


Abstract
Time-resolved diffraction and microscopy with femtosecond electron pulses provide four-dimensional recordings of atomic motion in space and time. However, the limited coherence of electron pulses, reported in the range of 2–3 nm, has so far prevented the study of complex organic molecules with relevance to chemistry and biology. Here we characterize the coherence of femtosecond single-electron pulses that are generated by laser photoemission. We show how the absence of space charge and the minimization of the source size allow the transverse coherence to be extended to 20 nm at the sample position while maintaining a useful beam diameter. The extraordinary coherence is experimentally demonstrated by recording single-electron diffraction snapshots from a complex organic molecular crystal and identifying more than 80 sharp Bragg reflections. Further optimization affords promise for coherences of 100 nm. These advances will allow time-resolved imaging of functional dynamics in biological systems, uniting picometre and femtosecond resolutions in a compact, table-top instrumentation.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Recording 'movies' of atomic motion during chemical and biological processes in molecular systems requires the simultaneous resolution of spatial structures and temporal dynamics on picometre and femtosecond scales, respectively [1–3]. Ultrafast electron or x-ray diffraction techniques are useful for this purpose [2, 4]. In a pump–probe approach, laser pulses initiate the reaction and femtosecond electron or x-ray pulses are used to record diffraction snapshots of transient structures. For example, phase transformations in crystals [5], layer dynamics of graphite [6], decay and recovery of charge density waves [7] or oscillations in ferroelectric materials [8] were studied with femtosecond diffraction. The specimens in these investigations, however, were inorganic materials of limited complexity.
Most molecular systems in biochemistry have sizes of many nanometres. This is challenging for time-resolved diffraction techniques, because the transverse coherence of the probe beam must be large enough to cover the entire molecular structure. X-ray pulses from free electron lasers provide a remarkable degree of spatial coherence [9], but significant effort is required to achieve wavelengths sufficient for resolving interatomic distances [10–12]. In contrast to synchrotrons and free electron lasers, a femtosecond electron diffraction apparatus is a table-top experiment. At the typical energies of 30–300 keV, electrons have subatomic de Broglie wavelengths (0.07–0.02 Å), well suited for resolving multiple diffraction orders simultaneously. On the other hand, these small wavelengths make it difficult to obtain a large transverse coherence ([13] and equation (1) below). In femtosecond pulses carrying many electrons, the electron's fermionic nature and space charge effects further limit the transverse coherence [3, 13, 14]. In time-resolving electron microscopes, apertures are used to improve the coherence properties of the electron beam such that sharp diffraction patterns can be recorded [15], but this comes at the cost of electron flux, or temporal resolution if electron flux is to be maintained and Coulomb repulsion cannot be circumvented [14]. For unfiltered beams of femtosecond pulses, the reported values for the transverse coherence are 2.5 and 3 nm for beams with radii of σ = 150 and  , respectively [16, 17]. This is insufficient for functional systems with relevance to biology and typical sample dimensions.
, respectively [16, 17]. This is insufficient for functional systems with relevance to biology and typical sample dimensions.
2. Results and discussions
In contrast to the previously reported low coherence values in time-resolved electron diffraction experiments, we demonstrate here that femtosecond single-electron pulses [1, 17] in a sub-100 μm beam can reach coherences of 20 nm and offer the potential to approach the 100 nm frontier. Two consequences of the single-electron regime are decisive for this order-of-magnitude improvement over the reported state of the art [3, 14, 16, 17]. Firstly, the transverse velocity spread of laser-emitted single electrons is remarkably low in the absence of space charge. In dense pulses, where many electrons are concentrated to femtosecond duration [3], Coulomb forces cause a significant increase of radial spread, i.e. divergence [18], which reduces the transverse coherence. Such effects are absent in the single-electron regime employed here, where the transverse velocity spread is solely determined by the physics of the photoemission process [17]. Secondly, it is possible to minimize the photoemission area, without suffering from image charge effects that would pull denser packets back into the emission surface [4, 19]. Such constraints are also absent in the single-electron regime.
Figure 1(a) depicts the essential stages of the experiment; for details see appendix A. Ultraviolet laser pulses at a wavelength of 266 nm are focused tightly onto a gold photocathode by using a lens with a focal length of f = 20 mm within the vacuum system, resulting in an optical focus with a radius of 1–4 μm (see appendix A). On average, one tenth of an electron is emitted per laser pulse. Accumulation of the electron flux over many laser shots forms the electron beam. An electrostatic field accelerates the electrons to 30 keV, corresponding to a de Broglie wavelength of 0.07 Å. Fluctuations in the arrival time of single electrons on target caused by the statistical distributions of electron energy and emission time at the photocathode result in an estimated effective pulse duration of ∼300 fs (full-width at half-maximum) for our experimental conditions [17]. Shorter pulses can be obtained by optimizing the laser's photon energy and the acceleration field [17], and/or by using a microwave cavity for pulse compression [20]. A magnetic lens [21] focuses the beam through the diffraction sample onto a phosphor screen [14]. No apertures or filters are applied. This geometry with a converging beam was chosen in order to produce a sharp diffraction pattern on the screen while maintaining a reasonably small beam at the sample for maximum diffraction yield.

Figure 1. Femtosecond pulses of single electrons are not subject to Coulomb forces and can be made highly coherent. (a) Experimental arrangement. (b) Shape of the electron beam as calculated from the source parameters.
Download figure:
Standard image High-resolution imageThe quality of a diffraction pattern and thus the ability to resolve large and complex structures depends critically on the transverse coherence of the electron beam at the sample, i.e. the ability of laterally separated parts of the scattered wave to interfere [22]. For an electron beam, the transverse coherence length can be defined by analogy with optics, using a criterion of 88% for the visibility of interference [23, 24]. This definition is related to approaches based on the van Cittert–Zernike theorem under the assumption of an incoherent source [23, 24]. Accordingly, the transverse coherence length  at a beam waist is
at a beam waist is

with the uncorrelated angular spread  (half-angle divergence), the uncorrelated transverse velocity spread σv⊥, the longitudinal velocity v0 and the de Broglie wavelength λ [3, 14, 16]. Written in terms of transverse velocity spread, we obtain
(half-angle divergence), the uncorrelated transverse velocity spread σv⊥, the longitudinal velocity v0 and the de Broglie wavelength λ [3, 14, 16]. Written in terms of transverse velocity spread, we obtain

where  is the electron mass [17, 19]. Note that differing definitions are sometimes used in the literature, e.g. in [24], but the 88% criterion leading to equation (1) is widely accepted in the field of ultrafast electron diffraction [3, 16, 17, 19]. The coherences measured so far (2–3 nm) were quantified according to this definition [16, 17].
is the electron mass [17, 19]. Note that differing definitions are sometimes used in the literature, e.g. in [24], but the 88% criterion leading to equation (1) is widely accepted in the field of ultrafast electron diffraction [3, 16, 17, 19]. The coherences measured so far (2–3 nm) were quantified according to this definition [16, 17].
In order to determine the coherence properties of our electron beam, we measured its radius on the screen as a function of the focal length of the magnetic lens; the source parameters were then fitted using transfer matrices for the beam propagation (see appendix B). This analysis provides a source radius of  , a transverse velocity spread at the source of
, a transverse velocity spread at the source of  and a beam radius at the sample of
and a beam radius at the sample of  (standard deviations). By evaluating equation (2) for our beam parameters at the source, we obtain a transverse coherence length of
(standard deviations). By evaluating equation (2) for our beam parameters at the source, we obtain a transverse coherence length of  . The electron-optical system magnifies the transverse coherence length and the macroscopic beam size by the same factor: the ratio of coherence length to beam diameter (global degree of coherence) is conserved by this transformation [25, 26]. Figure 2 depicts this relation. At the sample, we therefore expect
. The electron-optical system magnifies the transverse coherence length and the macroscopic beam size by the same factor: the ratio of coherence length to beam diameter (global degree of coherence) is conserved by this transformation [25, 26]. Figure 2 depicts this relation. At the sample, we therefore expect
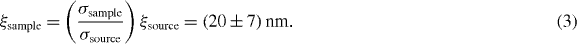
In principle, the transverse coherence length can be made as large as desired by magnifying the beam accordingly. However, the electron flux contributing to diffraction is significantly reduced if the beam becomes larger than the sample, whose size is limited. This cannot be compensated for by increasing the beam current, because the single-electron regime needs to be maintained right from the source in order to completely avoid the detrimental effects of space charge to pulse duration and coherence properties. The best diffraction efficiency at maximum coherence is achieved if the beam is just large enough to cover the entire sample, like in our experiment. In this regime, the source's coherence ξsource and its size σsource become the decisive parameters.

Figure 2. Global degree of coherence. The ratio of transverse coherence (dark blue) to beam diameter (light blue) is preserved in the single-electron regime. Maximization of the source's coherence and minimization of its size hence increase the coherence at the sample, even in the case of a converging or diverging beam.
Download figure:
Standard image High-resolution imageThe prediction of equation (3),  , was verified by recording diffraction from N-(triphenylmethyl)-salicylidenimine (figure 3(a)), a molecular switch showing an intra-molecular reaction upon photo-excitation: a proton is transferred from oxygen to nitrogen [27]. From single crystals of this material, we produced 50 nm thick films by ultramicrotomy (figure 3(b)). The film is slightly larger than the electron beam and entirely single crystalline. The crystal structure is triclinic with lattice parameters of
, was verified by recording diffraction from N-(triphenylmethyl)-salicylidenimine (figure 3(a)), a molecular switch showing an intra-molecular reaction upon photo-excitation: a proton is transferred from oxygen to nitrogen [27]. From single crystals of this material, we produced 50 nm thick films by ultramicrotomy (figure 3(b)). The film is slightly larger than the electron beam and entirely single crystalline. The crystal structure is triclinic with lattice parameters of  ,
,  and
and  , as well as
, as well as  ,
,  and γ = 87.08°. Figure 3(c) shows the diffraction pattern obtained by integrating over ∼108 single electrons from ∼109 laser shots over 5 min. The intensity pattern of the 85 identifiable Bragg spots spans two orders of magnitude and reflects the material's complex structure factor. The sharpness of the spots, i.e. the ratio between spot width and separation, indicates that a significant number of adjacent unit cells must have contributed coherently to the diffraction pattern [23, 26]. For 50 Bragg spots, we evaluated the σ -width in the [001] and [100] directions. All spots, including the central beam, have almost the same width. The average values on the screen are σ001 = (76 ± 13) μ m at a spot separation of s001 = (3226 ± 6) μ m and
and γ = 87.08°. Figure 3(c) shows the diffraction pattern obtained by integrating over ∼108 single electrons from ∼109 laser shots over 5 min. The intensity pattern of the 85 identifiable Bragg spots spans two orders of magnitude and reflects the material's complex structure factor. The sharpness of the spots, i.e. the ratio between spot width and separation, indicates that a significant number of adjacent unit cells must have contributed coherently to the diffraction pattern [23, 26]. For 50 Bragg spots, we evaluated the σ -width in the [001] and [100] directions. All spots, including the central beam, have almost the same width. The average values on the screen are σ001 = (76 ± 13) μ m at a spot separation of s001 = (3226 ± 6) μ m and  at a spot separation of
at a spot separation of  . We compare these numbers with diffraction patterns calculated from the theory of multi-slit interference [23]. Values similar to the measured sharpnesses are obtained if
. We compare these numbers with diffraction patterns calculated from the theory of multi-slit interference [23]. Values similar to the measured sharpnesses are obtained if  and
and  slits are coherently illuminated in the [001] and [100] directions, respectively. Taking into account the spacings of the lattice planes of our crystal,
slits are coherently illuminated in the [001] and [100] directions, respectively. Taking into account the spacings of the lattice planes of our crystal,  and
and  , we conclude that the transverse coherence of our beam spans (15 ± 3) and (18 ± 3) nm , respectively.
, we conclude that the transverse coherence of our beam spans (15 ± 3) and (18 ± 3) nm , respectively.

Figure 3. Diffraction experiment with highly coherent single-electron pulses. (a) Structure of the molecular crystal used for diffraction. (b) Free-standing, single-crystalline thin film on a gold grid. (c) Diffraction snapshot obtained with a sequence of single-electron pulses. The sharpness of the spots evidences the extraordinary coherence.
Download figure:
Standard image High-resolution imageWe note that with the molecular crystal in place, the central spot and the Bragg reflections are about 40% larger than the direct beam without the sample, which suggests that the sharpness of the diffraction pattern is determined by the coherence properties of the electron beam and not by its focused spot size on the detector. Additional effects like inelastic scattering, inhomogeneities of the sample or vibrations during the measurement may also have contributed to the broadening of the Bragg spots. Therefore, our estimates of the transverse coherence from the diffraction pattern, (15 ± 3) and  , are lower limits.
, are lower limits.
3. Conclusions and outlook
We conclude that two different types of measurements, by beam characterization and by diffraction, demonstrate the high transverse coherence length of our femtosecond electron pulses, opening the door for biochemical applications. Our scheme reveals the potential for substantial further optimization towards even higher coherences. In the presented experiments, the laser's photon energy was not perfectly matched to the photocathode's work function, producing an excess spread of energy and velocity [17]. If, for example, the transverse velocity spread  is reduced by a factor of two, this will double the initial coherence
is reduced by a factor of two, this will double the initial coherence  and at the same time halve the beam's divergence. Allowing the same size of the beam at the sample, and designing the magnetic lens accordingly, we hence expect an about two-fold increase of the coherence, i.e. about 40 nm. In addition, one can further reduce the source size by focusing more tightly. This is possible in the single-electron regime without reducing the total beam current. These two improvements afford promise for reaching transverse coherences of the order of 100 nm for transverse sample dimensions of the order of 100 μm.
and at the same time halve the beam's divergence. Allowing the same size of the beam at the sample, and designing the magnetic lens accordingly, we hence expect an about two-fold increase of the coherence, i.e. about 40 nm. In addition, one can further reduce the source size by focusing more tightly. This is possible in the single-electron regime without reducing the total beam current. These two improvements afford promise for reaching transverse coherences of the order of 100 nm for transverse sample dimensions of the order of 100 μm.
We note that the inherent transverse velocity spread of our single-electron pulses could be smaller than calculated from the beam's divergence, for example if part of the observed divergence of the beam resulted from an inhomogeneous broadening of the emission directions on an uneven surface [28, 29]. Cho et al [22] reported on field emitter sources with a coherence exceeding the predictions from the beam shape. This partial coherence was related to the delocalization of the initial electronic states within the material and its preservation during tunnelling in field emission. Similarly, photoelectric emission was also reported to directly promote the initial electronic states into vacuum [30, 31]. Combining these two observations indicates that photo-emitted electron pulses could have a higher coherence than suggested by beam parameter measurements, depending on the purity, temperature and morphology of the cathode material. In order to verify this assumption, more direct techniques than the evaluation of crystalline diffraction patterns will have to be employed. These could, for example, be based on interferometry [22].
Recently, metallic nanotips were investigated as a source of highly coherent electron pulses [32, 33]. They can produce ultrashort pulses from emission areas of few square nanometres upon pulsed-laser irradiation [34–36]. In addition, as mentioned above, field emission tips were reported to have partial coherence with measured transverse coherence lengths in the range of 5–35 nm [22].2 Despite these advantages, delivering femtosecond electron pulses from nanotip electron sources to the location of the diffraction sample remains challenging [33]. The strong curvature of the emission area causes a large divergence of the electron beam. Collimation with electron-optical lenses can introduce some significant distortions in the time domain [21]. In addition, the acceleration field is very inhomogeneous, deteriorating the pulse duration [33]. Simulations suggest that these difficulties can be overcome by specially designed electron optics [33], but they are intrinsically absent from flat, extended emitters as used in the present work.
In conclusion, the experimentally demonstrated transverse coherence length of about 20 nm at a beam radius of 77 μm represents an approximately tenfold increase over reported results and now covers a substantial number of organic systems of great biological relevance. Even larger coherences of ∼100 nm are anticipated by future optimizations. Femtosecond single-electron diffraction at picometre wavelengths hence delivers, all at once, the indispensable resolutions, flux and coherence for time-resolved, atomic-scale recordings of functional motion with a table-top approach.
Acknowledgments
We thank Aleksandra Lewanowicz for providing molecular crystals and corresponding x-ray data as well as Jom Luiten for several helpful discussions about beam parameters. We acknowledge funding from the ERC (grant '4D imaging'), the International Max Planck Research School of Advanced Photon Science, the Munich-Centre of Advanced Photonics, and the Rudolf-Kaiser Foundation.
Appendix A.: Experimental details
The laser system we use is a long-cavity titanium:sapphire oscillator [37], providing 60 fs pulses at a central wavelength of 800 nm (Femtolasers GmbH). A pulse picker (Bergmann Messgeräte Entwicklung KG) is applied to reduce the repetition rate to 2.56 MHz for the reported experiments. An alignment system (TEM Messtechnik GmbH) stabilizes the laser beam. We have two possibilities for the generation of ultraviolet pulses for electron emission, either third-harmonic generation (used here) or a tuneable source based on continuum generation followed by frequency doubling [17]. The photocathode is a sapphire substrate coated with 1 nm of chromium for adhesion and a 20 nm gold layer for photoemission. The  lens is placed at a fixed location inside the vacuum system and additional optics outside of the vacuum chamber is used to optimize the focus on the cathode. Measurement of the focus with a beam camera provides an upper limit for the radius of 4 μm, whereas a lower limit of 1 μm was calculated from Gaussian beam propagation. The emitted photoelectrons as accelerated in a static field of approx. 3.5 kV mm−1. The anode has a 2 mm hole and does not clip the electron beam. We placed the magnetic lens at a distance of 4.7 cm from the anode and applied a current of 2.04 A. Three coil pairs around the entire experiment are actively compensating for external magnetic distortions (Müller-BBM GmbH). Five linear and rotational stages (HUBER Diffraktionstechnik GmbH & Co. KG) are used to place and orient the sample into the electron beam along the required zone axis. Diffraction is detected with a phosphor screen and a CMOS camera with 4096 × 4096 pixels (Tietz Video and Image Processing Systems GmbH). Single crystals of N-(triphenylmethyl)-salicylidenimine with an edge length of about 0.5 mm were embedded in epoxy resin and were cut in the ultramicrotome (Leica Microsystems GmbH) with a 35° diamond knife (Diatome AG). Gold meshes with 2000 lines per inch were used for support.
lens is placed at a fixed location inside the vacuum system and additional optics outside of the vacuum chamber is used to optimize the focus on the cathode. Measurement of the focus with a beam camera provides an upper limit for the radius of 4 μm, whereas a lower limit of 1 μm was calculated from Gaussian beam propagation. The emitted photoelectrons as accelerated in a static field of approx. 3.5 kV mm−1. The anode has a 2 mm hole and does not clip the electron beam. We placed the magnetic lens at a distance of 4.7 cm from the anode and applied a current of 2.04 A. Three coil pairs around the entire experiment are actively compensating for external magnetic distortions (Müller-BBM GmbH). Five linear and rotational stages (HUBER Diffraktionstechnik GmbH & Co. KG) are used to place and orient the sample into the electron beam along the required zone axis. Diffraction is detected with a phosphor screen and a CMOS camera with 4096 × 4096 pixels (Tietz Video and Image Processing Systems GmbH). Single crystals of N-(triphenylmethyl)-salicylidenimine with an edge length of about 0.5 mm were embedded in epoxy resin and were cut in the ultramicrotome (Leica Microsystems GmbH) with a 35° diamond knife (Diatome AG). Gold meshes with 2000 lines per inch were used for support.
Appendix B.: Characterization of the electron beam
The electron beam profile at the detector was recorded for a range of focusing strengths of the magnetic lens with currents between 0 and 2.3 A. These beam profiles were fitted by elliptical Gaussians and corrected for the point spread function of our detector (0.8 pixels, pixel size  ). We can perceive some astigmatism of the magnetic lens and some ellipticity of the electron beam, but these effects are negligible. Thus, geometrically averaged beam radii were used in the succeeding analysis. The beam propagation was modelled by the transfer matrix technique [38] based on the geometry of the experiment, including the defocusing effect of the anode hole [39]. The constant acceleration between cathode and anode was taken into account by propagating the electron beam with its final velocity over twice the distance, which is a non-relativistic approximation. We further assume the spatial and transverse momentum distributions at the cathode to be uncorrelated. Applying a least-squares fit to the beam radius at the detector as a function of the magnetic lens current provided reliable values for the parameters of the electron source, as reported above (figure B.1). The measurement errors given above reflect the statistical errors of the fit. We refrain from a detailed analysis of systematic errors because of the good quality of the fit. Moreover, a significant increase in the error for the transverse coherence length is not to be expected since it is already dominated by the accuracy of the source radius.
). We can perceive some astigmatism of the magnetic lens and some ellipticity of the electron beam, but these effects are negligible. Thus, geometrically averaged beam radii were used in the succeeding analysis. The beam propagation was modelled by the transfer matrix technique [38] based on the geometry of the experiment, including the defocusing effect of the anode hole [39]. The constant acceleration between cathode and anode was taken into account by propagating the electron beam with its final velocity over twice the distance, which is a non-relativistic approximation. We further assume the spatial and transverse momentum distributions at the cathode to be uncorrelated. Applying a least-squares fit to the beam radius at the detector as a function of the magnetic lens current provided reliable values for the parameters of the electron source, as reported above (figure B.1). The measurement errors given above reflect the statistical errors of the fit. We refrain from a detailed analysis of systematic errors because of the good quality of the fit. Moreover, a significant increase in the error for the transverse coherence length is not to be expected since it is already dominated by the accuracy of the source radius.

Figure B.1. Beam radius  (standard deviation) on the detector as a function of the magnetic lens current. The measurement values (dots) are fitted using a transfer matrix technique (red line). This permits the determination of the source's radius and divergence.
(standard deviation) on the detector as a function of the magnetic lens current. The measurement values (dots) are fitted using a transfer matrix technique (red line). This permits the determination of the source's radius and divergence.
Download figure:
Standard image High-resolution imageThe retrieved source parameters were used to calculate the full propagation of the electron beam from the source to the detector for various magnetic lens currents (solid lines in figure B.2) by the same transfer matrix technique as described above. The resulting electron beams agree well with those calculated by numerically propagating 5 × 106 non-interacting electrons through our experimental geometry. The validity of our analysis was further supported by comparing the calculated beam radius with knife edge measurements at the position of the diffraction sample (diamonds in figure B.2) and with the measured beam radii at the detector (squares in figure B.2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure B.2. Retrieved beam shapes for different strengths of the magnetic lens (solid), compared to results of knife edge scans at the sample position (diamonds) and to measured beam radii at the screen (squares).
Download figure:
Standard image High-resolution image{kind=link}
Appendix C.: Reversibility and resolution
Ultrafast single-electron diffraction is restricted to the study of reversible processes, because multiple pump–probe cycles are required to obtain a diffraction pattern. Typically, 107–109 electrons are needed for each pump–probe delay. The exact number of electrons is determined by the amount of structural change, the desired spatial resolution and the correspondingly required signal-to-noise ratio. A high experimental repetition rate is desirable to minimize the total acquisition time, but a limitation consists in the time it takes the sample to revert back to the original state, in order to be ready for the next pump–probe cycle. In addition, many biological samples may only withstand a limited number of pump pulses before photodamage occurs. Depending on the type of molecule and desired resolution, it might hence be required to exchange the sample several times during a pump–probe experiment with the coherent single-electron pulses presented here.
Footnotes
- 2
We note that in [22] a different visibility criterion is used in the definition of the coherence length, making the direct comparison with the values in the present work difficult.