
Abstract
Scientific simulation codes are public property sustained by the community. Modern technology allows anyone to join scientific software projects, from anywhere, remotely via the internet. The phonopy and phono3py codes are widely used open-source phonon calculation codes. This review describes a collection of computational methods and techniques implemented in these codes and shows their implementation strategies as a whole, aiming to be useful for the community. Some of the techniques presented here are not limited to phonon calculations and may therefore be useful in other areas of condensed matter physics.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Supported by the exponential growth of computer power, computer simulations and scientific software development are essential for condensed matter physics and materials science. Results obtained from computer simulations are getting more realistic and scientific software development more robust.
Since many scientific codes are distributed under open-source licenses, it is easy to start using scientific software and perform computations. Participation in scientific software development has also become easier with the availability of open-source compilers and source code editors. Collaborative development by members at different worksites or locations, i.e. distributed development, has become quite popular for scientific software these days. Anyone can contribute to open-source software projects, and the contributions are usually united on internet hosting services for software development. Documentation of scientific software is an important part of software development, not only for users but also for scientific software developers. For users, it explains how to use the software. For developers, it describes how to read and write the code.
Development of key software can be a project that lasts for many years. Once a key code is developed, it may be used for an unexpectedly long time. However, computer architectures are constantly evolving. The way to achieve high-performance computing may therefore change drastically in a relatively short time, on the order of a decade. For example, it is always required to follow the increase in the number of cores in processors. Indeed, algorithms and data structures must be optimized for a concurrent use of many cores in a multi-core processor. Increasing memory space usually allows more flexible software design. To adapt the code to those changes, the documentation is important to share the meaning of the software design choices among users and developers.
The phonopy and phono3py codes are scientific software developed to perform phonon calculations. A variety of physical properties can be calculated using them: the phonon spectrum, the dynamical structure factor, and the lattice thermal conductivity (LTC), to mention just a few. The computational method is based on the supercell approach. In the computer implementations, various numerical methods and techniques are employed. Some of those implemented in the phonopy and phono3py codes are covered in this review. Our motivation for writing this review is to provide essential information to understand the codes in depth and to invite scientific software developers in the phonopy and phono3py projects.
Although this review is written targeting scientific software developers, it may be also useful for expert users of the phonopy and phono3py codes. The computational methods and techniques underlying the phonon calculations are described as implemented to the phonopy and phono3py codes.
In section 2, the representation of the crystal structure and the crystal symmetry are presented in a crystallographic way. The reciprocal space of the crystal and the crystal symmetry in the reciprocal space are also described. Then, the phonon coordinates are introduced. The concept of Brillouin zone (BZ) required for the phonon calculation is briefly defined.
In section 3, the geometry of the supercell structure model is described. The supercell geometry is associated with the primitive cell by an integer matrix and how to construct the supercell geometry using the integer matrix is explained.
In section 4, the transformation between the supercell force constants and the dynamical matrices is presented. Since the phonopy and phono3py codes employ the supercell approach, the force constants elements are limited to within the supercell. The transformation, therefore, needs to be performed with special care. Such details are given in this section.
In section 5, the treatment of long range dipole–dipole interaction in the dynamical matrix, as implemented in the phonopy code, is explained. The implementation is mainly based on [1], and its application to the supercell approach is reviewed.
In section 6, the traditional and generalized regular grids used to sample reciprocal space discretely are defined. The symmetry treatment of those grid points, used in the phonopy and phono3py codes, is also presented.
In section 7, a linear tetrahedron method is described using the formulation as implemented in the phonopy and phono3py codes. The implementation is mainly based on [2, 3]. The method is then applied to the three-phonon scattering.
In section 8, a scheme to generate random atomic displacements in the supercell at finite temperatures is presented. This is achieved by superpositions of displacements corresponding to randomly sampled harmonic oscillators.
In section 9, a band unfolding technique implemented in the phonopy code following [4] is explained. The formulation specific to the phonon calculation with the supercell approach is provided.
2. Crystal and symmetry
2.1. Crystal structure
As shown in figure 1, a crystal model is defined by a collection of unit cells periodically repeated in the three directions of space. Each unit cell contains atoms. We normally choose a conventional set of basis vectors to span the unit cell,  , so that it naturally represents the crystal symmetry along with the choice of the origin of atomic positions [5]. Positions of atoms are represented either in Cartesian coordinates or in crystallographic coordinates. Denoting the crystallographic coordinates of an atom as
, so that it naturally represents the crystal symmetry along with the choice of the origin of atomic positions [5]. Positions of atoms are represented either in Cartesian coordinates or in crystallographic coordinates. Denoting the crystallographic coordinates of an atom as  , its position R is given by
, its position R is given by

In the above equation, if the basis vectors are represented using column vectors containing their Cartesian components, then  becomes a matrix, and R a column vector containing the Cartesian coordinates of the atomic position vector,
becomes a matrix, and R a column vector containing the Cartesian coordinates of the atomic position vector,

In the following we will use both representations. Depending on the context,  can therefore be considered as a row vector containing three elements of a vector space, or a 3 by 3 matrix.
can therefore be considered as a row vector containing three elements of a vector space, or a 3 by 3 matrix.

Figure 1. Crystal structure model of β-Si3N4. See also figure 2 for the space group symmetry. (a) Unit cell containing atoms. (b) Basis vectors of the unit cell. (c) Crystal lattice. The filled circle symbols show the lattice points. In this model, the unit cell is assumed to be primitive.
Download figure:
Standard image High-resolution imageIt is assumed that elements of
x
are in the interval  to describe the structure. However it is often the case that we have to compare two positions of atoms which may be in different unit cells, possibly after a space group operation. For example, to identify two coordinates
x
and
to describe the structure. However it is often the case that we have to compare two positions of atoms which may be in different unit cells, possibly after a space group operation. For example, to identify two coordinates
x
and  which correspond to the same location within the unit cell, but are possibly shifted by a lattice vector, it is convenient to bring each element of the difference
which correspond to the same location within the unit cell, but are possibly shifted by a lattice vector, it is convenient to bring each element of the difference  into the interval
into the interval  and then confirm that the length of
and then confirm that the length of  is smaller than a tolerance ε. This is implemented in the phonopy and phono3py codes everywhere by rounding components of
is smaller than a tolerance ε. This is implemented in the phonopy and phono3py codes everywhere by rounding components of  to the nearest integer (
to the nearest integer ( ) and checking
) and checking ![$\left|(\mathbf{a}, \mathbf{b}, \mathbf{c}) [\Delta\boldsymbol{x} - \text{nint}(\Delta\boldsymbol{x})] \right| \lt \epsilon$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn12.gif) .
.
2.2. Space group operation
Various symmetries of crystals and phonons are used in the phonopy and phono3py codes. The most important one is the space group symmetry. A space group operation is composed of a (proper or improper) rotation  and a translation τ. It is often written with a composite notation
and a translation τ. It is often written with a composite notation  which sends position R to
which sends position R to

Using crystallographic coordinates this is written as

or

where
S
can be shown to be a unimodular matrix.
w
contains the components of the translation vector in the crystallographic basis. If Cartesian coordinates were used instead, the matrix representing the rotation would be orthogonal. Representing the space group by  , the crystallographic point group is given by
, the crystallographic point group is given by  . An example of space group operation is shown in figure 2. The crystal structure overlaps with itself after a rotation of 60∘ along the z axis and a
. An example of space group operation is shown in figure 2. The crystal structure overlaps with itself after a rotation of 60∘ along the z axis and a  shift. Therefore
shift. Therefore

must be a space group operation. It means that any atom in the unit cell at point x is sent by the space group operation to a new point

The new point  can be located out of the original unit cell, but an atom with the same atomic type must be found at this new location.
can be located out of the original unit cell, but an atom with the same atomic type must be found at this new location.

Figure 2. Symmetry operations applied on the unit cell of β-Si3N4 whose space-group type is  (No. 176). The left and middle figures illustrate identity and six-fold screw axis operations. Their notations follow the international tables for crystallography volume A [5]. The right figure shows the crystal structure viewed from the top (z-axis) that depicts the six-fold screw axis passing through the origin O.
(No. 176). The left and middle figures illustrate identity and six-fold screw axis operations. Their notations follow the international tables for crystallography volume A [5]. The right figure shows the crystal structure viewed from the top (z-axis) that depicts the six-fold screw axis passing through the origin O.
Download figure:
Standard image High-resolution image2.3. Primitive cell
In most cases, a unit cell as described in section 2.1 is used as the input crystal structure model of a phonon calculation. The most plausible choice of the unit cell is either a primitive cell or a conventional unit cell. The former represents the minimum unit of periodicity of a crystal lattice. It is therefore the one used for Fourier expansions and is necessary for a reciprocal space representation of phonon properties. The Bloch theorem evidenced this lattice translational symmetry. The latter follows the crystallographic convention. Although it provides intuitive shapes of the unit cells (cubic, hexagonal, etc), it may contain several lattice points. Figure 3 shows the conventional unit cell and a primitive cell for silicon crystallized within the face-centred-cubic structure.

Figure 3. Conventional unit cell and primitive cell of silicon. The space group type is  (No. 227). Since the conventional unit cell contains four lattice points with eight atoms, the primitive cell is chosen to have two atoms by
(No. 227). Since the conventional unit cell contains four lattice points with eight atoms, the primitive cell is chosen to have two atoms by  .
.
Download figure:
Standard image High-resolution imageThe conventional unit cell structure is uniquely defined to follow the crystallographic convention, [5] within the freedom due to Euclidean normalizer [5]. On the other hand, the choice of the primitive cell is not unique. This is inconvenient for a systematic handling of crystal structure models in phonon calculations. Therefore, phonopy suggests a predefined transformation matrix  from the conventional unit cell to the primitive cell.
from the conventional unit cell to the primitive cell.  is used as
is used as

where  are the basis vectors of the primitive cell and
are the basis vectors of the primitive cell and  those of the conventional unit cell. The choices of the transformation matrices used in the phonopy and phono3py codes are shown in table 1. For example, the transformation matrix used for silicon in figure 3 is
those of the conventional unit cell. The choices of the transformation matrices used in the phonopy and phono3py codes are shown in table 1. For example, the transformation matrix used for silicon in figure 3 is  . These matrices are equivalent to those presented in table 2 of [6] although those are given for the reciprocal space basis vectors.
. These matrices are equivalent to those presented in table 2 of [6] although those are given for the reciprocal space basis vectors.
Table 1. Choices of transformation matrices  of equation (8) used in the phonopy and phono3py codes. The subscripts X of the matrices
P
X
indicate the centring types: A, B, C for the base centring types, I and F for the body and face centring types, respectively, and R for the (obverse) rhombohedral centring type.
of equation (8) used in the phonopy and phono3py codes. The subscripts X of the matrices
P
X
indicate the centring types: A, B, C for the base centring types, I and F for the body and face centring types, respectively, and R for the (obverse) rhombohedral centring type.
 , , |
 , , |
 , , |
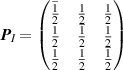 , , |
 , , |
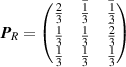 . . |
The crystal lattice is defined as the set of integer linear combinations of primitive lattice vectors. A lattice vector Rl can therefore be written as

In figure 1, if the unit cell is assumed to be primitive, then figure 1(c) shows the crystal lattice. The lattice point may then be chosen to coincide with the origin of each unit cell. Sometimes it is also useful to regard the crystal lattice as generated using the lattice translations of the space group,  , where
I
and
t
denote the identity operation and lattice translation, respectively.
, where
I
and
t
denote the identity operation and lattice translation, respectively.
2.4. Reciprocal space
The crystal structure, and the symmetry explained above, are defined in direct (real) space. In this section, crystallography is briefly summarized in reciprocal space. This is quite useful for our purpose since phonons are described in reciprocal space using wave vectors. It is convenient to introduce a reciprocal lattice whose basis vectors  are solutions of the equation
are solutions of the equation

This equation can be solved to give

where  . The reciprocal lattice points are then obtained from the integer linear combinations of reciprocal basis vectors,
. The reciprocal lattice points are then obtained from the integer linear combinations of reciprocal basis vectors,

Similarly to the atomic positions R, wave vectors q are defined from real linear combinations of the reciprocal lattice vectors,

If the qi
are restricted to the interval  , a primitive cell of reciprocal space is obtained.
, a primitive cell of reciprocal space is obtained.
If we consider a component of a Fourier expansion,  , it is transformed by an operation of the crystallographic point group to the function
, it is transformed by an operation of the crystallographic point group to the function

For this reason, the image of a wave vector through an operation of the crystallographic point group is defined to be

or



The set  with
S
in the crystallographic point group
with
S
in the crystallographic point group  is called the star of q.
is called the star of q.
2.5. Phonon coordinates
Atoms vibrate in the vicinity of their equilibrium positions in crystals. Instantaneous and equilibrium positions of atoms in crystals are denoted by  and
and  , respectively, where l and κ are used to label the lattice points and atoms in the primitive cell of l, respectively. Displacements of atoms are written as
, respectively, where l and κ are used to label the lattice points and atoms in the primitive cell of l, respectively. Displacements of atoms are written as  .
.
In this review, we define the dynamical matrix as [7]

α and mκ
denote the index of the Cartesian coordinates and the mass of the atom κ, respectively.  are the harmonic force constants, defined as the second derivative of the energy with respect to atomic positions, and evaluated at the equilibrium positions [7]. Phonon frequency
are the harmonic force constants, defined as the second derivative of the energy with respect to atomic positions, and evaluated at the equilibrium positions [7]. Phonon frequency  and eigenvector
and eigenvector  are obtained as the solution of the eigenvalue equation of the dynamical matrix in equation (19), which is written as
are obtained as the solution of the eigenvalue equation of the dynamical matrix in equation (19), which is written as

ν labels the phonon band index, and the composite index  is used to consider a phonon mode.
is used to consider a phonon mode.
Equation (20) can also be written in matrix form as

or, because  is hermitian,
is hermitian,

where  is the diagonal matrix whose diagonal elements are
is the diagonal matrix whose diagonal elements are  . Each column of
. Each column of  contains an eigenvector
contains an eigenvector  corresponding to a different band index ν. Elements of each column are ordered as
corresponding to a different band index ν. Elements of each column are ordered as  , where na is the number of atoms in the primitive cell.
, where na is the number of atoms in the primitive cell.
Because  , we have
, we have

and we can choose

Moreover eigenvalues and eigenvectors inherit symmetry properties of the force constants. It can be shown [8] that under a space group operation  the eigenvalues and eigenvectors transform as
the eigenvalues and eigenvectors transform as


where  represents the phonon eigenvector of atom κ in Cartesian coordinates, and
represents the phonon eigenvector of atom κ in Cartesian coordinates, and  is the image of atom κ under the space group operation.
is the image of atom κ under the space group operation.
An actual displacement can be described from a linear combination of phonon eigenvectors. This defines the phonon coordinates  as
as


where N is the number of lattice points in crystal.  is defined for the purpose of convenience in the following sections. The previous equation can be inverted to give
is defined for the purpose of convenience in the following sections. The previous equation can be inverted to give

2.6. BZ
Symmetry property of phonons in reciprocal space is best represented in the BZ [6, 8–12]. In the phonopy and phono3py codes, the BZ is defined as a Wigner–Seitz cell of the reciprocal lattice. To check if a q point belongs to the BZ we proceed as follows. First the basis vectors of the Niggli cell [13–16] are determined. The Niggli cell is a cell with the shortest possible reciprocal basis vectors. Then using integer linear combinations of those Niggli basis vectors, we search for the shortest  . Finally
. Finally  is used as the q point in the BZ.
is used as the q point in the BZ.
Three q points are needed for the three-phonon scattering considered in the phono3py code. In case one or more of those three points is on the BZ surface, we choose the translationally equivalent points on the BZ surface which minimize  .
.
As an example, the BZ of β-Si3N4 is presented in figure 4. The basal plane has the hexagonal shape and  is longer than
is longer than  and
and  because
because  is shorter than
is shorter than  and
and  . By definition,
. By definition,  ,
,  , and
, and  are located on the BZ surface. The high symmetry points and paths in the BZ have special symbols as shown in figure 4(b) [6].
are located on the BZ surface. The high symmetry points and paths in the BZ have special symbols as shown in figure 4(b) [6].

Figure 4. Brillouin zone (BZ) of β-Si3N4. See also figures 1 and 2 for the cell shape in direct space. (a) The first BZ. (b) BZs viewed from  axis direction. Symbols of the special points and paths follow the Bilbao crystallographic server [6].
axis direction. Symbols of the special points and paths follow the Bilbao crystallographic server [6].
Download figure:
Standard image High-resolution image3. Geometry of supercell model
3.1. Supercell construction
In the phonopy and phono3py codes, the supercell approach is employed. The supercell is defined by multiple primitive cells, so that the basis vectors of the supercell  can be represented as the image of the primitive cell basis vectors through an integer matrix
can be represented as the image of the primitive cell basis vectors through an integer matrix  ,
,

Like the unit cell, the supercells are arranged on the supercell lattice. The supercell lattice points are labeled by L, and located from the vectors RL .
The supercell model is constructed using  . The lattice points within the supercell are used to perform the summation found in equation (19). These lattice points can be elegantly obtained using the approach reported by Hart and Forcade [17]. The integer matrix
. The lattice points within the supercell are used to perform the summation found in equation (19). These lattice points can be elegantly obtained using the approach reported by Hart and Forcade [17]. The integer matrix  is first reduced to a diagonal integer matrix
is first reduced to a diagonal integer matrix  by the unimodular matrices
P
and
Q
, where we choose
by the unimodular matrices
P
and
Q
, where we choose  and
and  . This matrix decomposition can be the Smith normal form (SNF), however, it is unnecessary to strictly follow the definition of the SNF.
. This matrix decomposition can be the Smith normal form (SNF), however, it is unnecessary to strictly follow the definition of the SNF.
Using this transformation, equation (30) can be written

with


 and
and  define a new supercell and a new primitive cell, respectively. Because
define a new supercell and a new primitive cell, respectively. Because  , they have the same volumes as the former ones and generate the same lattices. The benefit of using those new primitive and supercell comes from the fact that
D
is diagonal. The new primitive and supercell lattice vectors are therefore collinear.
, they have the same volumes as the former ones and generate the same lattices. The benefit of using those new primitive and supercell comes from the fact that
D
is diagonal. The new primitive and supercell lattice vectors are therefore collinear.
We can easily find the lattice vectors located within the new supercell. If we write  , they are given by the vectors
, they are given by the vectors

with  . This can also be written
. This can also be written


Therefore, the lattice points within the original supercell have coordinates

along the supercell lattice vectors  . The
. The  operation is used to shift the lattice vectors from within the new supercell to within the original supercell.
operation is used to shift the lattice vectors from within the new supercell to within the original supercell.
In figure 5, an example of a supercell construction for a two-dimensional lattice is presented.  is computed as
is computed as

This gives


As can be seen in figure 5,  and
and  are mutually parallel, and the new supercell is simply built by
are mutually parallel, and the new supercell is simply built by  units of the new primitive cell. Since the lattices generated by the original or new basis vectors are the same, the lattice points inside the new supercell are simply brought into the original supercell by supercell lattice translations.
units of the new primitive cell. Since the lattices generated by the original or new basis vectors are the same, the lattice points inside the new supercell are simply brought into the original supercell by supercell lattice translations.

Figure 5. Lattices and basis vectors of primitive cell  , supercell
, supercell  , new primitive cell
, new primitive cell  , new supercell
, new supercell  , and auxiliary supercell
, and auxiliary supercell  . Circle and small filled circle symbols depict lattice points in the supercell and in the new supercell, respectively. The latter points can be brought by the supercell lattice translations to the former points uniquely.
. Circle and small filled circle symbols depict lattice points in the supercell and in the new supercell, respectively. The latter points can be brought by the supercell lattice translations to the former points uniquely.
Download figure:
Standard image High-resolution imageAnother way of constructing the supercell is to employ an auxiliary supercell,

which is defined by a diagonal integer matrix  . This gives
. This gives

We choose  to be an integer matrix. To satisfy it, in the implementation, the diagonal elements of
to be an integer matrix. To satisfy it, in the implementation, the diagonal elements of  are determined by making the smallest parallelepiped of
are determined by making the smallest parallelepiped of  that includes the parallelepiped of
that includes the parallelepiped of  . The lattice points within this auxiliary supercell are then
. The lattice points within this auxiliary supercell are then

with  . Their coordinates along the basis vectors of the auxiliary supercell are
. Their coordinates along the basis vectors of the auxiliary supercell are

while their coordinates along the basis vectors of the original supercell are

As before, we used the operation to shift the lattice points from within the auxiliary supercell to within the original supercell. Notice that every lattice point within the original supercell is obtained from
operation to shift the lattice points from within the auxiliary supercell to within the original supercell. Notice that every lattice point within the original supercell is obtained from  lattice points within the auxiliary supercell. Only one of them should be conserved.
lattice points within the auxiliary supercell. Only one of them should be conserved.
An example is shown in figure 5. Lattice points in the auxiliary supercell are brought into the supercell by the supercell lattice translations. But since  , each lattice point within the original supercell is obtained from two lattice points in the auxiliary supercell.
, each lattice point within the original supercell is obtained from two lattice points in the auxiliary supercell.
For backward compatibility, to preserve indices of atoms in the supercell, it is the latter way of the supercell construction which is used as default in the phonopy and phono3py codes. However, the latter algorithm is much slower than the former, and therefore the former should be used for very larger supercells.
3.2. Commensurate points
From equations (30) and (10), we have

 are the reciprocal basis vectors associated with the supercell. q points given by integer linear combinations of the reciprocal supercell basis vectors are called commensurate with the supercell because they fulfil
are the reciprocal basis vectors associated with the supercell. q points given by integer linear combinations of the reciprocal supercell basis vectors are called commensurate with the supercell because they fulfil  for any supercell lattice vector RL
.
for any supercell lattice vector RL
.
For practical purposes, we are interested in the commensurate points located within the reciprocal primitive cell. Since equation (46) has the same form as equation (30), the same approaches as those explained in section 3.1 can be used for the generation of the commensurate points within the reciprocal primitive cell.
4. Transformation between force constants and dynamical matrices
In equations (19) and (20), force constants are transformed to phonon eigenvalues and eigenvectors. In principle, in equation (19) the summation over lʹ is performed for all lattice vectors. However, in the supercell approach, it is not  which is obtained from numerical simulations, but
which is obtained from numerical simulations, but  , [18] with lʹ restricted to the supercell. It means that the force constants are only known within a restricted region of the crystal, the supercell, and within this region, they are contaminated by the periodic repetitions of the supercell. This also means that the supercell force constants
, [18] with lʹ restricted to the supercell. It means that the force constants are only known within a restricted region of the crystal, the supercell, and within this region, they are contaminated by the periodic repetitions of the supercell. This also means that the supercell force constants  are periodic under a supercell lattice translation while the phase factor
are periodic under a supercell lattice translation while the phase factor  is not, except when q is a wave vector commensurate with the supercell. There is therefore an ambiguity about where to choose the atom
is not, except when q is a wave vector commensurate with the supercell. There is therefore an ambiguity about where to choose the atom  . For commensurate wave vectors, it could be chosen in any periodic repetition of the supercell containing atom
. For commensurate wave vectors, it could be chosen in any periodic repetition of the supercell containing atom  without changing the value of the phase factor. However, for other values of q, this value would be changed, and therefore this choice matters.
without changing the value of the phase factor. However, for other values of q, this value would be changed, and therefore this choice matters.
In the phonopy and phono3py codes, following [18], the phase factors given by the shortest vectors of  are chosen, where RL
are the positions of the supercell lattice points L. This is implemented by searching L by
are chosen, where RL
are the positions of the supercell lattice points L. This is implemented by searching L by

and the results are stored and used many times in the calculation. Since multiple L can be found for each pair of atoms (0κ) in the primitive cell and ( ) in the supercell cell, equation (47) gives a set of L. The dynamical matrix of the supercell is computed from the supercell force constants
) in the supercell cell, equation (47) gives a set of L. The dynamical matrix of the supercell is computed from the supercell force constants  averaging the phase factors of
averaging the phase factors of  as follows, [18]
as follows, [18]

Equation (19) can be inverted to compute force constants from N dynamical matrices, [19]

 can be obtained from known eigenvectors and eigenvalues, as in equation (22), or calculated by equation (48).
can be obtained from known eigenvectors and eigenvalues, as in equation (22), or calculated by equation (48).  can be obtained from
can be obtained from  by restricting the above q sum to the wavevectors
by restricting the above q sum to the wavevectors  commensurate with the supercell. Equation (49) can also be used to transform supercell force constants in a different supercell shape by oversampling
commensurate with the supercell. Equation (49) can also be used to transform supercell force constants in a different supercell shape by oversampling  points. Indeed, we obtain
points. Indeed, we obtain

where subscripts of variables XL and XS mean those defined in different supercell shapes, e.g. larger (L) and smaller (S) supercells, respectively. The original supercell force constants  are given in the smaller supercell. Commensurate points
are given in the smaller supercell. Commensurate points  are sampled with respect to the larger supercell. The transformed supercell force constants
are sampled with respect to the larger supercell. The transformed supercell force constants  are given in the larger supercell. A possible application is to embed anharmonic contribution obtained through self-consistent harmonic approximation using a smaller supercell into the harmonic force constants of a larger supercell [20].
are given in the larger supercell. A possible application is to embed anharmonic contribution obtained through self-consistent harmonic approximation using a smaller supercell into the harmonic force constants of a larger supercell [20].
5. Non-analytical term correction
Long-range dipole–dipole interactions are difficult to capture in a supercell approach. Therefore it is treated with the help of a model, the so-called non-analytical term correction (NAC) [1, 21–23].
At the commensurate points, this contribution is already included in equation (48) via the supercell force constants. However, at general q points it is not. Gonze and Lee [1] have formulated this contribution to the dynamical matrix, and in the phonopy and phono3py codes only the reciprocal space term of the dipole–dipole interaction contribution to the dynamical matrix is calculated. It is given by

where β and γ indicate the Cartesian coordinates. If the polarization is called P, then  are the Born effective charges at zero electric field,
are the Born effective charges at zero electric field,  .
.  is the high-frequency static dielectric constant tensor. Λ is a parameter adjusted together with the cutoff radius used for the summation over G. A translational invariance condition can be applied to equation (51) by [1]
is the high-frequency static dielectric constant tensor. Λ is a parameter adjusted together with the cutoff radius used for the summation over G. A translational invariance condition can be applied to equation (51) by [1]

As said already, at the commensurate points, this dipole–dipole contribution is already included in equation (48) via the supercell force constants. Therefore, equation (51) is used for the interpolation of the dynamical matrix at general q points. The procedure is reported in [1], which is described shortly as follows. At the commensurate points  , the short-range dynamical matrix is calculated as
, the short-range dynamical matrix is calculated as

Next, the short-range supercell force constants  are obtained from
are obtained from  using equation (49), then the short-range dynamical matrix
using equation (49), then the short-range dynamical matrix  is calculated at the general q point by equation (48), i.e.
is calculated at the general q point by equation (48), i.e.

Finally, the dynamical matrix at the general q point with NAC is obtained by

An example of the application of NAC to wurtzite-type AlN is shown in figure 6. As can be seen, the correction is significant near the Γ point. It also shows the directional dependence near the Γ, in the direction of K and A, due to the non-spherical symmetry of  and
and  of AlN.
of AlN.

Figure 6. Calculated phonon band structure of wurtzite-type AlN using the  supercell with (solid curves) and without (dashed curves) non-analytical term correction (NAC). Circle and square symbols depict phonon frequencies at
supercell with (solid curves) and without (dashed curves) non-analytical term correction (NAC). Circle and square symbols depict phonon frequencies at  , where filled and open symbols indicate phonon modes calculated with and without NAC, respectively. Wave vector path was selected by Seek-path [24, 25].
, where filled and open symbols indicate phonon modes calculated with and without NAC, respectively. Wave vector path was selected by Seek-path [24, 25].
Download figure:
Standard image High-resolution image6. Regular grid in reciprocal primitive cell
When phonon properties at q points need to be integrated over the BZ, a technique often used is to discretize the reciprocal space. Usually, reciprocal primitive cells are uniformly sampled by a regular grid such as [26], even if other choices are possible [27]. Traditionally, the regular grid is defined by evenly dividing the reciprocal basis vectors, as shown in figure 7(a). However, other regular grids may be chosen, as shown in figure 7(b). For example, the grid shown in figure 7(b) is defined by dividing evenly the reciprocal basis vectors of the conventional unit cell instead. This is a type of generalized regular grid, [28–31] as will be explained later.

Figure 7. Different Γ-centred regular grids in plane reciprocal primitive cell ( ). Filled circle symbols depict the grid points. Open circle symbols show the grid points equivalent to some other grid points by periodicity. Shaded area indicates a microzone [2] and
). Filled circle symbols depict the grid points. Open circle symbols show the grid points equivalent to some other grid points by periodicity. Shaded area indicates a microzone [2] and  and
and  give the basis vectors of the microzone. (a) Traditional regular grid. (b) A type of generalized regular grid [28–31].
give the basis vectors of the microzone. (a) Traditional regular grid. (b) A type of generalized regular grid [28–31].
Download figure:
Standard image High-resolution image6.1. Traditional regular grid
Traditionally, the volume of the reciprocal primitive cell is divided into uniform microzones so that the basis vectors of the reciprocal primitive cell are simply integer multiples of the basis vectors of each microzone  . The equation which defines the microzone basis vectors is then
. The equation which defines the microzone basis vectors is then

with  .
.
A 2D example of this microzone is shown in figure 7(a). The q points of the grid points are represented by integer linear combinations of the microzone basis vectors plus a possible rigid shift  . Therefore their coordinates in the
. Therefore their coordinates in the  basis are
basis are

To conserve symmetry, ni
and si
are chosen so that the microzone lattice with the shift is invariant under the crystallographic point group  .
.
6.2. Generalized regular grid
A generalized regular grid is defined using a conventional unit cell related to the primitive cell by equation (8). The reciprocal conventional unit cell is therefore

Microzones can be defined considering that their basis vectors are integer divisions of the reciprocal basis vectors of the conventional unit cell. This is written as

A 2D example of this microzone is shown in figure 7(b). From equations (58) and (59), we have

The grid matrix  is an integer matrix.
is an integer matrix.  is the number of translationally nonequivalent grid points in the reciprocal primitive cell. When
is the number of translationally nonequivalent grid points in the reciprocal primitive cell. When  is not a diagonal matrix, the grid generated by equation (60) is a generalized regular grid, otherwise we obtain the traditional regular grid as described in section 6.1. Notice however that the generalized regular grid may be transformed into the traditional regular grid of some reciprocal cell using an SNF kind of decomposition, as explained in section 3.1. This is shown in the next section.
is not a diagonal matrix, the grid generated by equation (60) is a generalized regular grid, otherwise we obtain the traditional regular grid as described in section 6.1. Notice however that the generalized regular grid may be transformed into the traditional regular grid of some reciprocal cell using an SNF kind of decomposition, as explained in section 3.1. This is shown in the next section.
6.3. Indexing of grid points
The integer matrix  can be reduced to a diagonal integer matrix such as
can be reduced to a diagonal integer matrix such as  as has been employed in section 3.1. This property of the matrix decomposition is similarly used to index grid points by integer numbers of
as has been employed in section 3.1. This property of the matrix decomposition is similarly used to index grid points by integer numbers of  [17] in the phono3py code. Note that
[17] in the phono3py code. Note that  . Equation (60) is rewritten as
. Equation (60) is rewritten as

Denoting


we have

Since
Q
is a unimodular matrix,  and
and  generate the same reciprocal primitive lattice. Similarly
generate the same reciprocal primitive lattice. Similarly  and
and  generate the same microzone lattice due to the unimodular matrix
generate the same microzone lattice due to the unimodular matrix  .
.
Equation (64) has the same form as equation (56) with  . Therefore the q points of the grid points are calculated similarly as equation (57) but in the basis
. Therefore the q points of the grid points are calculated similarly as equation (57) but in the basis  ,
,

Notice that in the above equation we use  rather than si
. In fact for practical purposes, it may be convenient to define the grid shift
rather than si
. In fact for practical purposes, it may be convenient to define the grid shift  in the basis
in the basis  . Therefore we have
. Therefore we have  .
.
The indexing of the grid points in the reciprocal primitive cell  is a trivial task. Indeed, in the phonopy and phono3py codes, each grid point is bijectively mapped to an integer p by
is a trivial task. Indeed, in the phonopy and phono3py codes, each grid point is bijectively mapped to an integer p by

With this definition,  .
.
The q points generated this way are located within the reciprocal cell  . They could be shifted to the reciprocal cell
. They could be shifted to the reciprocal cell  by reciprocal lattice vector translations if needed. Notice however that to obtain the integer p through equation (66) for a general integer triplet
by reciprocal lattice vector translations if needed. Notice however that to obtain the integer p through equation (66) for a general integer triplet  , modulo
, modulo  is required to locate the point within the cell
is required to locate the point within the cell  . Indeed,
. Indeed,  and
and  indicate different locations in q space, although they are equivalent points due to periodicity.
indicate different locations in q space, although they are equivalent points due to periodicity.
6.4. Symmetry of generalized regular grids
For a Γ centred grid ( ), the coordinates of a q point in the basis
), the coordinates of a q point in the basis  are given by
are given by

where  and the reciprocal lattice vector
G
is chosen to bring qi
in the interval
and the reciprocal lattice vector
G
is chosen to bring qi
in the interval  . If the regular grid is a traditional one, we simply have
. If the regular grid is a traditional one, we simply have  . As shown by equation (18), the image of this q point through an operation of the crystallographic point group is given by
. As shown by equation (18), the image of this q point through an operation of the crystallographic point group is given by

where we have added a reciprocal lattice vector  to shift
to shift  of the image point in the interval
of the image point in the interval  . If
. If  belongs to the grid we have defined, for all
belongs to the grid we have defined, for all  in the crystallographic point group, we will say that the grid is invariant under the crystallographic point group. If it is so,
in the crystallographic point group, we will say that the grid is invariant under the crystallographic point group. If it is so,  can be written as
can be written as

with  . Comparing equations (68) and (69), we obtain
. Comparing equations (68) and (69), we obtain


Because
Q
and  are unimodular, and
D
contains the number of divisions along
are unimodular, and
D
contains the number of divisions along  , the last term is always a reciprocal lattice vector. The matrix
, the last term is always a reciprocal lattice vector. The matrix

is the matrix representation of  in the
in the  basis. Its determinant is always 1 or −1, but for
basis. Its determinant is always 1 or −1, but for  to be integer, it has to have integer entries, and therefore be unimodular. This is checked in the phono3py code, and if it is true for all
to be integer, it has to have integer entries, and therefore be unimodular. This is checked in the phono3py code, and if it is true for all  in the crystallographic point group, the regular grid follows the crystallographic point group, and we consider it is properly defined. The last equation can also be written as
in the crystallographic point group, the regular grid follows the crystallographic point group, and we consider it is properly defined. The last equation can also be written as

and an equivalent strategy is to check that both sides are unimodular.
Introducing the subgrid shift
s
(see section 6.3) can further break the symmetry of the regular grid. In equation (67)
m
is replaced by  and
and  by
by  in equation (69). Requiring the shift to be invariant
in equation (69). Requiring the shift to be invariant  under an operation of the crystallographic point group,
under an operation of the crystallographic point group,  , we obtain the condition
, we obtain the condition

against all  in the crystallographic point group.
in the crystallographic point group.
6.5. Double grid for subgrid shift
To satisfy the crystallographic symmetry, the subgrid shift si
is normally chosen to be either 0 or  . It is better to treat grid point arithmetic by integers for the computer implementation and its performance. This is realized by doubling
m
and
s
. This well-known technique, e.g. presented in [3], is directly usable for the generalized regular grid in section 6.2. As a choice, we define it by
. It is better to treat grid point arithmetic by integers for the computer implementation and its performance. This is realized by doubling
m
and
s
. This well-known technique, e.g. presented in [3], is directly usable for the generalized regular grid in section 6.2. As a choice, we define it by

and the q points are given like in equation (67) by

The symmetry operation is implemented as

The index of the grid point  is obtained by equation (66) after recovering
is obtained by equation (66) after recovering  using equation (75),
using equation (75),

In the phono3py code, an integer vector of  is used for the implementation, instead of
s
, to avoid floating point arithmetic.
is used for the implementation, instead of
s
, to avoid floating point arithmetic.
6.6. Grid points in BZ
In this section, the BZ grid points are defined to include different q points on the BZ surface that may be equivalent grid points by reciprocal lattice translations, in addition to the grid points inside the BZ.
For a Γ centred grid, in the  basis, each BZ grid point is represented by the integer triplet
basis, each BZ grid point is represented by the integer triplet

where  is chosen to minimize the norm of
is chosen to minimize the norm of  . Therefore,
. Therefore,  is written as
is written as

Multiple  can be found when the grid point is on the BZ surface.
can be found when the grid point is on the BZ surface.
In the phono3py code, equation (80) is implemented as follows. As the first step, reduced basis vectors of the reciprocal primitive cell are obtained by using the Niggli reduction or any reduction scheme. This is written using the change-of-basis matrix  as
as

where  is unimodular. This gives
is unimodular. This gives


The second line defines the reciprocal lattice vector G . G is divided into two pieces,

Nearest integers of  are stored in
are stored in  , which is formally written as
, which is formally written as

to bring the following  closer to the origin,
closer to the origin,

 that minimizes
that minimizes  is searched in
is searched in  . Finally,
. Finally,  is obtained as
is obtained as

As written above,
m
and p are in one-to-one correspondence and multiple  can be found for each
m
or p. In the phono3py code,
can be found for each
m
or p. In the phono3py code,  is calculated and stored at the initial step.
is calculated and stored at the initial step.
With the subgrid shift,
m
in the equations above are replaced by  . Either with or without the subgrid shift,
. Either with or without the subgrid shift,  is determined using the double grid technique presented in section 6.5. From stored
is determined using the double grid technique presented in section 6.5. From stored  , the BZ q points are obtained by
, the BZ q points are obtained by

6.7. Irreducible grid points
Using symmetry properties of phonons, e.g. equation (25), the computation required for the phonon calculations can be reduced. Indeed, many phonon properties result from the BZ integration of functions of a single q variable. Collecting the values of those functions for the q points in the irreducible part of the BZ only may speed up the calculation and also save memory space. When the BZ integration is performed on a regular grid, the irreducible q points are obtained from the irreducible grid points. In this section, we explain how to obtain those irreducible grid points, where it is assumed that the regular grid satisfies the symmetry as described in section 6.4.
A 2D example of BZ is presented in figure 8. A symmetry operation acts on a q point as  . The star of q is the set of those points written as
. The star of q is the set of those points written as  , where one q point represents its star. The irreducible BZ is the set of representative q points of all stars in the BZ. In figure 8(a), the 2D BZ has the eight-fold symmetry of
, where one q point represents its star. The irreducible BZ is the set of representative q points of all stars in the BZ. In figure 8(a), the 2D BZ has the eight-fold symmetry of  , and the irreducible BZ is depicted by the shaded area.
, and the irreducible BZ is depicted by the shaded area.

Figure 8. (a) 2D Brillouin zone (BZ). Shaded area is a choice of the irreducible BZ. (b)  regular grid on the BZ including the BZ surface. Filled and open circle symbols show the grid points belonging to the irreducible BZ under the
regular grid on the BZ including the BZ surface. Filled and open circle symbols show the grid points belonging to the irreducible BZ under the  symmetry and the other grid points, respectively. (c) The irreducible grid points may not be located in a connected space.
symmetry and the other grid points, respectively. (c) The irreducible grid points may not be located in a connected space.
Download figure:
Standard image High-resolution imageIn a phonon calculation, q points are sampled on a regular grid, and the irreducible grid points are defined as the grid points that belong to the irreducible BZ as shown in figure 8(b). In practical calculations, the irreducible grid points considered may however not be located in a connected space, as shown in figure 8(c). This simply means that the selected representative point in the star is located somewhere else.
In the phonopy and phono3py codes, the irreducible grid points are obtained as follows. Each grid point indexed by p is represented in the double grid of equation (75). All symmetry operations  of the crystallographic point group are applied to the grid point as given by equation (77). The grid point index of the rotated grid point,
of the crystallographic point group are applied to the grid point as given by equation (77). The grid point index of the rotated grid point,  , is recovered successively applying equations (78) and (66). The minimum value in
, is recovered successively applying equations (78) and (66). The minimum value in  is chosen as the irreducible grid point. The mappings of the grid point indices
is chosen as the irreducible grid point. The mappings of the grid point indices ![$p \xrightarrow[]{\{\mathcal{S}\}} \underline{p}$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn226.gif) are stored for all grid points in
are stored for all grid points in  , and the unique elements of
, and the unique elements of  give the irreducible grid points of the regular grid. It is important to perform these operations only by integers for computational efficiency.
give the irreducible grid points of the regular grid. It is important to perform these operations only by integers for computational efficiency.
6.8. q point triplets
In the phono3py code, triplets of q points,  , are considered for the computation of the phonon–phonon interaction. This interaction is therefore not a function of a single q variable. The number of grid points
, are considered for the computation of the phonon–phonon interaction. This interaction is therefore not a function of a single q variable. The number of grid points  becomes large when dense sampling is used, e.g.
becomes large when dense sampling is used, e.g.  in the study of [20]. Then, the number of triplets of q points becomes
in the study of [20]. Then, the number of triplets of q points becomes  , which is a really huge number for numerical computations and also storing in the memory of computers.
, which is a really huge number for numerical computations and also storing in the memory of computers.
Due to lattice translational symmetry, elements of the three phonon interaction strength can be non-zero only if  [32, 33]. This is used to reduce the computation of the interactions for all triplets
[32, 33]. This is used to reduce the computation of the interactions for all triplets  to all pairs of points
to all pairs of points  . To satisfy this condition, the Γ centred regular grid is used in the phonon–phonon interaction calculation of the phono3py code. Moreover, the crystallographic point group operations, time-reversal symmetry, and permutation symmetry for each pair of q and
. To satisfy this condition, the Γ centred regular grid is used in the phonon–phonon interaction calculation of the phono3py code. Moreover, the crystallographic point group operations, time-reversal symmetry, and permutation symmetry for each pair of q and  are used to reduce further the number of interactions to be computed. However, even doing so, the number of the combination of two q points is still large. Therefore, in this section, we describe the strategy used to make those computations practical.
are used to reduce further the number of interactions to be computed. However, even doing so, the number of the combination of two q points is still large. Therefore, in this section, we describe the strategy used to make those computations practical.
As mentioned in section 6.7, many phonon related properties are written as sum of functions of a single variable q, such as  . For example, under the relaxation time approximation, the LTC computed in the phono3py code can be written as
. For example, under the relaxation time approximation, the LTC computed in the phono3py code can be written as

where  and
and  denote the phonon mode heat capacity and group velocity, respectively,
denote the phonon mode heat capacity and group velocity, respectively,


In equation (90),  and
and  denote the reduced Planck constant and the Boltzmann constant, respectively, and T is the temperature.
denote the reduced Planck constant and the Boltzmann constant, respectively, and T is the temperature.  in equation (89) is the phonon lifetime, and the reciprocal of
in equation (89) is the phonon lifetime, and the reciprocal of  is calculated from the phonon–phonon interaction strength in the phono3py code. The explicit equation used for
is calculated from the phonon–phonon interaction strength in the phono3py code. The explicit equation used for  is given at equation (111).
is given at equation (111).
The LTC of equation (89) is conveniently computed iterating over irreducible q points. Indeed, the mode heat capacity and the lifetime have the same symmetry as the phonon band structure, equation (25),


and the derivative of equation (25) gives

Therefore, the summation in equation (89) can be reduced to a summation over the irreducible grid points. Denoting  the irreducible points, we obtain
the irreducible points, we obtain

where the last factor is the number of branches in the star of  ,
,  , divided by the cardinality of the crystallographic point group,
, divided by the cardinality of the crystallographic point group,  .
.
When each calculation of  is computationally demanding, the computation of
is computationally demanding, the computation of  at different
at different  points may be distributed over multiple or many computer nodes. Therefore, it is convenient to have a set of q point triplets at fixed
points may be distributed over multiple or many computer nodes. Therefore, it is convenient to have a set of q point triplets at fixed  in
in  . As shown in equation (95), in this strategy,
. As shown in equation (95), in this strategy,  is chosen from the irreducible BZ. Now, because
is chosen from the irreducible BZ. Now, because  must be kept fixed, it is not the full set of operations of the crystallographic point group which should be used to reduce the summation over
must be kept fixed, it is not the full set of operations of the crystallographic point group which should be used to reduce the summation over  , but a subgroup which lets
, but a subgroup which lets  invariant. This subgroup is known as the point group of
invariant. This subgroup is known as the point group of  ,
,

Consequently,  is chosen in the irreducible part of the BZ defined by
is chosen in the irreducible part of the BZ defined by  . Finally,
. Finally,  is computed as
is computed as  where G is a reciprocal lattice vector chosen to shift
where G is a reciprocal lattice vector chosen to shift  within the BZ of the same origin. For phonons, one of
within the BZ of the same origin. For phonons, one of  and
and  is chosen due to the permutation symmetry.
is chosen due to the permutation symmetry.
In the phono3py code, q,  , and
, and  are taken from the BZ of the same origin. When some of them are on the BZ surface, they are chosen among their translationally equivalent points on the BZ surface so that the triplet minimizes
are taken from the BZ of the same origin. When some of them are on the BZ surface, they are chosen among their translationally equivalent points on the BZ surface so that the triplet minimizes  of
of  .
.
This triplet search is implemented using the irreducible grid points and the BZ grid points described in sections 6.7 and 6.6, respectively. The q points  ,
,  , and
, and  are represented by the BZ grid points
are represented by the BZ grid points  ,
,  , and
, and  , respectively. As shown in figure 9(a),
, respectively. As shown in figure 9(a),  is chosen in the irreducible grid points. The point
is chosen in the irreducible grid points. The point  breaks the crystallographic point-group
breaks the crystallographic point-group  . Using
. Using  given by equation (72), the point group of
given by equation (72), the point group of  is obtained as
is obtained as

As shown in figure 9(b),  is sampled from the irreducible grid points of
is sampled from the irreducible grid points of  . The third grid point is given by
. The third grid point is given by  Finally,
Finally,  ,
,  , or
, or  may be shifted to minimize
may be shifted to minimize  if some of them are on the BZ surface as explained for
if some of them are on the BZ surface as explained for  above.
above.

Figure 9. A strategy to choose a q point triplet. The same 2D Brillouin zone (BZ) as figure 8 is used. The star symbols depict three q points in the BZ under the constraint of  . (a)
q
is chosen from the irreducible grid points of
. (a)
q
is chosen from the irreducible grid points of  symmetry.
q
works as the stabilizer of the subgroup of
symmetry.
q
works as the stabilizer of the subgroup of  symmetry. (b)
symmetry. (b)  is chosen in the irreducible grid points of the subgroup. (c)
is chosen in the irreducible grid points of the subgroup. (c) 
 where
G
of the shortest
where
G
of the shortest  , i.e.
, i.e.  in this example, is chosen.
in this example, is chosen.
Download figure:
Standard image High-resolution image7. Tetrahedron method
Once implemented, tetrahedron methods are easy to use and robust. Moreover, when the code is written in a modular way, it can be reusable for different kinds of BZ integrations. The phonopy and phono3py codes employ a linear tetrahedron method where several techniques are picked and mixed from the various reports, notably, by MacDonald et al [2] and Blöchl et al [3]
The implementation in the phonopy and phono3py codes is structured as schematically illustrated in figure 10. The bottom of the structure is the regular grid. The parallelepiped formed by the basis vectors of the reciprocal primitive cell is divided into many smaller parallelepiped microzones on the regular grid. Every microzone has the same shape, defined by the basis vectors  , as presented in section 6. In the case of the traditional regular grid, equation (56) defines the microzones to be considered, while for the generalized regular grid equation (59) is used. Each of the microzones is divided into six tetrahedra in the same way as shown in figure 11.
, as presented in section 6. In the case of the traditional regular grid, equation (56) defines the microzones to be considered, while for the generalized regular grid equation (59) is used. Each of the microzones is divided into six tetrahedra in the same way as shown in figure 11.

Figure 10. Implementation of the linear tetrahedron method in the phonopy and phono3py codes. This routine is provided as a module that returns integration weights. In the module, each layer depends on the lower layers. Crystal symmetry is treated outside of this module.
Download figure:
Standard image High-resolution image
Figure 11. A scheme to divide a parallelepiped microzone into six tetrahedra with the same volume [3]. The eight vertices of the parallelepiped are shared by 6, 6, 2, 2, 2, 2, 2, and 2 tetrahedra, respectively.
Download figure:
Standard image High-resolution imageThe integral over the BZ to be performed is then regarded as a sum of contributions from those tetrahedra. The function to be integrated is linearly interpolated within each tetrahedron (see section 7.2) and therefore the integration within each tetrahedron can be done analytically. To interpolate linearly a tridimensional function one needs four values. Each tetrahedron has four vertices. Therefore the values of the function to be integrated on the vertices of the tetrahedra are used to build the interpolations.
Different recipes can be used to implement this program. We follow [2] as well as [3] to obtain integration weights for grid points rather than for the tetrahedra themselves. Indeed, as it will be shown, it is possible to rearrange the contributions from tetrahedra to contributions from the grid points. This is useful to make the tetrahedron method easy use, and to make it similar to smearing methods. In addition, this way, the symmetry of the regular grid described in section 6.4 is applied straightforwardly to the integration weights.
7.1. Division into six tetrahedra
The scheme to divide the microzone into six tetrahedra follows [3] reported by Blöchl et al We choose a shortest main diagonal of the parallelepiped  . Then the six tetrahedra are selected sharing this main diagonal as shown in figure 11.
. Then the six tetrahedra are selected sharing this main diagonal as shown in figure 11.
7.2. Functions
In this section we introduce the functions  ,
,  ,
,  , and
, and  to be computed using the linear tetrahedron method. The notation roughly follows [2]. Moreover, the band index ν is not written explicitly, since each band can be treated independently. It is therefore understood that a sum over the bands should be performed at the end of the calculation.
to be computed using the linear tetrahedron method. The notation roughly follows [2]. Moreover, the band index ν is not written explicitly, since each band can be treated independently. It is therefore understood that a sum over the bands should be performed at the end of the calculation.
The density of states  is written as
is written as


The definition is written at the first line, the approximation obtained from the linear tetrahedron method at the second line and an obvious definition of gi
, the contribution of tetrahedron i, at the third line. Here i is an index running throughout the 6N tetrahedra, and  is the volume of a tetrahedron. The values of frequency at the vertices are assumed to be arranged in ascending order,
is the volume of a tetrahedron. The values of frequency at the vertices are assumed to be arranged in ascending order,  .
.
The integrated density of states, or number of states function,  , is written following the same conventions,
, is written following the same conventions,


Weighted density of states frequently appears in phonon calculations. They are defined by

where  is a function of q and we used the notation
is a function of q and we used the notation  at the vertex k of the tetrahedron labelled by i.
at the vertex k of the tetrahedron labelled by i.  are given in appendix
are given in appendix
Finally, the integral of  over frequency,
over frequency,  , is written as
, is written as

where  are given in appendix
are given in appendix
In appendix  ,
,  , are stated from [2]. To derive those formulae, the calculation of
, are stated from [2]. To derive those formulae, the calculation of  is considered first, and
is considered first, and  is obtained through differentiation.
is obtained through differentiation.  and
and  are just special cases for F = 1. The
are just special cases for F = 1. The  function in
function in  shows that computing the contribution from one tetrahedron is related to the estimation of the volume
shows that computing the contribution from one tetrahedron is related to the estimation of the volume  , which can be described from an intersection of a tetrahedron with a plane
, which can be described from an intersection of a tetrahedron with a plane  , since
, since  is now assumed to be a linear function of q. As shown in figure 12 the volume will assume different shapes depending on the value of ω. Different formulae are therefore obtained for the different cases to be considered.
is now assumed to be a linear function of q. As shown in figure 12 the volume will assume different shapes depending on the value of ω. Different formulae are therefore obtained for the different cases to be considered.

Figure 12. Different linear cuts by ω plane of a tetrahedron. (a)  , (b)
, (b)  , and (c)
, and (c)  [2].
[2].
Download figure:
Standard image High-resolution image7.3. Integration weights
In equation (102),  is expressed as a summation over 6N tetrahedra and four vertices. Following Blöchl et al in [3], this can be rearranged as a sum over grid points. This scheme hides the cumbersome handling of tetrahedra from outside the linear tetrahedron method module as shown in figure 10. Once integration weights, that are described in this section, are computed, the data structure is shared with smearing methods and the symmetry handling is made easy.
is expressed as a summation over 6N tetrahedra and four vertices. Following Blöchl et al in [3], this can be rearranged as a sum over grid points. This scheme hides the cumbersome handling of tetrahedra from outside the linear tetrahedron method module as shown in figure 10. Once integration weights, that are described in this section, are computed, the data structure is shared with smearing methods and the symmetry handling is made easy.
 can be rewritten as
can be rewritten as

where p denotes the grid points (see section 6.3), and ip
is the index of a tetrahedron in the 24 tetrahedra that are sharing the grid point p (see figure 11 for  ). The composite index
). The composite index  indicates a grid point, and
indicates a grid point, and  selects the vertex which is identical to the grid point p in the four vertices of the tetrahedron ip
.
selects the vertex which is identical to the grid point p in the four vertices of the tetrahedron ip
.
Since  is only non-zero when
is only non-zero when  is the point p, we write
is the point p, we write  . The summation (104) becomes
. The summation (104) becomes

where

 has therefore been expressed as a summation over grid points. This integral is given by the weighted sum of the values of the function F at points p, Fp
, with weights wp
given by equation (106).
has therefore been expressed as a summation over grid points. This integral is given by the weighted sum of the values of the function F at points p, Fp
, with weights wp
given by equation (106).
A 2D illustration of the summation (105) is presented in figure 13. Figure 13(a) shows that each parallelogram is cut in two triangles, and the four vertices of each parallelogram are shared by 2, 2, 1, and 1 of those triangles. Six triangles share the grid point marked by the circle symbol as shown in figure 13(b). This shows that only six terms are summed up in the summation (106) because of  . For the tetrahedra in 3D, 24 terms are summed up at each grid point in the summation (106).
. For the tetrahedra in 3D, 24 terms are summed up at each grid point in the summation (106).

Figure 13. 2D schematic illustration of rearrangement of summation (104). (a) Each parallelogram is cut into two triangles. Four vertices of the parallelogram are shared by 2, 2, 1, and 1 triangles, respectively. (b) Six triangles (shaded) that share a grid point (circle symbol) contribute to the integration weight of the grid point.
Download figure:
Standard image High-resolution imageTo compute equation (106), it is convenient to represent the positions of grid points using shifts from the grid point p,  . The 2D example is depicted in figure 14. The main diagonal
. The 2D example is depicted in figure 14. The main diagonal  is chosen. For this main diagonal, the six triangles that share the central point are uniquely determined. They are represented by the following
is chosen. For this main diagonal, the six triangles that share the central point are uniquely determined. They are represented by the following  of the apices:
of the apices:  .
.

Figure 14. 2D schematic illustration of grid point shifts  from the focused grid point (centre) to the neighbours.
from the focused grid point (centre) to the neighbours.
Download figure:
Standard image High-resolution imageFor the linear tetrahedron method, there are four choices for the main diagonal, and for each choice of the main diagonal, the  of the vertices of the 24 tetrahedra shared by a grid point are predetermined. Therefore, the datasets of the 24 tetrahedra of the four main diagonals are predetermined and hard-corded in the phonopy and phono3py codes as
of the vertices of the 24 tetrahedra shared by a grid point are predetermined. Therefore, the datasets of the 24 tetrahedra of the four main diagonals are predetermined and hard-corded in the phonopy and phono3py codes as  integer array whose elements are in
integer array whose elements are in  for the regular grid.
for the regular grid.
For the generalized regular grid, the shifts −1 or 1 correspond to the coordinates of the neighbouring points in the  basis. The coordinates of those shifts are however easily obtained in the
basis. The coordinates of those shifts are however easily obtained in the  basis used in the calculation,
basis used in the calculation,  , since
, since

where equation (63) is used in the last equation.
7.4. Use of symmetry of grid points
The symmetry of grid points can be applied to the linear tetrahedron method. The symmetry is used to prepare the input dataset of a grid point required by the linear tetrahedron method module as illustrated in figure 10. Once the input dataset is made, symmetry information is unnecessary for the linear tetrahedron method module.
In this section, we denote  and the mapping of the grid point
and the mapping of the grid point ![$p \xrightarrow[]{\{\mathcal{S}\}} \underline{p}$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn349.gif) defined in section 6.7 is written as
defined in section 6.7 is written as  . Under the condition
. Under the condition  , which is satisfied by the phonon frequencies, the integration weights fulfil the relation
, which is satisfied by the phonon frequencies, the integration weights fulfil the relation

This is an approximation since the set of 24 tetrahedra may not follow the crystallographic point group, i.e. the rotated tetrahedron may not be mapped to any of the 24 tetrahedra. However, equation (108) is a very good approximation.
When Fp
has the symmetry  , equation (105) can be written as
, equation (105) can be written as

The right hand side of Approx. (109) can therefore be computed from the values at irreducible grid points only. Defining the multiplicity of the irreducible points as  , the right hand side of Approx. (109) is written as
, the right hand side of Approx. (109) is written as

Here  is equivalent to
is equivalent to  in equation (95), i.e. the number of branches in the star of
in equation (95), i.e. the number of branches in the star of  .
.
7.5. Triplets integration weights
As shown in [33], the phonon linewidth computed by the phono3py code, due to phonon–phonon interactions, is given by


If we consider q to be fixed, the summations to be performed over the BZ have the form

where  is an arbitrary function of q,
is an arbitrary function of q,  and
and  .
.  is a function of
is a function of  and
and  . For example, in equation (111), to compute the contribution from the first delta function we need to consider,
. For example, in equation (111), to compute the contribution from the first delta function we need to consider,  .
.
In the above equation,  means 1 when
means 1 when  otherwise 0. In equation (111) this factor is included in
otherwise 0. In equation (111) this factor is included in  [33]. For given q,
[33]. For given q,  , and
, and  in the BZ,
in the BZ,  is chosen so that
is chosen so that  is smallest. At fixed q, for given
is smallest. At fixed q, for given  , only a single
, only a single  contributes. This allows to eliminate the summation over
contributes. This allows to eliminate the summation over  , therefore we obtain
, therefore we obtain


This quantity has therefore the form of the function  we considered previously. To apply the linear tetrahedron method to
we considered previously. To apply the linear tetrahedron method to  we need to know the value of the functions
we need to know the value of the functions  and
and  at the vertices
at the vertices  around the point
around the point  . They are given by
. They are given by


It means that  for the corresponding neighbouring point of
for the corresponding neighbouring point of  . Using the values
. Using the values  as the input dataset of the linear tetrahedron method, the triplets integration weights are computed in the same way as written in section 7.3.
as the input dataset of the linear tetrahedron method, the triplets integration weights are computed in the same way as written in section 7.3.
8. Random displacement generation
Once phonon frequency  and eigenvector
and eigenvector  are obtained in a supercell, they can be used to generate atomic configurations relevant at finite temperatures. This can be achieved using equation (27) and writing
are obtained in a supercell, they can be used to generate atomic configurations relevant at finite temperatures. This can be achieved using equation (27) and writing

where both  and
and  are real variables. It is shown in [34] that they are real normal coordinates, fulfilling the equation of the harmonic oscillator. Their probability density is therefore a Gaussian,
are real variables. It is shown in [34] that they are real normal coordinates, fulfilling the equation of the harmonic oscillator. Their probability density is therefore a Gaussian,

with

where  is the Bose–Einstein distribution function.
is the Bose–Einstein distribution function.
Substituting equation (118) into equation (27) and remembering  , we obtain
, we obtain


Here, A is a set of q points commensurate with the supercell, where  becomes real (e.g. Γ point) or
becomes real (e.g. Γ point) or  , and B includes other commensurate q points in the one side of the BZ and
, and B includes other commensurate q points in the one side of the BZ and  . Namely, only one of q or −q should be included in the summation over
. Namely, only one of q or −q should be included in the summation over  [34].
[34].
Finally, generating random numbers following equation (119) and using them for  and
and  in equation (122), random displacements
in equation (122), random displacements  in a supercell can be generated systematically at a given temperature.
in a supercell can be generated systematically at a given temperature.
The random structures generated in this way can be used to investigate the impact of phonon excitation on various physical properties, such as electronic structure and magnetism, at finite temperatures. Indeed, the investigated quantity is computed for each random structure generated, and the average of values obtained for those random structures is computed to mimic the effect of temperature.
We note that this approach is physically valid only when all phonon modes are dynamically stable ( at all commensurate q points. This condition is usually satisfied in many compounds where lattice anharmonicity is not very strong. However, in some anharmonic systems, including high-temperature metastable phases, the harmonic approximation may yield unstable phonon modes.
at all commensurate q points. This condition is usually satisfied in many compounds where lattice anharmonicity is not very strong. However, in some anharmonic systems, including high-temperature metastable phases, the harmonic approximation may yield unstable phonon modes.
Even when all phonons are stable, their frequencies may deviate from experimental values significantly. In such cases, it is recommended to use anharmonic phonon frequencies and eigenvectors at finite temperatures for equations (120) and (122). The anharmonic frequencies and eigenvectors renormalized at finite temperatures can be obtained, for example, by performing a self-consistent phonon calculation using either the real-space stochastic approaches, [35–40] which are often termed the stochastic self-consistent harmonic approximation (SSCHA), or the momentum-space implementation which uses anharmonic force constants [41, 42]. An application of SSCHA performed with random displacements generated by the phonopy code coupled with the ALM force constants calculation code [43] is found in [20].
9. Phonon band unfolding
To draw a phonon band structure for a system with defects computed in a supercell approach, band unfolding technique may be useful. Since even one point defect breaks the periodicity of a crystal, a certain approximation is necessary to achieve this representation. In this section, an implementation of the method proposed by Allen et al [4] is explained.
In figure 15, a 2D schematic illustration of a supercell with a vacancy is presented. It is analyzed by presuming the corresponding perfect supercell with the same lattice vectors. Equation (30) gives the relationship between the basis vectors of the primitive cell and those of the supercell, which indicates that the perfect supercell contains  primitive cells. The reciprocal basis vectors are given by equation (46). The BZ volume of the primitive cell is N times larger than that of the supercell as shown in figure 16.
primitive cells. The reciprocal basis vectors are given by equation (46). The BZ volume of the primitive cell is N times larger than that of the supercell as shown in figure 16.

Figure 15. 2D schematic illustration of a supercell with a vacancy (left) and the corresponding perfect supercell (right). The atoms are depicted by open circle symbols. The vacancy is marked by "V". The perfect supercell contains  primitive cells whose lattice points (
primitive cells whose lattice points ( ) are indicated by filled circle symbols. Positions of all atoms in the defective supercell can deviate slightly from the equilibrium positions of the perfect supercell. At unfolding, the phase shift of
) are indicated by filled circle symbols. Positions of all atoms in the defective supercell can deviate slightly from the equilibrium positions of the perfect supercell. At unfolding, the phase shift of  is multiplied to the phonon eigenvector of the defective supercell at each primitive lattice translation as given in equation (135).
is multiplied to the phonon eigenvector of the defective supercell at each primitive lattice translation as given in equation (135).
Download figure:
Standard image High-resolution image
Figure 16. Unfolding of 2D Brillouin zone (BZ) of the supercell ( ) to
) to  BZs of the supercell (
BZs of the supercell ( ). Gray levels of shaded backgrounds indicate BZs of the supercell which belong to different
). Gray levels of shaded backgrounds indicate BZs of the supercell which belong to different  points.
points.
Download figure:
Standard image High-resolution imageFor this analysis to be possible, we assume there exists a one-to-one correspondence between the atoms and vacancy sites in the defective supercell and the atoms in the perfect supercell. The atoms and vacancies in the defective supercell can therefore be labelled by the composite index  in the same way as in the perfect supercell. It means that vacancy sites in the defective supercell must have respective atoms in the perfect supercell. The atoms in the defective supercell can nevertheless have slightly different positions than in the perfect supercell. The 2D schematic illustration we obtain is shown in figure 15.
in the same way as in the perfect supercell. It means that vacancy sites in the defective supercell must have respective atoms in the perfect supercell. The atoms in the defective supercell can nevertheless have slightly different positions than in the perfect supercell. The 2D schematic illustration we obtain is shown in figure 15.
We write the coordinates of lattice points in the supercell as

where
l
are integers, while  may not be.
may not be.
Let  be the jth lattice translation operator which is defined on a function as follows
be the jth lattice translation operator which is defined on a function as follows

In this section,
l
correspond to lattice vectors within the perfect supercell. There are N such lattice vectors and therefore N lattice translation operations.  maps a lattice vector with index li
in the perfect supercell to a lattice vector with index lk
. The mapping between indices is determined from
maps a lattice vector with index li
in the perfect supercell to a lattice vector with index lk
. The mapping between indices is determined from

where  means that the lattice points translated outside the supercell are brought back inside the supercell by a supercell lattice translation. The mapping obtained this way is stored in an N × N permutation matrix. In figure 15, the 2D supercell model is made of
means that the lattice points translated outside the supercell are brought back inside the supercell by a supercell lattice translation. The mapping obtained this way is stored in an N × N permutation matrix. In figure 15, the 2D supercell model is made of  primitive cells. For example, the translation by
primitive cells. For example, the translation by  brings l1 to l2. One more translation by
brings l1 to l2. One more translation by  brings l2 to l1 due to the periodicity of the supercell. In the following, the mapping
brings l2 to l1 due to the periodicity of the supercell. In the following, the mapping ![$l_i \xrightarrow[]{\hat{T}_j} l_k$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn413.gif) is written compactly as
is written compactly as  .
.
The first step of this method is to compute the phonon eigenvectors of the defective supercell. The Bloch wave of an eigenvector given by equations (19) and (20) is written as

where  is the phonon eigenvector of the defective supercell, with band index
is the phonon eigenvector of the defective supercell, with band index  , and at the wave vector
, and at the wave vector  in the BZ of the supercell. Eigenvector elements corresponding to vacancies are treated as zeros.
in the BZ of the supercell. Eigenvector elements corresponding to vacancies are treated as zeros.
We consider that  is applied to the Bloch wave as
is applied to the Bloch wave as

The first line of equation (127) may not be consistent with equation (124) since positions of atoms in the defective supercell can be displaced slightly from those in the perfect supercell.
The next step is to unfold the  point from the BZ of the supercell to the N
q points in the BZ of the primitive cell which are equivalent up to vectors of the reciprocal supercell lattice. This is written as
point from the BZ of the supercell to the N
q points in the BZ of the primitive cell which are equivalent up to vectors of the reciprocal supercell lattice. This is written as

where  is an integer linear combination of
is an integer linear combination of  . The schematic illustration for the BZ of the 2D supercell is shown in figure 16. The
. The schematic illustration for the BZ of the 2D supercell is shown in figure 16. The  points in the BZ of the supercell are shifted by
points in the BZ of the supercell are shifted by  inside the BZ of the primitive cell.
inside the BZ of the primitive cell.
Applying periodic boundary conditions on the supercell, the translation operations  form an Abelian group of order N with irreducible representations
form an Abelian group of order N with irreducible representations  . The great orthogonality theorem then gives the relation
. The great orthogonality theorem then gives the relation

This result is also proved algebraically in appendix

where  are the N vectors
are the N vectors  within the BZ of the primitive cell. We define the operator
within the BZ of the primitive cell. We define the operator

It can easily be shown that it is a projection operator, which selects the part of  that is also a Bloch function of the primitive lattice with wave vector
that is also a Bloch function of the primitive lattice with wave vector  . Indeed, in appendix
. Indeed, in appendix

Moreover, because from equation (130),  , the decomposition of
, the decomposition of  into the N functions
into the N functions  is unique,
is unique,

The projection by equation (131) and the decomposition by equation (133) indicate that the weight of state having the Bloch symmetry  of the primitive lattice in the supercell Bloch state
of the primitive lattice in the supercell Bloch state  is therefore defined as
is therefore defined as


It can easily be checked that  from equation (130) and the unit normalization of the phonon eigenvectors. Interestingly, this result does not require phonon eigenvectors of the perfect crystal. The second equation is implemented in the phonopy code though the third equation may be more intuitive.
from equation (130) and the unit normalization of the phonon eigenvectors. Interestingly, this result does not require phonon eigenvectors of the perfect crystal. The second equation is implemented in the phonopy code though the third equation may be more intuitive.  is the phonon eigenvector elements belonging to the lattice point
is the phonon eigenvector elements belonging to the lattice point  . The unfolding weight indicates its correlation with
. The unfolding weight indicates its correlation with  after the phase shift
after the phase shift  . In the example of figure 15, the phase shifts are integer multiples of π, since the 2D supercell model is made of
. In the example of figure 15, the phase shifts are integer multiples of π, since the 2D supercell model is made of  primitive cells.
primitive cells.
An example of the phonon band unfolding technique applied to Al with a vacancy is shown in figure 17. The corresponding perfect supercell (32 atoms) is constructed by  of the conventional unit cell (4 atoms). The primitive cell contains one atom and
of the conventional unit cell (4 atoms). The primitive cell contains one atom and


Figure 17. Phonon band unfolding of Al with a vacancy. Color scale indicates unfolding weights of phonon modes depicted by filled circle symbols, where those of the degenerated modes are summed. Phonon modes calculated for the defective supercell are presented by + (plus) symbols. Unshaded and shaded backgrounds show Brillouin zones of the supercell that belong to  of Γ and L, respectively. Only a part of the weights are unfolded into these two commensurate points among 32 commensurate points. Although this unfolding technique requires no reference states, for the comparison, the phonon band structure of the perfect supercell is shown by solid curves.
of Γ and L, respectively. Only a part of the weights are unfolded into these two commensurate points among 32 commensurate points. Although this unfolding technique requires no reference states, for the comparison, the phonon band structure of the perfect supercell is shown by solid curves.
Download figure:
Standard image High-resolution imageUnder fixed basis vectors of the supercell with a vacancy, internal atomic positions are relaxed. For the unfolding, q points are sampled along the BZ path L–Γ–L of the primitive cell (see figure 17). Those points can be uniquely decomposed as  , where
, where  belongs to the BZ of the supercell, and
belongs to the BZ of the supercell, and  is a reciprocal lattice vector of the supercell. For the above example,
is a reciprocal lattice vector of the supercell. For the above example,  is found to be Γ or L, depending on the position of q along the path L–Γ–L. Those two possibilities are represented using unshaded and shaded backgrounds in figure 17.
is found to be Γ or L, depending on the position of q along the path L–Γ–L. Those two possibilities are represented using unshaded and shaded backgrounds in figure 17.
The phonon modes of the defective supercell are calculated at those  points. The projection operator of equation (131) is
points. The projection operator of equation (131) is  dependent and only a fraction of the phonon modes are unfolded along the L–Γ–L path, because the selected
dependent and only a fraction of the phonon modes are unfolded along the L–Γ–L path, because the selected  points are only a subset of the commensurate
points are only a subset of the commensurate  points in the summation of equation (133). In figure 17, though we can see phonon frequencies are perturbed by the vacancy, the unfolded phonon band structure roughly follows the phonon band structure of the perfect supercell.
points in the summation of equation (133). In figure 17, though we can see phonon frequencies are perturbed by the vacancy, the unfolded phonon band structure roughly follows the phonon band structure of the perfect supercell.
Substitutional defects can be managed straightforwardly. Interstitial defects can be handled in the same way as the vacancy case by defining the corresponding supercell differently [44]. Suppose we have one interstitial defect in the supercell model. It is treated as a usual atom in the primitive cell of the corresponding perfect supercell. In the defective supercell, the interstitial atom is located in one primitive cell, and in the other primitive cells, the interstitial sites are treated as vacancies.
10. Conclusion
A scientific simulation code implements mathematical models that are described with formulae. In those formulae, the information is often not detailed enough for direct computer implementation, and the detailed knowledge can be implicit. Scientific software developers may have to learn it from skilled people around, otherwise find it independently. To ease the latter situation in the modern era of distributed software development, we aimed to provide details of the computational methods employed in the phonopy and phono3py codes, keeping the formalism as close as possible to the implementations.
Not all of the computational methods described in this review are specific to the phonon calculation or the supercell approach. For example, the handling of the regular grids and the linear tetrahedron method presented are applicable to electronic structure calculations. We therefore hope that this review will be a useful source of information for software developers in condensed matter science.
Acknowledgments
This work was supported by MEXT Japan through ESISM (Elements Strategy Initiative for Structural Materials) of Kyoto University and JSPS KAKENHI Grant Numbers JP21K04632 and JP21K03424. L C thanks the Institut Universitaire de France and the EXPLOR center hosted by the University de Lorraine.
Data availability statement
No new data were created or analysed in this study.
Appendix A: Functions used in the linear tetrahedron method
In this section we give explicit formulae for gi
, ni
,  , and
, and  [2]. They assume different forms depending on the position of ω in the sequence
[2]. They assume different forms depending on the position of ω in the sequence  . Also, they are expressed using
. Also, they are expressed using  and
and  which are defined as
which are defined as


gi , ni , Ii , and Ji satisfy the following relations:


It may be useful to remember


A.1.






A.2.

As shown in figure 12(a), the volume to consider is a tetrahedron with vertices located at q1,  ,
,  ,
,  , and
, and

Inside this volume,  is linearly interpolated, i.e.
is linearly interpolated, i.e.  ,
,  , and the contribution of ith tetrahedron to
, and the contribution of ith tetrahedron to  is approximated as
is approximated as ![$V_\mathrm{t} n^i [F(\mathbf{q}_1) + F(\mathbf{q}_\alpha) + F(\mathbf{q}_\beta) + F(\mathbf{q}_\gamma)]/4$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn465.gif) . By substituting it in equation (103), we obtain
. By substituting it in equation (103), we obtain


Using equations (A3) and (A4),



A.3.

In this case, the occupied part of the tetrahedron is the sum of the following three tetrahedra. Vertices of the first tetrahedron are q1, q2,  ,
,  , and its volume is
, and its volume is  . The vertices of the second tetrahedron are q1,
. The vertices of the second tetrahedron are q1,  ,
,  ,
,  , and its volume is
, and its volume is  . The vertices of the third tetrahedron are q1,
. The vertices of the third tetrahedron are q1,  ,
,  ,
,  , and its volume is
, and its volume is  . The locations of the vertices are depicted in figure 12(b). We obtain
. The locations of the vertices are depicted in figure 12(b). We obtain










A.4.

As shown in figure 12(c), the occupied part of the tetrahedron is the full tetrahedron minus the tetrahedron with vertices at q4,  ,
,  and
and  . Its volume is
. Its volume is  . Therefore, the contribution of i-th tetrahedron to
. Therefore, the contribution of i-th tetrahedron to  is approximated as
is approximated as ![$\{V_\mathrm{t}[F(\mathbf{q}_1) + F(\mathbf{q}_2) + F(\mathbf{q}_3) + F(\mathbf{q}_4)] - V_{4\beta\delta\gamma}^i [F(\mathbf{q}_4) + F(\mathbf{q}_\beta) + F(\mathbf{q}_\gamma) + F(\mathbf{q}_\delta)] \}/4$](https://content.cld.iop.org/journals/0953-8984/35/35/353001/revision2/cmacd831ieqn484.gif) . We obtain
. We obtain








A.5.

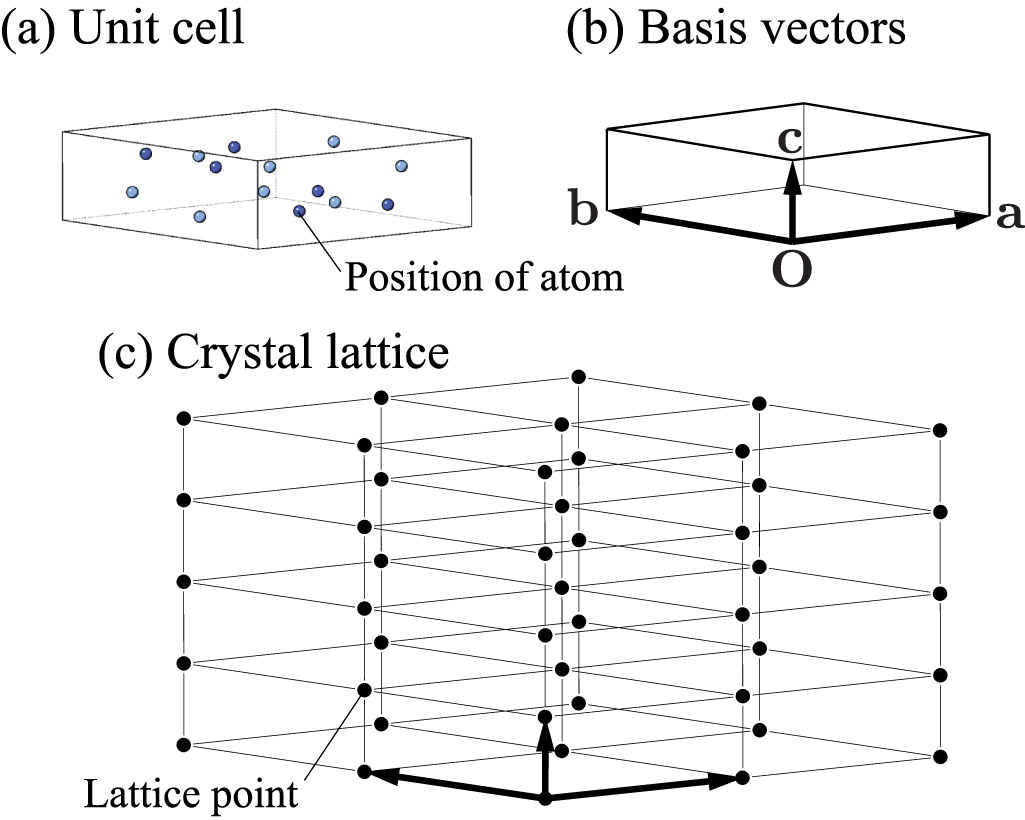{kind=link}
{kind=link}
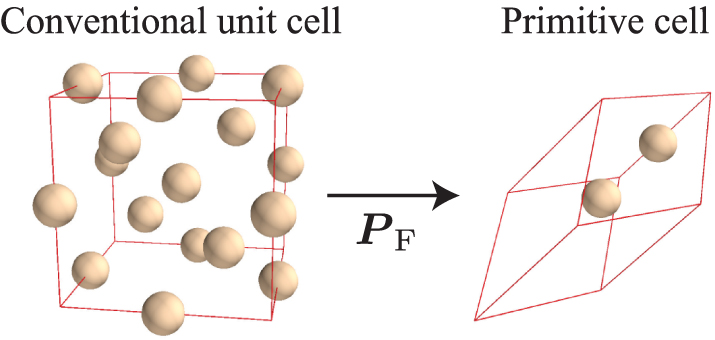{kind=link}
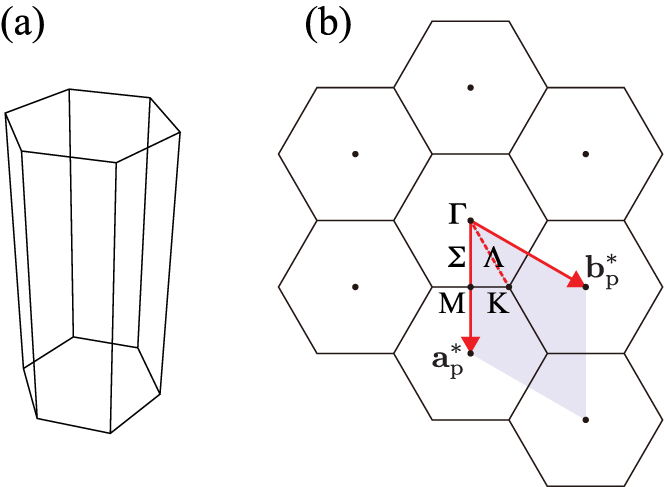{kind=link}
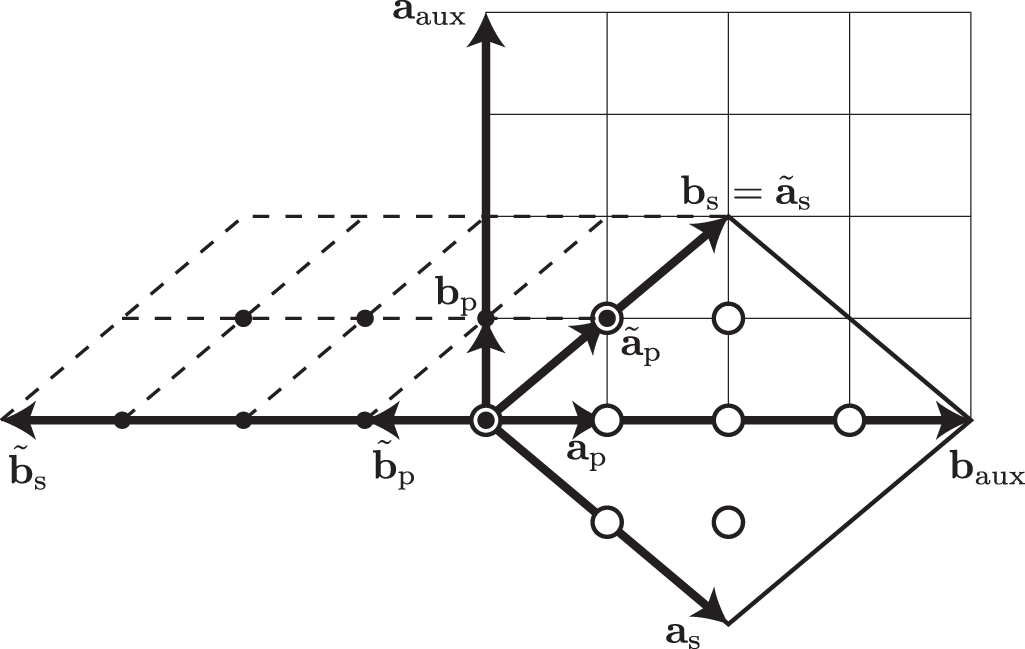{kind=link}
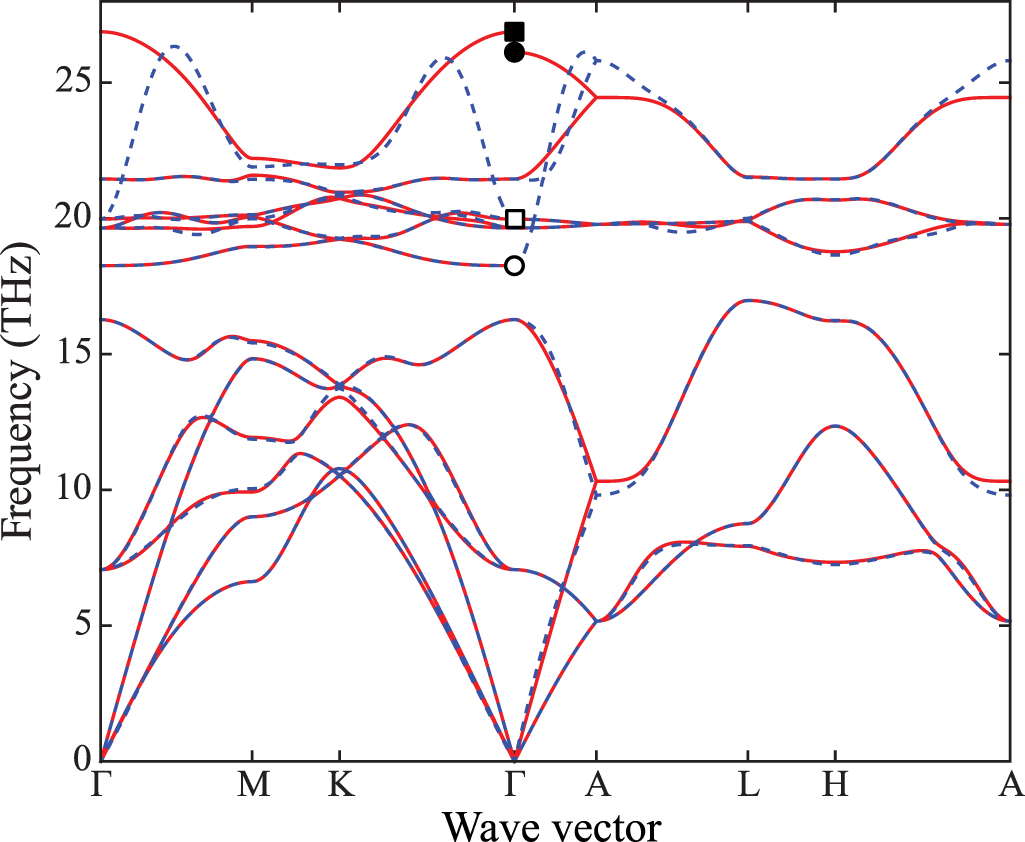{kind=link}
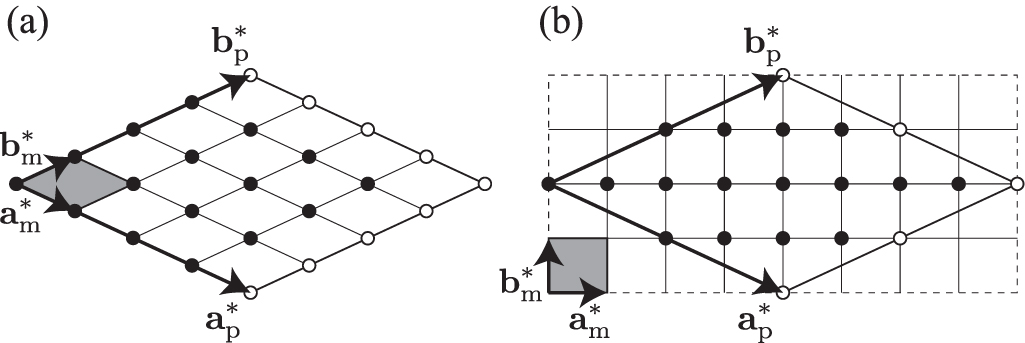{kind=link}
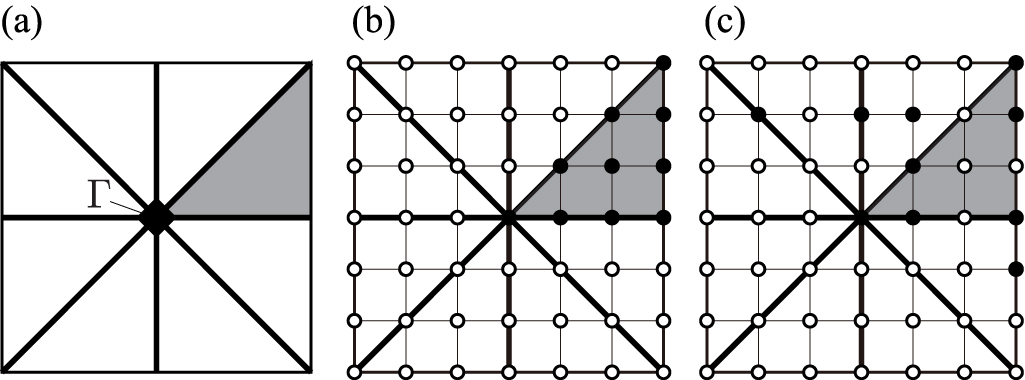{kind=link}
{kind=link}
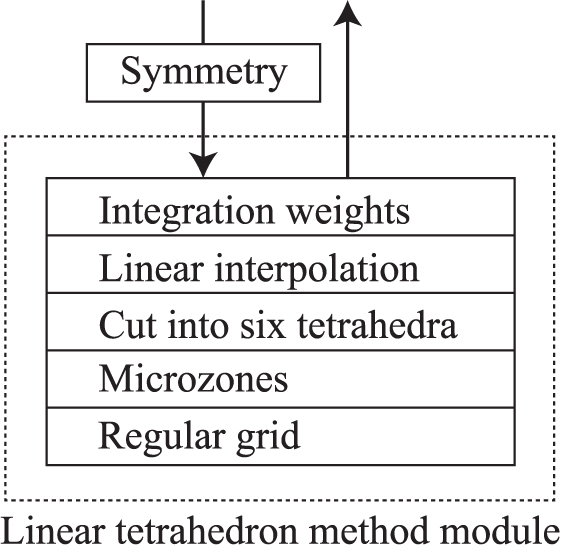{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
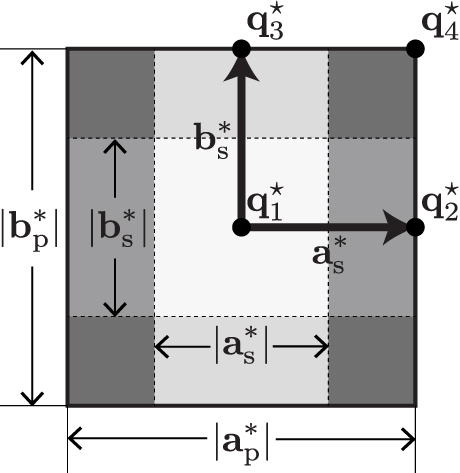{kind=link}
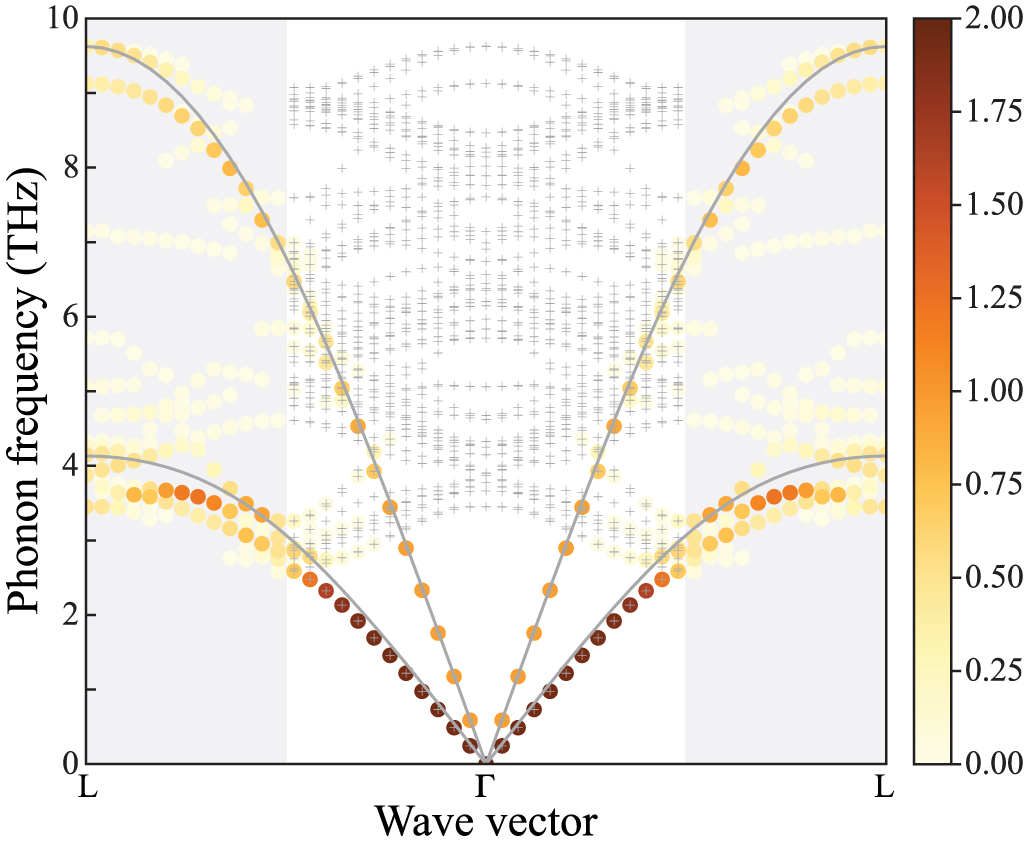{kind=link}
The tetrahedron is fully occupied, therefore,




Appendix B: Summation formulae
When the supercell lattice vectors are collinear to those of the primitive cell,

With  , it is proved in most textbooks that the set of lattice vectors
, it is proved in most textbooks that the set of lattice vectors


and their conjugated wave vectors



fulfil the summation formulae


where G is any reciprocal lattice vector, and RL any supercell lattice vector.
When the supercell is not collinear to the primitive cell, the set of lattice vectors,  , are those integer linear combinations of
, are those integer linear combinations of  located within the supercell
located within the supercell  , and the set of wave vectors,
, and the set of wave vectors,  , are those integer linear combinations of
, are those integer linear combinations of  located within the reciprocal cell
located within the reciprocal cell  . Since the above summations can not easily be split into the product of three geometric series, it is not obvious whether the above results are still valid.
. Since the above summations can not easily be split into the product of three geometric series, it is not obvious whether the above results are still valid.
However, we have seen in section 3.1 that SNF like transformations can always be used to obtain primitive cell and supercell which are collinear, and for which the summation formulae (B7) and (B8) are obviously valid. We have


and therefore



while the set of lattice vectors and wave vectors,


with  and
and  , fulfils,
, fulfils,


Since the same lattices are generated by the unimodular transformations (B11) and (B12), we can write


where RL
is a supercell lattice vector used to bring  within the original supercell
within the original supercell  , and G a reciprocal lattice vector used to bring
, and G a reciprocal lattice vector used to bring  to the original reciprocal cell
to the original reciprocal cell  . Since
. Since  ,
,  ,
,  are multiple of 2π, and
are multiple of 2π, and  , we obtain equations (B7) and (B8) for arbitrary supercell and primitive cell related by an integer matrix
, we obtain equations (B7) and (B8) for arbitrary supercell and primitive cell related by an integer matrix  . Notice however that the definition of the set of lattice vectors and wave vectors for which those summation formulae hold is now more general. The wave vectors are along the reciprocal vectors of the supercell and located within the reciprocal cell of the primitive lattice, while the lattice vectors are along the primitive lattice vectors and located within the supercell.
. Notice however that the definition of the set of lattice vectors and wave vectors for which those summation formulae hold is now more general. The wave vectors are along the reciprocal vectors of the supercell and located within the reciprocal cell of the primitive lattice, while the lattice vectors are along the primitive lattice vectors and located within the supercell.
Appendix C: Projection into primitive cell Bloch states
From the definition of the projection operator,




 may be outside the supercell, therefore we write
may be outside the supercell, therefore we write  , where RL
is a supercell lattice vector. Because
, where RL
is a supercell lattice vector. Because  has the periodicity of the supercell, and
has the periodicity of the supercell, and  , we obtain
, we obtain



