised in surface protection: cubic metastable Ti1–xAlxN. Instead of assuming vacuum, we have explored surface processes on Ti0.5Al0.5N(001) interacting with residual and environmental gases, namely O2, H2O and CO2, using ab initio molecular dynamics. Dissociative adsorption of O2 occurs on Ti sites, which are unusual sites as Al2O3 is more stable than TiO2. This may be understood based on the electronic structure. We suggest that an increased Ti–O bond strength relative to Al–O surface bond strength is the electronic origin for the early stages of TiO2 formation on Ti0.5Al0.5N(001). Another unexpected atomic mechanism is identified as O covers the surface: Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, generating vacancies and hence enables mobility at the interface. In the case of H2O and CO2, the dominating physical mechanism is dissociative adsorption.
Wider implications. These fundamental surface processes are relevant for the initial stages of oxidation, surface diffusion and nucleation of reaction layers upon exposure to residual and environmental gases.
 Figure. Ti0.5Al0.5N(001)–O2 interactions as a function of coverage. The highlighted region designates the outermost surface, which is used for all coverages.
Figure. Ti0.5Al0.5N(001)–O2 interactions as a function of coverage. The highlighted region designates the outermost surface, which is used for all coverages.
1. Introduction
A predictive modelling in solid-state physics requires a detailed knowledge of all underlying fundamental mechanisms on the nanoscale. For instance, surfaces should be considered when solids interact with another medium. However, very often these surface models are idealized, as surface structure and energetics are usually described in vacuum [1–3]. An important benchmark system in surface protection of tools is cubic metastable Ti1−xAlxN (space group  , prototype NaCl), which exhibits considerable improvements of physical and chemical properties with respect to its binary counterparts, cubic TiN and hexagonal AlN [4–6]. The metastable solubility limit of AlN in TiN, forming cubic rather than thermodynamically stable hexagonal Ti1−xAlxN, was experimentally determined to be in the range x = 0.5–0.9 [7–13]. Furthermore, Ti1−xAlxN has extensively been studied by ab initio calculations, probing its bonding, phase stability, structure and elastic properties [14–20]. It was suggested that the phase stability of Ti1−xAlxN does depend not only on the Al content but also on the Al distribution on the metallic sublattice [16, 21].
, prototype NaCl), which exhibits considerable improvements of physical and chemical properties with respect to its binary counterparts, cubic TiN and hexagonal AlN [4–6]. The metastable solubility limit of AlN in TiN, forming cubic rather than thermodynamically stable hexagonal Ti1−xAlxN, was experimentally determined to be in the range x = 0.5–0.9 [7–13]. Furthermore, Ti1−xAlxN has extensively been studied by ab initio calculations, probing its bonding, phase stability, structure and elastic properties [14–20]. It was suggested that the phase stability of Ti1−xAlxN does depend not only on the Al content but also on the Al distribution on the metallic sublattice [16, 21].
Synthesis of metastable Ti1−xAlxN can only be facilitated under non-equilibrium conditions, which is readily available in physical vapour deposition techniques such as sputtering and cathodic arc [7–13]. These non-equilibrium synthesis methods are governed by an interplay of thermodynamics and kinetics [22]. Fundamental understanding of elementary growth processes, such as surface diffusion and ion–surface interactions, is crucial for obtaining the required combination of physical and chemical properties for a particular application [22]. Interaction of Ti1−xAlxN with residual gas during synthesis as well as post-exposure has not been considered using ab initio methods. Recently, potential energy maps for Ti and Al adatoms on ordered and disordered Ti0.5Al0.5N(001) surfaces were calculated using ab initio methods and it was suggested that the surface disorder reduces Ti adatom mobility [23]. Furthermore, surface energetics is relevant for various processes, ranging from initial stages of oxidation to preferred crystallographic orientation [24–28]. Based on ab initio molecular dynamics (MD) data, it was argued that the oxidation of TiN(001) is facilitated by dissociative adsorption of O2 and consequent formation of crystalline TiO2 at 900 K [25]. No such data are available for Ti1−xAlxN. It is, however, known from annealing experiments that TiO2 preferably forms upon oxidation of Ti1−xAlxN [29, 30]. It was suggested that Ti outward diffusion is responsible for these observations [29, 30], but no understanding on the atomic and electronic structure levels is available. Generally, other residual and environmental gases, such as H2O, may also be relevant for synthesis, namely impurity incorporation, nucleation and surface diffusion, as well as for many physical properties such as ferroelectric, elastic and tribological [31–34]. Based on ab initio calculations, it was reported that molecular H2O adsorption occurs on TiN(001) and TiN(111) surfaces, while dissociative H2O adsorption is observed for TiN(110) [35]. As soon as AlN is incorporated into cubic TiN forming Ti1−xAlxN, these adsorption processes may be altered, but this has not yet been explored.
In this work, we use ab initio MD simulations to explore surface processes on Ti0.5Al0.5N(001) interacting with residual and environmental gases, O2, H2O and CO2. Dissociative adsorption of O2 induces preferred Ti–O bond formation and extraction of Ti from the TiAlN(001)/O interface, generating Ti vacancies at the interface. H2O gives rise to O–H and N–H dipoles. Adsorption of CO2 is responsible for C–O dipole formation and free atomic O triggers similar physics as the adsorption of O2. These data explain on the atomistic level several experimental observations, such as preferred TiO2 formation of Ti–Al–N surfaces [29, 30] and nucleation and growth of Ti–Al–O–N thin films [36].
2. Methodology
Ab initio MD simulations of Ti0.5Al0.5N(001) interacting with O2, H2O and CO2 were performed using the OpenMX code [37], based on the density functional theory [38] and basis functions in the form of linear combination of localized pseudoatomic orbitals [39]. The electronic potentials were fully relativistic with partial core corrections [40, 41] and the generalized gradient approximation was applied [42]. The basis functions used were generated by a confinement scheme [39, 43] and specified as follows: Ti5.0-s2p2d1, Al6.0-s2p2, N4.5-s2p1, O4.5-s2p1, H4.5-s2 and C4.5-s2p1. Ti, Al, N, O, H and C designate the chemical name, followed by the cutoff radius (Bohr radius units) in the confinement scheme and the last set of symbols defines primitive orbitals applied. The confinement radii as well as the basis set were carefully checked with respect to basic elemental data, such as equilibrium volume (or bond length in the case of free molecules) and bulk modulus. The energy cut off 2040 eV (150 Ryd) and the 96 × 96 × 240 grid within the real-space grid technique [44] were adjusted to reach the accuracy of 3 × 10−5 eV atom−1 (10−6 H atom−1). Canonical ensembles at 300 K were used to simulate Ti0.5Al0.5N(001) surface (six atomic layers, surface area 12.536 × 12.536 Å2, 216 atoms, random distribution of Al and Ti [15]) interacting with O2, H2O and CO2 gases as a function of coverage. The coverage was calculated based on ideal dissociative adsorption. These molecules were initially placed 3 Å from the Ti0.5Al0.5N(001) surface at ad hoc positions. The bottom layer of Ti0.5Al0.5N(001) slab was always frozen to mimic the infinitive bulk. The MD time step was 1.0 fs and the simulation time was 2000 fs for each coverage. The total MD time for all simulations performed was 54 000 fs. The adsorption energy (ΔEad) was calculated at 0 K as

where Eslab, Emolecule, ETiAlN, n and m designate the total energy of a Ti0.5Al0.5N(001) slab with adsorbed molecules, total energy of a molecule, total energy of the pristine Ti0.5Al0.5N(001) surface, number of adsorbed molecules and number of atoms in a molecule, respectively. All total energies at 0 K were obtained after full structural relaxations by minimizing the interatomic forces.
3. Results and discussion
We start the discussions on the Ti0.5Al0.5N(001) surface interaction with residual and environmental gases by considering the O2 case. Figure 1 shows the fundamental atomic process occurring during Ti0.5Al0.5N(001)–O2 interactions as a function of coverage. Dissociative adsorption of O2 occurs for all coverages probed. We observe the interaction of individual O atoms with surface metal atoms, but never with N. Ti sites are preferred over Al sites for the O2 coverage up to 0.17 monolayers (ML). At an O2 coverage of 0.17 ML, Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, which is induced by an increased Ti–O coordination (three O nearest neighbours) compared to the Ti–O coordination (one O nearest neighbour) at lower O2 coverages. This generates Ti vacancies in the Ti0.5Al0.5N(001)/O interface layer. This is consistent with the literature, since metal vacancies were suggested to form in Ti0.5Al0.5N [45] and isostructural Cr–Al–O–N [46]. We observe the apical Ti–O bond length of 1.66 Å at an O2 coverage of 0.06–0.11 ML, which is not consistent with TiO2 [47]. However, as soon as Ti escapes from the Ti0.5Al0.5N(001)/O interface layer at an O2 coverage of 0.17 ML, the Ti–O bond adopts the length of 1.87–1.96 Å, which is in turn consistent with TiO2 [47]. Hence, it is reasonable to assume that the O adsorption induces Ti extraction and the associated vacancy formation enables and constitutes the initial stage of TiO2 formation. At an O2 coverage of 0.22 ML, Al–O bonds begin to form, but it is only Ti that escapes from the Ti0.5Al0.5N(001)/O interface layer. This Al–O bond formation may be due to a decreased population of Ti atoms in the surface exposed to additional O2 molecules. In this case, there is a broader distribution of O–metal bond lengths, from 1.65 to 2.18 Å. This structural disorder (broad bond length distribution) was also observed for the oxidation of Al(111) [48]. At an O2 coverage of 0.44 ML, a Ti atom coordinated with five nearest O neighbours is observed. This is consistent with TiO2 [47]. Our data also support the suggestion by Gnoth et al [49] that surface oxidation of cubic Ti–Al–N thin films is caused by reaction with atmospheric oxygen and/or residual gas immediately after synthesis [49]. to Baben et al [50] showed that the incorporation of O into cubic Ti–Al–N is energetically favourable and Sjölen et al [36] have observed the incorporation experimentally. Hence, the above discussed phenomena are also relevant for nucleation and growth of Ti–Al–O–N thin films. This may be a general feature of cubic transition metal Al nitrides, as O incorporation is energetically also favoured for Sc-, V- and Cr-containing phases [51].

Figure 1. Ti0.5Al0.5N(001)–O2 interactions as a function of coverage. MD snapshots at 300 K are shown after 2000 fs at each coverage. Nearest neighbours are connected. The highlighted region designates the outermost surface, which is used for all coverages.
Download figure:
Standard image High-resolution imageWe continue to discuss the structure evolution as a function of coverage in terms of energetics. Figure 2 contains the corresponding adsorption energy data for O2 molecules. As the O2 coverage is increased from 0.06 to 0.50 ML, the adsorption energy increases from −5.8 to −3.5 eV per adatom. Clearly, the affinity of O towards Ti0.5Al0.5N(001) decreases with larger O2 coverages. This trend as well as the absolute values is consistent with the literature [52, 53].
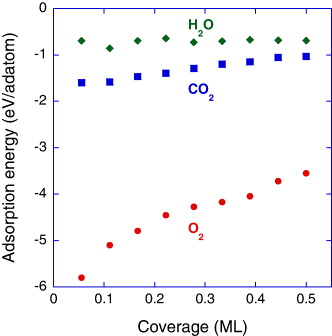
Figure 2. Adsorption energy for O2, H2O and CO2 on Ti0.5Al0.5N(001) as a function of coverage.
Download figure:
Standard image High-resolution imageThe fact that Ti–O bond formation is preferred at low O2 coverages is counter-intuitive as Al2O3 is known to be more stable than TiO2 [54]. This apparent inconsistency can be understood based on the electronic structure. Figure 3 shows the density of states for Ti0.5Al0.5N(001)–O2 interaction at a coverage of 0.06 ML. We explore two cases: (i) O adsorbed on Ti sites, which is preferred according to our ab initio MD data presented above and (ii) O adsorbed on Al sites. The general features in the density of states for Ti0.5Al0.5N(001) are consistent with bulk Ti0.5Al0.5N [18], so no detailed discussions are provided here. O 2p states are localized in the range of approx. −4 to −2 and −2 to 0 eV for O adsorbed on Ti and Al sites, respectively. The O 2p states are hybridized with Ti 3d in case of adsorption on the Ti sites and Al 3p states in case of adsorption on the Al sites (partial density of states analysis is not shown here). A shift to lower energies, as observed for the O adsorption on the Ti sites, is consistent with an increase in the bond energy and hence bond strength. This is consistent with the electronic structure of bulk Ti0.5Al0.5N with incorporated O [50]. We suggest that the increased Ti–O bond strength constitutes the electronic origin for the preferred Ti–O bond formation compared to (the initially expected) Al–O formation and represents the early stages of TiO2 formation on Ti0.5Al0.5N(001). These data explain on the electronic structure level the experimental observation of the selective oxidation of Ti on Ti–Al–N [29, 30] and TiAl [55] surfaces.
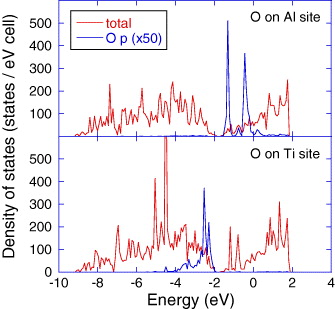
Figure 3. Density of states for Ti0.5Al0.5N(001)–O2 interaction at a coverage of 0.06 ML. Two partial densities of states for O 2p orbitals are shown: (i) O adsorbed on Ti sites and (ii) O adsorbed on Al sites. The Fermi level is set to 0 eV.
Download figure:
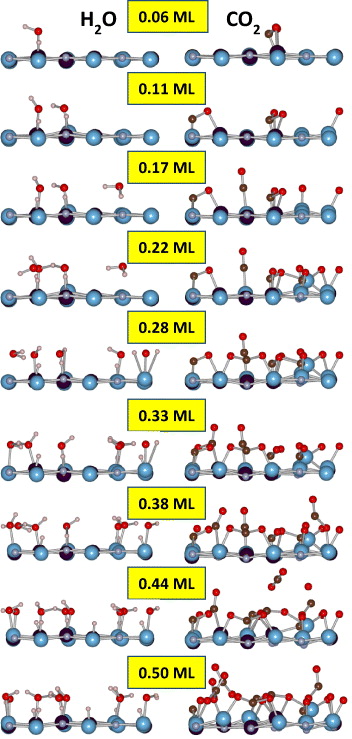
Standard image High-resolution imageAfter considering the Ti0.5Al0.5N(001) surface interaction with O2, we continue with the exposure to H2O and CO2. Figure 4 shows the fundamental atomic process occurring during Ti0.5Al0.5N(001)–H2O and Ti0.5Al0.5N(001)–CO2 interactions as a function of coverage. Dissociative adsorption of H2O is observed for the coverage of 0.06–0.11 ML. O–H and N–H dipoles are formed. O–H dipoles have a tendency to align laterally. This may be relevant for tribological properties of these surfaces [34, 56]. At an H2O coverage of 0.17 ML, a non-dissociated H2O molecule is observed. Molecular H2O adsorption was reported on TiN(001) [35]. On the other hand, dissociative adsorption of H2O on Al2O3(0001) occurs [57]. At H2O coverages larger than 0.22 ML, all H2O molecules dissociate. Clearly, dissociative adsorption is the dominating physical mechanism. Figure 2 also contains the corresponding adsorption energy data for H2O molecules. The average adsorption energy is −0.7 eV per adatom, which is the lowest value observed here. The adsorption energy appears not to be a function of H2O coverage. It seems that the affinity of H2O towards Ti0.5Al0.5N(001) is rather small.
{kind=link}
{kind=link}
{kind=link}
Figure 4. Ti0.5Al0.5N(001)–H2O and Ti0.5Al0.5N(001)–CO2 interactions as a function of coverage. MD snapshots at 300 K are shown after 2000 fs at each coverage. Nearest neighbours are connected and only outermost surfaces are shown.
Download figure:
Standard image High-resolution image{kind=link}
The last molecule interacting with Ti0.5Al0.5N(001) to be considered is CO2 (see figure 4). Non-dissociative adsorption of CO2 occurs for the lowest coverage of 0.06 ML. Already at a CO2 coverage of 0.11 ML, CO2 dissociates and C–O and Ti–O dipoles are formed. Evidently, surface oxidation can also be CO2 driven. The observed dissociative adsorption of CO2 on Ti0.5Al0.5N(001) is consistent with the interaction of CO2 and many metal surfaces, such as Al, Bi, Fe, Mg, Ni, Pd, Re, Rh and Ru [58]. At a CO2 coverage of 0.22 ML, Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, induced by an increased Ti–O coordination. Here, Al–O bonds already coexist. This is the same physical mechanism as in the case of Ti0.5Al0.5N(001)–O2 interaction, which is discussed above. Dissociative adsorption is continued for all coverages probed except of a CO2 coverage of 0.44 ML, where a non-dissociative adsorption of CO2 occurs at very large distance from the surface, namely ∼5 Å which is in the order of two interplanar distances. Since a relatively large CO2 coverage is reached, Coulomb repulsion may occur with already adsorbed CO2 molecules. However, already at a CO2 coverage of 0.50 ML, dissociative adsorption is continued including the dissociation of the non-dissociated molecule at a CO2 coverage of 0.44 ML. Clearly, dissociative adsorption is the dominating physical mechanism. Figure 2 also contains the corresponding adsorption energy data for CO2 molecules. As CO2 coverage is increased from 0.06 to 0.50 ML, the adsorption energy increases from −1.6 to −1.0 eV per adatom. This is the same trend as in the case of O2 adsorption and the affinity of CO2 towards Ti0.5Al0.5N(001) is in the range between those of O2 and H2O. This implies that the Ti0.5Al0.5N(001) surface would mainly be covered with O, while C-containing species should be seen as minor impurities.
4. Conclusions
Using ab initio MD simulations at 300 K, we have studied fundamental surface processes on Ti0.5Al0.5N(001) interacting with residual and environmental gases, O2, H2O and CO2. Dissociative adsorption of O2 occurs for all coverages probed. Surprisingly, Ti sites are preferred for an O2 coverage of 0.06–0.17 ML. This may be understood based on the electronic structure. We suggest that the increased Ti–O bond strength relative to the Al–O surface bond strength is the electronic origin for the early stages of TiO2 formation on Ti0.5Al0.5N(001), which explains the experimental observation of the selective oxidation of Ti on Ti–Al–N surfaces [29, 30] as well as nucleation and growth of Ti–Al–O–N thin films [36]. At an O2 coverage of 0.17 ML, Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, generating Ti vacancies. At a O2 coverage of 0.22 ML, Al–O bonds begin to form. In the case of H2O, dissociative adsorption, where O–H and N–H dipoles are formed, is a dominating mechanism for all coverages. O–H dipoles have a tendency to align laterally. At a H2O coverage of 0.17 ML, a non-dissociated H2O molecule is observed. This may be due to a competition of two mechanisms. Molecular H2O adsorption was reported on TiN(001) [35], while dissociative adsorption occurs on Al2O3(0001) [57]. In the case of CO2, non-dissociative adsorption occurs for the lowest coverage of 0.06 ML. Already at a CO2 coverage of 0.11 ML, CO2 dissociates and C–O and Ti–O dipoles are formed. At a CO2 coverage of 0.22 ML, Ti escapes from the Ti0.5Al0.5N(001)/O interface layer, induced by an increased Ti–O coordination. This is the same physical mechanism as in the case of Ti0.5Al0.5N(001)–O2 interaction. Larger CO2 coverages are dominated by dissociation. The identified physical mechanisms are relevant for various processes, ranging from initial stages of oxidation and surface diffusion to nucleation of reaction layers upon exposure to residual and environmental gases.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft within the Collaborative Research Centre SFB-TR 87.