
Abstract
Time differential perturbed angular correlation (TDPAC) of γ-rays spectroscopy has been applied in chemistry and biochemistry for decades. Herein we aim to present a comprehensive review of chemical and biochemical applications of TDPAC spectroscopy conducted at ISOLDE over the past 15 years, including elucidation of metal site structure and dynamics in proteins and model systems. β-NMR spectroscopy is well established in nuclear physics, solid state physics, and materials science, but only a limited number of applications in chemistry have appeared. Current endeavors at ISOLDE advancing applications of β-NMR towards chemistry and biochemistry are presented, including the first experiment on 31Mg2+ in an ionic liquid solution. Both techniques require the production of radioisotopes combined with advanced spectroscopic instrumentation present at ISOLDE.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The family of hyperfine interaction techniques, including NMR, NQR, ESR, μSR, time differential perturbed angular correlation (TDPAC), β-NMR, Mössbauer spectroscopy and several others, provides experimental characterisation of electronic and molecular structure and dynamics, as well as magnetic properties at the probe site of the molecule or material investigated. Some of these techniques, such as TDPAC of γ-rays and β-NMR spectroscopy, rely on radioactive probes for the detection of the hyperfine interaction. They benefit from the exceptional sensitivity of radiotracers in combination with a local spectroscopic probe elucidating properties of the material in question. This, however, requires both the production of radioisotopes, the presence of advanced spectroscopic instrumentation, and highly qualified staff at the same location. All these requirements are fulfilled at ISOLDE and at similar research infrastructure facilities around the world, providing a unique setting for application of such spectroscopic techniques.
Both TDPAC and β-NMR spectroscopy exploit the anisotropic emission of particles from spin oriented radionuclides. In TDPAC spectroscopy two γ-rays emitted in succession in the nuclear decay are detected, the first 'selecting' a spin aligned ensemble, and the second then emitted from this ensemble anisotropically with respect to the first. In β-NMR spectroscopy, the radioactive nuclei are spin oriented prior to implantation into the sample of interest, and the anisotropic emission of β-particles is the spectroscopic signal. The methodologies are presented in more detail in the following sections.
With this review we aim to provide an exhaustive compilation of published work on applications of TDPAC and β-NMR spectroscopy in chemistry or biochemistry conducted at ISOLDE since the latest ISOLDE laboratory portrait [1]. In addition, we include selected examples extending further back in time, which were not included in [1]. In some cases the division between chemistry and solid state physics may not be obvious, and we refer the interested reader to the review on solid state physics research at ISOLDE presented in this special issue of J. Phys. G.
2. TDPAC of γ-rays spectroscopy
2.1. The method
Radioactive nuclei can be used as probes of structural, dynamic, electronic, and magnetic properties of the hosting lattices or molecules. The local charge distribution and magnetic field may be explored via the hyperfine interaction with the electric quadrupole and magnetic dipole moment of excited nuclear states. The technique of TDPAC spectroscopy uses a γ-ray cascade, with an intermediate state with suitable nuclear properties, and a half-life from a few ns to μs. Due to angular momentum conservation in the nuclear decay, the probability of a γ-ray being emitted in a certain direction depends on the orientation of the nuclear spin, I. The nuclei initially have a random distribution of their spin orientation, and thus radiation is emitted isotropically in space. In order to observe an anisotropic angular distribution of radiation an ensemble of nuclei with aligned spin distribution is selected by detecting the first γ-ray of the cascade in a certain direction. The probability of the emission of the second γ-ray, with respect to the direction of the first, is therefore anisotropic, with anisotropy coefficients that depend on the properties of the nuclear decay cascade. This phenomenon was described by Hamilton [2] giving the theoretical basis of TDPAC, and the first experimental measurement was successfully performed in 1947 [3]. The influence of extranuclear (hyperfine) fields, perturbing the angular correlation of the two consecutive γ-rays, was presented by Goertzel [4] and Abragam and Pound [5]. Initially these studies aimed to determine nuclear parameters such as magnetic moments and electric quadrupole moments. Later the measurement of hyperfine fields were applied to explore local properties of specific hosts at the probe site, using the spectroscopic signals from radioactive probes implanted into or bound to the hosts [6]. The canonical reference for these techniques is Frauenfelder and Steffen (1965) [7]; applications in chemistry and biochemistry are compiled and presented in [8–12].
During the lifetime of the intermediate nuclear state, the angular correlation between the two γ-rays may be perturbed due to the hyperfine interaction of the nucleus with the magnetic field or electric field gradient (EFG) from the surrounding electronic charge distribution and nuclei. This perturbation is accounted for by the perturbation factor  where k may assume values
where k may assume values  where
where  and
and  are the angular momenta carried by
are the angular momenta carried by  and
and  respectively. The general expression for the time dependence of the angular correlation function for randomly oriented molecules or polycrystalline samples is:
respectively. The general expression for the time dependence of the angular correlation function for randomly oriented molecules or polycrystalline samples is:
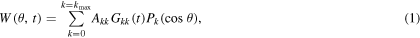
where  are the angular correlation coefficients and
are the angular correlation coefficients and  the Legendre polynomials of kth order. γ scintillation detectors and nuclear electronic modules are used to select and store histogram spectra of
the Legendre polynomials of kth order. γ scintillation detectors and nuclear electronic modules are used to select and store histogram spectra of  −
− coincidences, as a function of time between detection of
coincidences, as a function of time between detection of  and
and  These are recorded for detector angles, between the two γ-rays, typically of 90° and 180°. After subtraction of a constant background due to accidental coincidences originating from γ rays emitted from different nuclei, the spectra are combined to the geometric mean of all 90° and 180° spectra, W(90°, t) and W(180°, t), respectively. The function, R(t), the experimental equivalent of the perturbation function, is then formed as:
These are recorded for detector angles, between the two γ-rays, typically of 90° and 180°. After subtraction of a constant background due to accidental coincidences originating from γ rays emitted from different nuclei, the spectra are combined to the geometric mean of all 90° and 180° spectra, W(90°, t) and W(180°, t), respectively. The function, R(t), the experimental equivalent of the perturbation function, is then formed as:
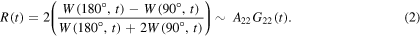
The analytical expression for the perturbation factor  is known [8, 13], and depends on the type of hyperfine interaction. Magnetic interactions are not treated further here, because practically all examples, vide infra, rely on nuclear quadrupole interactions (NQIs). For NQIs of randomly oriented molecules:
is known [8, 13], and depends on the type of hyperfine interaction. Magnetic interactions are not treated further here, because practically all examples, vide infra, rely on nuclear quadrupole interactions (NQIs). For NQIs of randomly oriented molecules:

The sum runs over transitions between sublevels of the hyperfine split intermediate nuclear level, and the amplitudes  and frequencies
and frequencies  depend on the NQI. The NQI is proportional to the product of the nuclear electric quadrupole moment, Q, and the EFG from the nuclear probe surroundings. It is usually characterised by two parameters,
depend on the NQI. The NQI is proportional to the product of the nuclear electric quadrupole moment, Q, and the EFG from the nuclear probe surroundings. It is usually characterised by two parameters,  and
and 
 (or
(or  ), representing the interaction strength, where
), representing the interaction strength, where  is the numerically largest component of the diagonalized EFG tensor and
is the numerically largest component of the diagonalized EFG tensor and  is the nuclear spin of the intermediate state. Occasionally, ω0 is used instead of
is the nuclear spin of the intermediate state. Occasionally, ω0 is used instead of  where
where  (integer spin) or
(integer spin) or  (half-integer spin).
(half-integer spin).  characterises the axial symmetry of the EFG and is defined by combining the axial tensor components as:
characterises the axial symmetry of the EFG and is defined by combining the axial tensor components as:  The observable frequencies
The observable frequencies  depend on the Eigenvalues of the interaction Hamiltonian and
depend on the Eigenvalues of the interaction Hamiltonian and  The expressions are lengthy, and may be found in work by Butz [14]. In the particular case of
The expressions are lengthy, and may be found in work by Butz [14]. In the particular case of  ω1 (observable) = ω0 and
ω1 (observable) = ω0 and  keeping in mind that ω3 always fulfills
keeping in mind that ω3 always fulfills  for the case of intermediate spin I = 5/2. The perturbation function
for the case of intermediate spin I = 5/2. The perturbation function  therefore carries the information of the EFG, and thus the characteristics of the local electronic and molecular structure at the probe site.
therefore carries the information of the EFG, and thus the characteristics of the local electronic and molecular structure at the probe site.
Molecules in solution undergo Brownian tumbling, which in a simple model may be represented by the rotational correlation time  For a spherical molecule,
For a spherical molecule,  depends on viscosity,
depends on viscosity,  absolute temperature,
absolute temperature,  and volume,
and volume,  of the molecule:
of the molecule:

where  is the Boltzmann constant. This motion may give rise to a time dependent EFG. For rapid rotational diffusion,
is the Boltzmann constant. This motion may give rise to a time dependent EFG. For rapid rotational diffusion,  the perturbation function is given by [11]:
the perturbation function is given by [11]:

While for slow molecular reorientation,  the anisotropy exhibits exponential damping so that the perturbation function is approximated by
the anisotropy exhibits exponential damping so that the perturbation function is approximated by  When
When  the interaction is considered static and G22(t) is described by the sum of cosine functions, vide supra.
the interaction is considered static and G22(t) is described by the sum of cosine functions, vide supra.
2.2. TDPAC isotopes
Selected PAC probes and their properties are presented in table 1.
Table 1. Selected isotopes used or potentially useful for TDPAC experiments in chemistry and biochemistry, indicating γ-ray cascade energies and intermediate nuclear state parameters: nuclear spin, I, half-life, t1/2, quadrupole moment, Q, and magnetic moment μ. The nuclear data were obtained from the most recent data published on Atomic Data and Nuclear Data Tables [15]; μ values were collected from 'table of nuclear magnetic dipole and electric quadrupole moments', Atomic Data and Nuclear Data Tables [16]; Q values were collected from 'table of nuclear electric quadrupole moments', Atomic Data and Nuclear Data Tables [15]. Data presented for (a) 68Cu are from [17], for (b) 111Cd from [18] and for (c) the half-life of 172Yb from [19, 20]. IT—isomeric transition, EC—electron capture, β− (electron) emission.
| Parent isotope | t1/2 | Decay | Probe isotope | γ1–γ2 E (keV) | I | t1/2 (ns) | Q (b) | μ (μN) |
|---|---|---|---|---|---|---|---|---|
| 68mCu | 3.75 min | IT | 68Cu | 526–84 | 2+ | 7.84(8) | (−)0.110(3)(a) | (+)2.857(6)(a) |
| 99Mo | 2.7 d | EC | 99Tc | 740–181 | 5/2+ | 3.61(7) | unknown | +3.48(4) |
| 111Ag | 7.45 d | β− | 111Cd | 97–245 | 5/2+ | 84.5(4) | +0.68(2)(b) | −0.7656(25) |
| 111mCd | 48 min | IT | 111Cd | 151–245 | 5/2+ | 84.5(4) | +0.68(2)(b) | −0.7656(25) |
| 111In | 2.8 d | EC | 111Cd | 171–245 | 5/2+ | 84.5(4) | +0.68(2)(b) | −0.7656(25) |
| 172Lu | 6.7 d | EC | 172Yb | 91–1094 | 3+ | 8.14(17)(c) | −2.9(3) | +0.65(4) |
| 181Hf | 42.4 d | β− | 181Ta | 133–482 | 5/2+ | 10.8(1) | +2.28(2) | +3.29(3) |
| 199mHg | 42 min | IT | 199Hg | 374–158 | 5/2− | 2.46(3) | +0.95(7) | +0.88(3) |
| 204mPb | 67 min | IT | 204Pb | 912–375 | 4+ | 265(6) | 0.44(2) | +0.225(4) |
The isobaric series of 111mCd/Cd (t1/2 = 48 min), 111In/Cd (t1/2 = 2.8 d) and 111Ag/Cd (t1/2 = 7.45 d) can be used together to perform complementary and comparable analysis on the chemistry of different parent elements, which are all measured at the same intermediate state (I = 5/2 and 245 keV) of 111Cd. Cd, In and Ag are well produced at ISOLDE [21] but, for practical purposes when chemistry is required, the 111Ag isotope is better produced by irradiating 110Pd with thermal neutrons at a reactor. The 111Ag production at ISOLDE presents the disadvantage of having 111In as isobaric contaminant. The optimisation of the Ag/In ratio depends on the efficiency of the Ag+ LASER ion source [22] and on the reduction of the surface ionised In+ contaminant, still an ongoing R&D project. Currently used LaBr3(Ce) detectors have enough energy resolution to well separate the photo-peaks of the start γ rays of 97 keV and 171 keV, respectively obtained from the decays of 111Ag and 111In. However, Compton events from the 171 keV γ ray, which are distributed down to low energies, overlap with the 97 keV γ from 111Ag, producing a TDPAC measurement that contains information from both Ag and In. This problem can be diminished by chemical separation of Ag from In or, alternatively, by starting experiments ∼one week after collection of the radioactive isotopes, when more than 80% of 111In atoms have decayed. We further point to the fact that the particular case of 111In decaying by electron-capture leaves the 111Cd probe ion in a highly charged metastable state, out of equilibrium, so that the inner electron orbitals are filled up from outer shells and upon recombination, the ion loses quite a number of electrons due to Auger processes. In a solid state sample, if the number of electron carriers and electron mobility are high enough, it may be rapidly compensated (t < ns) by electrons from its neighbourhood. If the recombination process is slow (t > ns), i.e., occurring within the TDPAC measurement time window, it may influence the observable perturbation function. To disentangle doubtful cases, 111mCd/Cd (t1/2 = 48 min) can be used since there is no element transformation during the nuclear decay. However, the longer 2.8 d half-life of 111In, in comparison with the 111mCd 48 min half-life, allows the same sample, and same sample preparation, to be studied while changing experimental conditions, e.g., temperature, illumination, and pressure.
Additionally, when aiming for heavy metal TDPAC nuclear probes, two isotopes are well produced at ISOLDE, 199mHg/Hg (t1/2 = 42 min) and 204mPb/Pb (t1/2 = 67 min). The angular correlation of the γ–γ cascade 374–158 keV of the 199mHg/Hg probe has been successfully used for chemistry and biochemistry studies at ISOLDE for decades [13]. More recently, 204mPb/Pb has also appeared as a probe in chemical applications of TDPAC spectroscopy [23]. It may become useful because of its greater sensitivity to deviations of the EFG from axial symmetry than other usual TDPAC probes [24]. Its long intermediate state half-life of 260 ns, Q = 0.44(2) b and μ = +0.225(4) μN [25] should allow performing γ–γ TDPAC measurements for very weak EFGs, characterised by low frequencies of the perturbation function. However, the high spin of the intermediate level, I = 4, gives rise to rather complicated R(t) spectra, with multiple frequencies due to the many possible transitions between nuclear sublevels, and due to line broadening, it may be difficult to carry out unambiguous interpretation of the experimental data.
At ISOLDE other interesting TDPAC isotopes, still not used in chemistry or biochemistry, have been characterised recently, such as the rare-earth 172Lu/Yb (t1/2 = 6.7 d) decaying by electron capture [20]. In 2014, a proof of principle experiment using 68mCu/Cu (t1/2 = 3.75 min) isotope for TDPAC spectroscopy [17] was conducted, aiming towards new experiments in both solid state physics and chemistry.
We additionally wish to point to two interesting cases—still barely produced at ISOLDE—but currently produced by neutron activation at nuclear reactors that may deserve future investment in on-line production methods. The first concerns the 181Hf/Ta (t1/2 = 42.4 d) decay to excited levels of 181Ta through the emission of a β− particle. A suitable cascade with high γ anisotropy A22 ∼ −0.3, an intermediate half-life state of 10.8 ns and high values of Q = +2.36(5) b and μ = +3.24(5) μN [26], make it an excellent TDPAC probe used in solid state physics since the early seventies. The other promising candidate is 99mTc, currently used as a medical radiotracer. This idea is motivated by the fact that in addition to the 82% 99Mo (t1/2 = 2.7 d) decaying to 99mTc, there are 16% of 99Mo atoms decaying via a suitable two γ-ray TDPAC cascade with 3.6 ns half-life intermediate state of 99Tc. The quadrupole moment is still unknown, but it was revealed to be large enough for TDPAC measurements [27], and may be characterised in dedicated future experiments. So far, the production of 99Mo relies on neutron activation of U targets and from the necessary chemical separation that is currently used in 99Mo/99mTc generators, meaning that these experiments must rely on proper collaborations with the authorised medical centres conducting radiotracer diagnosis.
Finally, TDPAC experiments are also performed at the ISOLDE using isotopes produced elsewhere, if the half-lives are longer than a few days, exploiting the large collection of TDPAC instruments present at ISOLDE, and the local expertise in design and application of these instruments.
2.3. Instrumentation at ISOLDE
Four 6-detector TDPAC, used in chemistry and biochemistry, are currently present at ISOLDE. Three of these are analogue instruments equipped with BaF2 detectors [28] and one is a digital setup equipped with LaBr3(Ce) detectors [29]. The temperature of the samples can be controlled from −20 °C to 50 °C using a Peltier element, or experiments may be conducted with the sample in a Dewar in, for example, liquid nitrogen (77 K).
Since the mid-nineties, a simple vacuum collection chamber has been used at ISOLDE, designed for biophysics and aqueous sample preparations. It mainly consists of a removable sample holder made of a small copper support with a Teflon cup containing typically 150 μl highly pure (frozen) water. At the centre of the vacuum chamber there is a cold finger copper rod, onto which the sample holder is mounted. The cold finger is immersed into a liquid nitrogen bath, maintaining the sample holder at low temperature, thus limiting water sublimation during the vacuum implantation. Once the desired activity is obtained, the small sample holder is removed from the collection chamber, and transported to the chemistry laboratory for further sample preparation.
2.4. Applications in chemistry and biochemistry
In this chapter we provide an overview on the diverse applications of TDPAC spectroscopy in chemistry (sometimes bordering on solid-state physics) and in biochemistry, performed under the umbrella of the ISOLDE collaboration since (or not included) in the previous Laboratory Portrait, published in 2000 [1].
2.4.1. Coordination compounds of HgII formed with mercaptans, amino acid derivatives and short-chain peptides
199mHg is one of the most used TDPAC isotopes. The majority of studies presented in this section date back to the late 1990s, however, they were not elaborated on in the previous ISOLDE Laboratory Portrait [1].
As simple model systems for the biologically relevant S–Hg–S coordination environment, mercury mercaptides (Hg(S(CH2)xCH3)2, x = 0, 1, 2) were investigated by Soldner et al [30]. The HgII coordination geometry was monitored via the detection of the NQI by TDPAC and quantum chemical calculation. All TDPAC spectra showed one unique NQI. Relatively high frequencies and small asymmetry parameters are generally observed for a linear or distorted linear coordination of HgII by two thiols, with a negligibly small contribution to the EFG from distant azimuthal atoms or molecules. However, a larger asymmetry parameter and additional line broadening was observed for HgII-methylmercaptide suggesting a more significant contribution of such distant azimuthal donors.
Distorted linear two-fold coordination by thiolates are common for HgII, but at the metal site of the MeR protein threefold coordination by the thiolate groups of three cysteines is observed [31], and there are also examples of fourfold Hg–S-coordination as found e.g. for tert-butyl-mercaptide [32]. These coordination modes are easy to differentiate by the NQI of 199mHg monitored by TDPAC. A typical example reflecting the efficiency of the technique in the distinction of two-, three- and fourfold coordination is the study of the HgII-coordination of the various penicillamine enantiomers [33, 34]. Rather interestingly, tricoordinate planar HgII-structures are present with all enantiomers of this therapeutic agent, even below ligand-to-metal ratios of 2:1. Indeed, threefold coordination was observed to be the preferred binding mode at any D-penicillamine : HgII ratios.
In order to study the interaction of cysteine containing peptide chains with HgII-ion, terminally non-protected Ala-Ala-Cys-Ala-Ala (AACAA) free and resin bound oligopeptides were investigated by Tröger et al [35]. The 199mHg-TDPAC spectra recorded for HgII−AACAA by using only the trace amounts of radioactive 199mHg ('infinite dilution') and in samples of AACAA:HgII > 10 stoichiometry exhibit high frequency, νQ = 1.52 GHz and a relatively small asymmetry parameter of η = 0.19 [35]. Furthermore, the observed frequency is similar to that of Hg(cysteine)2 (νQ = 1.41 GHz, η = 0.16) [32], suggesting the presence of only two-coordinated HgII-complexes. At a HgII−AACAA 1:2 ratio, the recorded spectra display a broad signal and a significantly lower frequency (∼0.72 GHz) [35], indicating a higher coordination number. Two NQIs with line broadening were identified for the resin bound AACAA with 'infinite dilution' of HgII. A low frequency NQI presumably reflecting HgII sites with coordination numbers above 2, and a high frequency component very similar to the HgII-complex formed with the non-immobilised AACAA.
Ctortecka et al later extended these investigations to C-terminally protected oligopeptides, AAXAA–NH2, containing histidine (H or His) and tyrosine (Y or Tyr) residues, respectively (X = H or Y) [36]. Histidine and tyrosine might bind HgII by their neutral imidazole ring and deprotonated OH-group, respectively. Two (or more) NQIs are required to adequately fit the TDPAC data recorded for the HgII−AAHAA–NH2 and HgII−AAYAA–NH2 complexes, contrary to the HgII−AACAA complex, which displayed only one NQI at low HgII:peptide stoichiometry, vide supra. This may reflect the high affinity of HgII for thiolates, giving exclusive bis-thiolato-HgII complexes for HgII−AACAA, while more complex speciation is observed for the other two peptides.
2.4.2. Peptides modelling the metal-binding sites of metalloproteins
Several alpha-helical cysteine-substituted derivatives of the TRI peptide family (Ac-G-(LaKbAcLdEeEfKg)4-G-NH2) [37] have been synthesised to create self-assembling de novo designed oligopeptides providing a scaffold for systematic elucidation of metal ion binding sites in metalloproteins. The HgII-interaction of TRI L9C, a ligand with a Cys replacing the ninth Leu residue of the original sequence, was studied via UV–visible, 199Hg NMR and 199mHg TDPAC spectroscopies [38]. In the absence of metal ions, the peptide forms double stranded coiled coils below pH ∼ 4 and three-stranded coiled coils above pH ∼ 6. HgII-ions were not shown to have a major influence on the aggregation of the peptides when the peptide/HgII ratio was above 3:1, in contrast to double Cys-substituted TRI peptide derivatives, where the three-stranded aggregates were stabilised by the addition of CdII or HgII ions [39]. The coordination geometry of HgII and the number of metal-bound thiolate donors of TRI L9C were affected by pH and the applied TRI L9C per HgII ratios (see figure 1).
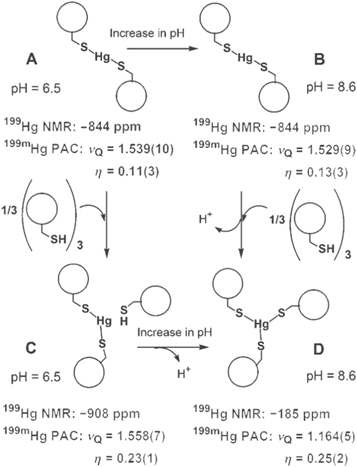
Figure 1. Model of metal site structures for TRI L9C and HgII at different peptide/HgII ratios and different pH values [38]. The model is derived from 199mHg TDPAC and 199Hg NMR data determined for the dominant species under the indicated conditions, denoted by A–D. The original figure was published in [38] John Wiley & Sons. Copyright © 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced with permission.
Download figure:
Standard image High-resolution imageExperiments performed with a 3–12-fold peptide excess over HgII at pH = 8.7 suggested the binding of three Cys-thiolates from the interior of the three-stranded coiled-coil (figure 2) (νQ = 1.164 GHz and η = 0.25). These values very closely resemble those determined for the HgII-loaded MerR protein (νQ = 1.16 GHz and η = 0.22 [40]), implying that the metal binding site provided by this self-assembled three-stranded helical coiled coil is a good structural mimic of the MerR protein.
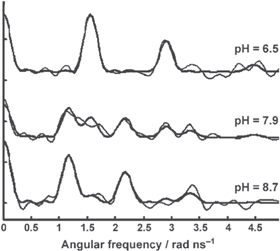
Figure 2. 199mHg TDPAC spectra recorded for TRI L9C (125–130 μM) and HgII (10.8 μM) at three different pH values [38]. The thin and bold lines represent the Fourier transform of the experimental data and the fit, respectively. The original figure was published in [38] John Wiley & Sons. Copyright © 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced with permission.
Download figure:
Standard image High-resolution imageUnder similar conditions, but in the presence of only two-fold ligand excess relative to HgII, fitting of the recorded 199mHg TDPAC spectrum resulted in parameters (νQ = 1.529 GHz and η = 0.13) that are typical for (distorted) linear bis-thiolato HgII-complexes. Very similar TDPAC parameters were fitted for the TRI L9C–HgII 2:1 sample at a lower pH (pH = 6.6) suggesting that a [Hg(TRI L9C)2] species is present in a broad pH range. The decrease of pH in the TRI L9C–HgII 12:1 system (pH = 7.9) was accompanied by the appearance of a new signal in the TDPAC spectrum, reflecting the presence of a second NQI (see figure 2). This NQI became, indeed, dominant at pH = 6.5 and the TDPAC parameters (νQ = 1.558 GHz and η = 0.23) again most likely reflect a [Hg(RS)2] type species. Surprisingly, these values are slightly but significantly different from those found for the TRI L9C–HgII 2:1 system (see data in figure 1). The notable difference between the asymmetry parameters shows a larger deviation from an axially symmetric EFG suggesting a larger distortion of the linear coordination geometry in this [Hg(TRI L9C)2(TRI L9C–H)] species where the Cys-residue of the third strand is presumably still protonated (structure C in figure 1).
As an important achievement in de novo protein design, an antiparallel three-helix bundle, formed by a single oligopeptide chain of a modified α3D protein (α3DIV), was constructed [41]. Three Cys residues of α3DIV, each located at one of the three helices, form a potential metal binding site at the C-terminal end of the bundle. This 3-Cys site possesses high affinity for HgII but also for CdII and PbII ions. Similar to TRI L9C and other Cys-substituted TRI peptides [42–44], HgII binding to α3DIV displays a clear pH-dependence with a pKa value of 7.1 which is interpreted as the transformation of a HgS2(SH) type complex (νQ = 1.48 GHz and η = 0.15) to a HgS3 type trigonal species (νQ = 1.11 GHz and η = 0.40). This study, together with those of the TRI peptide family, illustrates the benefits using 199mHg TDPAC and 199Hg NMR as complementary spectroscopic tools in the investigation of the coordination environment of HgII in solution.
The highly flexible Cys-Xaa-Xaa-Cys amino acid sequence (where Xaa stands for amino acids other than cysteine) is a metal ion chelating unit often found in loops of a variety of metalloproteins where the protein scaffold plays an important role in positioning the Cys-units for ideal metal ion chelation [45]. A specific short tetrapeptide sequence, Ac-Cys-dPro-Pro-Cys-NH2 (CdPPC) was used for the modelling of the HgII-binding of such loops [45]. The β-turn inducing dPro-Pro unit was introduced into the sequence to force constraints in the oligopeptide chain and to preorganize the two Cys-residues for metal ion binding. Amongst other methods, 199mHg TDPAC was utilised to analyse the HgII coordination geometry under slightly sub-stoichiometric (0.9 eq. of HgII per ligand) conditions at various pH-values. The parameters for the single NQI observed at pH = 5.1 (νQ = 1.39 GHz and η = 0.09) correspond to those observed for nearly linear HgS2-type model complexes ([Hg(Cys)2], νQ = 1.41 GHz and η = 0.15 [32]). The slightly smaller νQ value may be due to longer Hg–S bond lengths as was also suggested by computational modelling [45]. It is interesting to note that the νQ = 1.53 GHz value found for the bis-thiolate bound complexes of the TRI L9C peptide (e.g. [Hg(TRI L9C)2]), vide supra, suggests the opposite, i.e. shorter Hg–S bond lengths [38]. An increase or decrease of Hg–S bond length values, relative to those of more flexible simple model complexes, might be due to strain imposed on the metal site by the CdPPC peptide and in the two- or three-stranded coiled coil assemblies of TRI L9C [38], respectively.
The metal ion responsive bacterial copper efflux regulator CueR protein regulates the intracellular level of CuI (as well as AgI or AuI) but shows no response in the presence of the divalent metal ions ZnII and even HgII [46], in spite of the similar donor ligand and coordination geometry preferences of the latter ion to those of CuI and AgI. As part of a research project aiming at the better understanding of the molecular details of this interesting selectivity, the mono-(AgI) and divalent metal ion (ZnII, HgII) binding of model peptides encompassing the metal binding loop of the bacterial copper-efflux regulator CueR protein (Ac-SCPGDQGSDCPI-NH2–MBL-VC) and a histidine containing variant of the native sequence (Ac-SCHGDQGSDCSI-NH2–HS) were investigated and compared [47, 48]. Analysis of the 199mHg TDPAC spectra recorded at different pH values in the equimolar systems of HgII and the ligands, revealed the existence of one dominant NQI with parameters typical for bis-thiolate coordinated HgII-complexes, independently of the applied pH and ligand. The similarity of the TDPAC-features for the spectra recorded for HgII–MBL-VC at pH 9.5 (νQ = 1.42 GHz and η = 0.13) [48] and for HgII–HS at pH 8.0 (νQ = 1.43 GHz and η = 0.13) [47] implies that the histidine residue of the latter sequence does not play a major role in HgII-binding. Interestingly, the lower pH TDPAC-spectra obtained for both systems reflect slightly broader lines and also a somewhat lower frequency in case of MBL-VC (νQ = 1.36 GHz) [48] which may indicate the presence of a small fraction of species with different structures and/or a very weak binding of an additional donor ligand in the HgII–MBL-VC complex.
2.4.3. Probing the metal binding sites of metalloproteins and the influence of non-native metal ions on protein structure
TDPAC spectroscopy was applied to study the metal-binding features of various metalloproteins using the 199mHg isotope as a spectroscopic probe substituting the native metal ions at the probed metal sites. Studies have been performed with the HgII-substituted rubredoxin [49] and in the systems of HgII/CdII and various zinc-finger model oligopeptides [50], the HgII-binding of the human metallochaperone HAH1 [51] as well as the 23-mer oligopeptide fragment cleaved off near the C-terminus of the transmembrane protein BRI2 (BRI2-23) [52]. The first two of these examples are discussed here and the latter two in the following sections.
Studies on various divalent metal ion substituted forms of rubredoxin, bearing a well-defined tetrathiolate metal binding site, were executed to model thiolate-rich metal binding sites of other proteins [53]. In addition to UV–vis, CD and MCD spectroscopy, the HgII–rubredoxin system was investigated by 199mHg TDPAC spectroscopy. Experiments were performed at pH = 8.0 and at a temperature of t = 4 °C (solution saturated with sucrose in order to reduce rotational diffusion) and at low pH (pH = 2.5) both at t = 4 °C and in the frozen state (t = −30 °C, with no sucrose added) [49]. Surprisingly, the three spectra showed rather distinct spectral features (figure 3) in spite of similar CD signatures obtained at pH 2.5 and 8.0. The three TDPAC spectra recorded under different conditions were fitted by including 4 different NQIs. One of these NQIs seems to be present under any conditions accounting for ca 20% of the signal, and the NQI parameters may reflect a diagonal Hg-XY type species with unknown non-thiolate ligands where the metal ions are probably kinetically trapped in this less favoured state. The other three NQIs (1: ω1 = 0.09 Grad s−1 — η = 0; 2: ω1 = 1.13 Grad s−1 — η = 0.21; ω1 = 1.34 Grad s−1 — η = 0.20) may represent distorted 4-, 3- and 2-thiolate coordinated metal binding sites. Interestingly, the spectrum measured in the frozen solution is strongly dominated by the features of a bisthiolate coordinated HgII species, the three NQIs are present in comparable proportion at t = 4 °C and pH 2.5, while at pH = 8.0 the 4-coordinated species becomes the major component (with a relative proportion of ca 70%), reflecting either kinetic or thermodynamic differences between the samples [49].

Figure 3. 199mHg TDPAC time spectra of HgII-rubredoxin (Rd) (left) and their Fourier transforms (right) recorded under the indicated conditions (note that there appears to be a minor typographical error in panels (A) and (B), where pH should presumably be 2.5) [49]. The points (left) or thin lines (right) represent the experimental data while the fits are shown by the bold lines. The original figure was published in [49]. 2000 © SBIC 2000. Reproduced with permission of Springer.
Download figure:
Standard image High-resolution imageIn a study of three oligopeptides, modelling the classical C2H2 zinc fingers, double zinc fingers of the C3H type and the zinc C4 sites, the metal site structures were investigated at various metal ion:peptide stoichiometries for ZnII, CdII or HgII [50]. 199mHg spectra recorded with the C3H model in the presence of 0.1, 1 and 2 equivalents of HgII revealed a pronounced change in the coordination sphere of the metal ion. A tetrathiolate-coordinated metal centre is suggested by the very low frequency signal observed under substoichiometric conditions which indicates metal ion bridging either intra- or intermolecularly. In the HgII–peptide 1:1 system the low frequency NQI is still present together with a new set of signals characterised by higher frequencies. The second equivalent of HgII induced further spectral changes and the fit of the spectrum required the involvement of a third NQI. The increasing HgII–peptide ratio thus induces structural changes in par with the more and more limited access to the Cys donor atoms and the appearance of species with non-C4 type coordination environment. Indeed, experiments performed with CdII and the three model ligands (and even studies with the ZnII–C2H2 system) indicate, similarly, that the zinc fingers are prone to misfold in the presence of low equivalents of metal ions, leading to the appearance of various structural states different from the fully loaded species [50]. These results challenge the general view on the mechanism of action of metal ion regulated proteins, i.e. such systems might operate in a more complex way than simply switching between a loaded and unloaded state [50].
2.4.4. Exchange dynamics at protein metal binding sites
The coordination site of the human copper chaperone HAH1 was probed by studying the interaction of the protein with HgII ions [51]. HAH1 is responsible for the delivery of CuI ions from the cytosol to the ATPases ATP7A or ATP7B located in the Golgi membrane [54]. HAH1 binds metal ions at a conserved Met-Thr/His-Cys-Xaa-Xaa-Cys motif and is proposed to transfer CuI to one of the very similar Met-Xaa-Cys-Xaa-Xaa-Cys sites of the ATPases. The work of Luczkowski et al aimed at the better understanding of the molecular mechanism of this transfer process and thus the HgII-binding of HAH1 was studied via the combination of UV–visible, 199Hg NMR (I = 1/2−, stable) and 199mHg TDPAC spectroscopies in the pH-range of 7.5–9.4 [51].
A central part of the work was the comparison of the pH-dependent change of the 199Hg NMR (figure 4, left) and 199mHg TDPAC (figure 4, right) spectral features obtained for the systems containing 0.5 equivalents of HgII relative to the HAH1 protein. The 199Hg NMR spectrum recorded at pH = 7.5 shows one single peak at −819 ppm, reflecting a HgS2 type two-coordinate species while a different resonance appearing at −348 ppm is observed at pH = 9.4. At pH = 8.5 both resonances are present with comparable intensities. In line with the NMR results the 199mHg TDPAC spectrum at pH = 7.5 exhibits the signature of a single HgS2 complex, however, at pH = 8.5 the appearance of a second NQI, possessing a major peak at 0.5 rad ns−1 results in a loss of amplitude and broadening of the most intense HgS2-related signal (see the right panel in figure 4). The TDPAC spectrum recorded at pH 9.4 indicates the coexistence of at least 2 (but maybe even more) different NQIs, one of which may be attributed to a distorted HgS4 structure while the second is possibly a tricoordinate T-shaped HgS3 species. The exchange between the two-coordinate HgS2 type complex and the higher coordination number species is slow both on the TDPAC and the NMR timescale (ms or slower) resulting in separate NMR signals at pH = 8.5. Contrary to this, the exchange rate at pH 9.4 between the presumably 3- and 4-coordinate structures is fast enough to result in the coalescence of the NMR signals leading to a single resonance at −348 ppm (figure 4, right), falling in the intercept of the ppm ranges typical for T-shaped HgS3 and HgS4 complexes, respectively, but slow or intermediate on the TDPAC timescale (μs–ns) [51]. The main conclusions of the work are that (i) the increase of pH leads to a HgII-promoted association of two HAH1 molecules, i.e. HgS3 and HgS4 type metal ion bridged structures; (ii) these species are presumable adequate models of the intermediate states of the metal ion transfer between the chaperone and ATPase proteins.

Figure 4. 199Hg NMR spectra (left) and Fourier transform of the experimental 199mHg TDPAC data (right) for the samples of HgII–HAH1 1:2 at different pH values [51]. In the right panel, the two major 199mHg TDPAC signals and their change with pH are indicated by the arrows: pH 7.5 (solid line), 8.5 (dashed line), and 9.4 (dotted line). The original figure was published in [51]. Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced with permission.
Download figure:
Standard image High-resolution image2.4.5. Metal ion promoted folding/misfolding and association of proteins
The oligopeptide fragment BRI2-23, cleaved off by proteolytic enzymes from the C-terminal extracellular part of the transmembrane protein BRI2, was demonstrated to have an inhibitory effect on the aggregation of amyloidβ [55], the peptide related to Alzheimer's disease. Several transition metal ions (e.g. Cu, Zn, Fe) are believed to play various roles in neurodegeneration, however, the metal binding ability of BRI2-23, possessing several potential metal coordinating amino acid residues, including two cysteines at position 5 and 22, is largely unexplored. In this multi-method study, HgII and AgI ions were used to probe the CuI-binding characteristics of BRI2-23 and the effect of the metal ion on the folding and aggregation of the oligopeptide molecules [52]. BRI2-23 inherently possesses a disordered, random coil structure and despite the relatively large distance between the two Cys residues, HgII binding was observed in a broad pH-range, when the metal ion–peptide ratio was not more than 0.5:1. 199mHg TDPAC experiments indicated the presence of a single, bis-thiolate bound HgII complex under these conditions at pH = 3.0. However, with increasing the HgII–peptide ratio, especially at a higher pH (pH = 7.4), more complex spectra were observed, indicating the coexistence of species with higher coordination numbers. The results of TDPAC, NMR and MD simulations led to a conclusion that at a 0.7:1 metal-to-peptide ratio metal ion bridged oligomeric species occur. Aggregation might be promoted by the β-sheet formation that is enforced by the coordination of the metal ions (even in the presence of 0.5 equivalents of HgII or AgI). As a consequence, the authors speculate that metal ion binding of BRI2-23 may diminish or eliminate the inhibitory effect of the peptide on Aβ aggregation or might even lead to a reverse action [52].
2.4.6. In vivo applications of TDPAC spectroscopy
In the study on the HgII-binding of a C3H zinc finger model system, discussed in section 2.4.3, Heinz et al also performed experiments where HgII was added to samples of the oligopeptide pre-loaded by ZnII ions [50]. One equivalent of HgII was added to solutions containing ZnII and the C3H model in 1:1 and 2:1 ratios. In contrast to the findings for the ZnII-free system, vide supra, the TDPAC features of both samples are nearly identical and demonstrate very clear signals, typical for linear HgS2 structures (νQ = 1.43 GHz and η = 0.17–0.19) [50]. In addition, 199mHg (and 111mCd) TDPAC experiments with living barley plants and a sample containing homogenised barley leaves were also performed. For the former experiment, a 5–7 day seedling of the plant was removed with intact roots and soaked in a solution containing 199mHgII for 10 min, followed by data collection. The sample of homogenised barley leaves was prepared by shock-freezing the leaves in liquid nitrogen, followed by grinding. The mixture of non-radioactive and radioactive HgII was added to freshly thawed ground barley leaves and after 10 min of incubation, data collection was started. Rotational correlation times showed that the metal ions were bound to high molecular weight or immobilised components and the spectra were stable in time, i.e. stable binding occurred after the 10 min incubation time. The recorded spectra are, indeed, almost identical to those measured in the C3H model systems in the presence of ZnII (νQ = 1.46 GHz and η = 0.14 (whole plant) and νQ = 1.43 GHz and η = 0.21 (ground barley leaves)) [50]. These data suggest that HgII may bind to already pre-loaded metal sites, a finding which may have relevance for mechanisms of HgII-toxicity. At the same time, this experiment also reflects the applicability of TDPAC spectroscopy in the investigation of in vivo samples.
2.4.7. 204mPb-TDPAC applied to a coordination compound
Vibenholt et al applied 204mPb-TDPAC to study the NQI in a molecular crystal of (tetraphenylarsonium lead(II) isomaleonitriledithiolate ![$({[{\rm{A}}{\rm{s}}{\rm{P}}{{\rm{h}}}_{4}]}_{4}[{\rm{P}}{{\rm{b}}}_{2}{({\rm{i}}-{\rm{m}}{\rm{n}}{\rm{t}})}_{4}])$](https://content.cld.iop.org/journals/0954-3899/44/6/064003/revision2/jpgaa666bieqn43.gif) ) [24]. The recorded experimental data showed only one signal indicating that a single PbII coordination geometry is present in the sample. Density functional theory (DFT) calculations of the EFG at the PbII site were carried out for both monomeric and dimeric models of the molecular crystalline structure, demonstrating that the PbII coordination geometry is best described as
) [24]. The recorded experimental data showed only one signal indicating that a single PbII coordination geometry is present in the sample. Density functional theory (DFT) calculations of the EFG at the PbII site were carried out for both monomeric and dimeric models of the molecular crystalline structure, demonstrating that the PbII coordination geometry is best described as ![${[{\rm{P}}{{\rm{b}}}_{2}{({\rm{i}}-{\rm{m}}{\rm{n}}{\rm{t}})}_{4}]}^{4-}$](https://content.cld.iop.org/journals/0954-3899/44/6/064003/revision2/jpgaa666bieqn44.gif) dimers and not the monomeric
dimers and not the monomeric ![${[{\rm{P}}{\rm{b}}{({\rm{i}}-{\rm{m}}{\rm{n}}{\rm{t}})}_{2}]}^{2-},$](https://content.cld.iop.org/journals/0954-3899/44/6/064003/revision2/jpgaa666bieqn45.gif) contributing to a discussion of this topic in the literature. This work represents a step from applications of 204mPb TDPAC to simple inorganic Pb(II) containing compounds towards biomolecular model systems.
contributing to a discussion of this topic in the literature. This work represents a step from applications of 204mPb TDPAC to simple inorganic Pb(II) containing compounds towards biomolecular model systems.
2.4.8. Online 68mCu TDPAC spectroscopy
Isotopes with lifetimes too short to allow for chemical sample preparation (which typically takes at least 15 min), may be applied in TDPAC spectroscopy by direct implantation into the sample of interest, and mounting a TDPAC instrument around the implantation site at the beam line, see figure 5. This approach, referred to as online experiments, was applied at ISOLDE in 2014 for 68mCu-TDPAC experiments using the 68mCu/68Cu decay (6−, 721 keV, 3.75 min) [17], see table 1. In the context of biochemistry this was motivated by the fact that a number of Cu-dependent proteins carry out essential metabolic functions for example in photosynthesis and respiration, and that CuI is difficult to observe using most standard spectroscopic techniques. The Cu isotope with the most favourable properties for the TDPAC application was found to be 68mCu, since it provides a cascade of γ-rays with suitable energies (637 and 84 keV), the half-life of the 2+ intermediate state falls into the TDPAC time scale (7.84 ns), the angular anisotropy expected is rather high (15%) and the 68mCu available production yields at ISOLDE are satisfactory when a UCx target is irradiated with the 1.4 GeV proton beam.
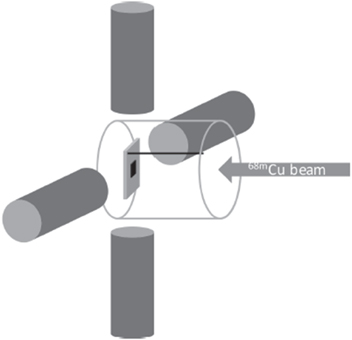
Figure 5. Experimental setup for online 68mCu-TDPAC measurements at VITO-ISOLDE. The transparent cylinder corresponds to the quartz finger, the sample and the sample holder are placed in the geometrical centre and the four cylindrical LaBr3 detectors surround the implantation spot.
Download figure:
Standard image High-resolution imageThe 68Cu beam [56] was ionised to CuI using the resonance ionisation laser ion source and directed to the VITO beamline [57]. At the end of the VITO beam line, a collection chamber was placed allowing for direct optical view of the sample holder, and hence the sample, that was mounted in the geometrical centre of the experimental setup as shown in figure 5. The setup consisted of four LaBr3 detectors with good energy resolution (11.6% at 84.1 keV and 3.0% at 637 keV) and time resolution of 800 ps for the selected cascade. The four detectors were positioned in a plane perpendicular to the direction of the ion beam and each detector was placed at 90° or 180° with respect to each other. Multiple host materials were tested and depending on the sample and the beam conditions the collection and detection times varied from two to four hours per sample. A Cu2O sample was used to probe the quadrupole interaction at Cu sites, while magnetic Ni and Co foils were chosen to measure the magnetic hyperfine interactions.
These are the first online PAC experiments at ISOLDE with a copper isotope. The results led to determination of the relevant nuclear moments of the isotope. The relatively small nuclear electric quadrupole moment (∣Q(2+, 84.1 keV)∣ = 0.110(3) b) [17], combined with the relatively short half-life of the intermediate nuclear level (7.84 ns), makes it difficult to record just one oscillation in the experimental PAC data, even in the presence of a large EFG. Thus 68mCu may not be useful for elucidation of CuI coordination geometries in metalloproteins.
3. β-NMR spectroscopy
3.1. The method
β-NMR relies on the anisotropic emission of β particles in the decay of spin-polarised nuclei [58]. Application of a radiofrequency (rf) field at a resonance frequency leads to transitions between nuclear sublevels and thus to loss of spin polarisation and a decrease in the decay asymmetry. The spin polarisation is high (between 1% and close to 100%), compared to about 0.001% in conventional NMR, since it does not rely on the differential occupation of energy levels of probe nuclei in thermal equilibrium. Signal detection is also very efficient, because it relies on counting β particles (see figure 6), which can be performed with around 10% efficiency (resulting from close to 100% detector efficiency and 10%–20% detector opening angle). For these reasons β-NMR offers ∼10 orders of magnitude increase in sensitivity compared to conventional NMR.
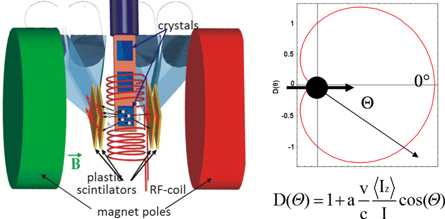
Figure 6. Principle of signal detection in β-NMR. Left: the anisotropically emitted β-particles are detected in two sets of β detectors (plastic scintillators). Right: the angular distribution of β-particles emitted by a spin polarised ensemble. a is the nuclear asymmetry parameter, v/c is the β-particle speed divided by the speed of light (close to 1), 〈Iz〉/I is the spin polarisation degree, and Θ is the angle of emission with respect to polarisation axis.
Download figure:
Standard image High-resolution imageInitially an ion beam of spin-polarised nuclei is implanted into the material of interest. As the direction of emission of the β-particles depends on the nuclear spin direction, the β-particles are emitted anisotropically [58]. The decay anisotropy is proportional to the degree of spin polarisation and to a so-called 'asymmetry parameter', which depends on the spins of the nuclear states involved in the β-decay. In analogy to conventional NMR spectroscopy, the probe nuclei in the system of interest are placed in an external magnetic field and exposed to radio-frequency electromagnetic radiation. At the resonance frequency, transitions between magnetic sub-states occur, giving rise to a loss of nuclear spin polarisation, and thus, a loss of anisotropy in the β-particle emission.
β-NMR spectroscopy is well-established in nuclear-structure and material-science investigations. In nuclear-structure studies it provides high-precision values of magnetic dipole and electric quadrupole moments as well as the nuclear spin of short-lived nuclei [59]. In condensed-matter studies, β-NMR is a sensitive probe of the local electronic structure and magnetic properties [26], and may even be applied for nanoscale depth profiling [60–62].
There are several ways to polarise short-lived nuclei for β-NMR studies [59]. Fragmentation reactions are used at the GANIL, MSU-NSCL, and RIKEN facilities. Although in principle they can be used for many chemical elements and for very short-lived isotopes, they are affected by rather low polarisation (usually 1%–10%) and have very high energy (around 100 MeV/nucleon) and large energy spread. A system for low-temperature nuclear orientation is used e.g. at ISOLDE, but it requires cooling the samples to very low temperatures. Polarisation has been also created via passage through tilted foils, both at several facilities in Japan (e.g. TRIAC or Osaka University) and at ISOLDE, where the technique has been recently revived at optimal beam energy (100–300 keV/nucleon) and the first proof-of-principle experiment has already taken place [63]. Relatively high beam energy and good beam quality could make the implantation into a liquid sample relatively straight forward, but the achieved polarisation is still low (several %) and it requires the use of a post-accelerator. The last polarisation method relies on optical pumping of relatively slow (30–60 keV) and good quality radioactive beams. Such studies were performed at ISOLDE, using the collinear laser spectroscopy setup, COLLAPS, providing many successful β-NMR spectra for nuclear-structure studies, e.g. 8,9,11Li [64]; 11Be [65]; 26–29Na [66]; 29,31,33Mg [67–69]. Another actively used system based on optical pumping is located at the TRIUMF facility in Canada [70]. It uses laser-polarised 8Li beam for condensed matter studies [60–62, 71]. Also 9,11Li were used for nuclear structure studies [72] and recently 31Mg was added to the polarised beams [73].
The optical pumping method is suitable for biochemistry studies, since it gives very high degrees of spin polarisation (10%–100%), it does not require stopping or extreme cooling of the ion, and it has provided very good and reproducible results for over a dozen isotopes. The method uses the atom/ion interaction with circularly polarised laser beam [59] and the resulting electron polarisation is then transferred to the nucleus via the hyperfine interaction. The beam of radioactive ions is brought into resonance with laser light by Doppler tuning their energy. After passing the polarisation section, the beam is implanted into the NMR sample placed in a magnetic field. There, a continuous RF field applied to a coil around the sample leads to transitions between nuclear sublevels at the resonance frequency, and β detectors placed at 0° and 180° record the decrease in spin polarisation as the change in β-decay asymmetry (see figure 6).
High degrees of polarisation may be achieved by optical pumping, and the β-decay asymmetries recorded at ISOLDE have approached 40% (28Na [66]). The polarisation depends on the chemical element and the used atomic or ionic excitation scheme with the main losses due to decays into hyperfine states that cannot be excited again and due to spin rotation in the magnet's stray field. The minimum required production rate is around 103 ions s−1, reached both at ISOLDE and TRIUMF for 11Li [64], which is also the shortest-lived nucleus (t1/2 = 9 ms) used in β-NMR. The widths of resonances are in the 1–10 kHz range (with Larmor frequencies of several MHz), mostly due to RF power broadening. The widths are acceptable for present applications, but they can be lowered with lower RF power and with other excitation schemes (e.g. via 2-photon excitation) resulting in widths as low as 30 Hz that have been achieved [74–76]. Chemical shift resolution below 100 ppm has been demonstrated [71] and in liquids the resolution may be even better because the rapid tumbling of molecules averages anisotropic (direction-dependent) components to zero.
In the following we will focus on the experimental efforts towards β-NMR studies in chemistry and biochemistry being undertaken at ISOLDE.
3.2. Isotopes
The criteria dictating which β-decaying radio-isotopes are suitable for β-NMR is the half-life, production rate, the achievable polarisation, and β-asymmetry factor. The half-life should be comparable to or shorter than the spin-lattice relaxation time, ensuring that most β-decays happen while the nuclei are polarised. When recording relaxation times, the half-life should be longer, so that there are enough β counts to properly fit the exponential decay in β-asymmetry. The production rate should be high enough that a spectrum with adequate signal-to-noise ratio can be recorded ideally during a few minutes or even faster, avoiding long term drifts in magnetic field leading to broadening and shifts in resonance position. The degree of polarisation, when using laser excitation to achieve it, is connected to the electronic structure (e.g. Mg+ is much easier to polarise than neutral Mg) and depends on the strength of the transitions accessible with lasers. The β-decay asymmetry factor (ranging between −1 and +1) depends on the spin change in the nuclear decay. In practice most isotopes studied so far have t1/2 between tens of ms and several s, production rates above 105 ions s−1, and recorded β-decay asymmetry (i.e. the product of polarisation and asymmetry factor) above 1%.
The nuclei which have been polarised with lasers (at ISOLDE and at TRIUMF) are presented in the following table (the reference list is not exhaustive).
| Isotopes | Host | Application | Reference |
|---|---|---|---|
| ISOLDE | |||
| 8,9,11Li | Solid | Nuclear structure | [64] |
| 11Be | Solid | Nuclear structure | [65] |
| 22, 26–31Na | Solid | Nuclear structure | [66] |
| 21,29,31,33Mg | Solid | Nuclear structure | [67–69, 77] |
| 31Mg | aLiquid | Chemistry | [78] |
| TRIUMF | |||
| 8Li | Solid, polymer | Material science | [60–62, 71] |
| 9,11Li | Solid | Nuclear structure | [72] |
| 11Be | Solid | Material science | [79] |
| 20,21,26–31Na | Nuclear structure | [80–85] | |
| 31Mg | aSolid | Material science, chemistry | [73] |
β-NMR studies in solution are not trivial, due to the technical challenge of having a liquid sample at the end of a vacuum beam line. The TRIUMF group succeeded in β-NMR studies on polymers, however without the need to go to high pressures [86], while at ISOLDE we recorded the first β-NMR spectrum of 31Mg in an ionic liquid [78]. In addition, in 2016 Japanese groups reported the use of 12C(p, n) transfer reaction to obtain an energetic (20 MeV/u) spin-polarised 12N, which allowed recording the first β-NMR signal in water [87].
The only isotope which has been laser-polarised and used to record a β-NMR resonance in a liquid sample so far is 31Mg. A Mg isotope was chosen because Mg2+ is a metal ion of considerable interest in biology participating in practically all ATP chemistry and in the folding of RNA. In addition, 31Mg: (i) has a suitable half-life (230 ms) and spin 1/2 (and thus is not sensitive to quadrupole interactions which could broaden the resonance and could lead to shorter relaxation times); (ii) it is well polarised and gives high β-decay asymmetries; (iii) is rather well produced at ISOLDE (105 ions s−1) and thus NMR resonances in a solid can be recorded within a few minutes; and (iv) 29Mg has spin 3/2 and also gives good β-NMR response, thus the influence of I > 1/2 spin (for which the quadrupole interaction comes into play) on the NMR spectra can be studied.
Other metal ions of interest for chemistry and biochemistry are Cu+ and Zn2+, because of their importance in all living organisms and the difficulties in studying them with conventional spectroscopic techniques including NMR. Two accepted ISOLDE proposals planned at the dedicated VITO beamline will be devoted to the study of the degree of polarisation in several Cu isotopes [88] and to β-NMR studies in chemistry using 31Mg [89].
The properties of Mg, and Cu isotopes suitable for optical pumping and β-NMR in liquids are given below.
| Isotope | Half-life | Spin | Magnetic moment | Quadrupole moment | ISOLDE yield | Max β-asymmetry at ISOLDE | Reference |
|---|---|---|---|---|---|---|---|
| 29Mg | 1.2 s | 3/2 | +0.9780(6) μN | −0.16(4) b | 6 × 106 | 3% | [68, 90] |
| 31Mg | 230 ms | 1/2 | −0.88355(15) μN | — | 1.5 × 105 | 8% | [67, 68] |
| 58Cu | 3.2 s | 1 | +0.570(2) μN | −0.15(3) b | 3 × 105 | Not yet polarised | [91] |
| 74Cu | 1.6 s | 2 | −1.068(3) μN | +0.26(3) b | 5 × 105 | Not yet polarised | [92] |
| 75Cu | 1.2 s | 5/2 | +1.0062(13) μN | −0.269(16) b | 1 × 105 | Not yet polarised | [92, 93] |
3.3. Instrumentation at ISOLDE
The first β-NMR spectrum of a liquid sample was recorded using the ISOLDE setup for collinear laser spectroscopy, COLLAPS, which is normally used for nuclear-structure studies. Since then the initiative was taken to build a separate beamline—called VITO—devoted, among others, to β-NMR studies on liquid samples [57].
The main elements of the COLLAPS experimental setup used for proof-of-principle β-NMR in liquids are shown in figure 7. The 30–60 keV singly-ionised beam of interest (e.g. 29,31Mg or 8,9,11Li) from ISOLDE is deflected slightly and gets overlapped with the laser beam. The tuning of ions into resonance with laser light that allows optical pumping is made by changing the ion velocity by a set of electrodes. If laser excitation is more favourable on neutral atoms, the ions are neutralised via charge exchange with an alkali vapour (most often Na or K). The ions (or atoms) continue travelling through a 1 m straight section where they are in resonance with circularly polarised laser light and where their spins get polarised along the beam momentum (for ions this region is offset by 100–200 V compared to the rest of the system). The magnetic field of the non-superconducting NMR magnet (up to 0.5 T) is perpendicular to the beam momentum and thus the spins rotate by 90° in the magnet's fringe field, before they reach a sample, usually a crystal or metal plate, placed in the middle of the magnet. In this process some of the polarisation can get lost, especially when working with atoms, which are still in resonance with the laser light. The sample is surrounded by an RF coil, to which continuous RF signals are applied. The ions/atoms path from the production site up to the NMR sample takes place in a high vacuum environment of 10−5–10−6 mbar. Outside the NMR chamber which for solid-state samples is also under high vacuum two sets of thin β detectors are placed in front of each of the magnet poles—these register the β decays and allow calculation of the β-decay asymmetry.

Figure 7. Schematic of ISOLDE proof-of-principle experiment for β-NMR with liquid samples [78], see the text for details. Reproduced from [78] John Wiley & Sons. © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Download figure:
Standard image High-resolution imageIn the proof-of-principle liquid β-NMR experiment [78], after being polarised, the beam passed via the prototype differential-pumping section, which in several stages brought the pressure from 10−5 mbar up to 1 mbar. The second part of the prototype was the NMR chamber which could host both solid and liquid samples with the possibility to change the pressure inside it. The ion beam had to travel for only 2–3 mm through the region of high pressure between a pinhole and a 3 mm liquid drop at the end of a capillary, which allowed reaching reasonable beam transmissions. The sketch of the experimental setup is shown in figure 7.
Within this initiative, work is presently underway on the experimental setup which will allow to polarise with lasers the ions and atoms of interest, detect their polarisation via β-decay asymmetry and in addition, to use these beams for solid-state and liquid NMR. A new differential pumping system and user-friendly liquid NMR chamber are also being prepared. The details of the already existing experimental setup are presented in another contribution to this Journal special volume [94].
This first stage of the setup has been successfully commissioned using radioactive 26Na atoms (t1/2 = 1.1 s) in two short runs in September and October 2016. In the next stages of the project we aim to reionise the neutral beams and to bend the beam of polarised nuclei by 90°, while keeping the direction of their spins. This will decouple the path of the laser light from the beam direction and from the sample position. In this way one can avoid undesired repumping between atomic states when the spins get rotated in the magnet's stray field and the laser beam does not interact with the liquid sample. At the same time other ways of bridging the large pressure difference between beam line and sample will be investigated.
3.4. Applications in chemistry and biochemistry
3.4.1. First NMR resonances of 31Mg implanted in 1-ethyl-3-methyl-imidazolium acetate (EMIM-OAc)
In the ISOLDE proof-of-principle experiment the 31Mg+ beam was implanted in a solid MgO crystal (used as a reference) and then in the ionic liquid EMIM-OAc [78], chosen because (i) it has a very low vapour pressure, and thus can be used to study the polarisation behaviour of the beam when passing gases introduced in the final section of the setup; (ii) it is expected to form oxygen-rich environments for Mg ions which are common in aqueous solutions and binding sites in Mg-dependent proteins; and (iii) ionic liquids are becoming more and more widely used as alternative solvents to water in chemistry. The laser excitation scheme (UV at 280 nm) is suited for Mg+ and not Mg2+ ions. However, once the beam was implanted into the liquid, it was expected to quickly oxidise to the stable oxidation state Mg2+ which is relevant in biochemical investigations.
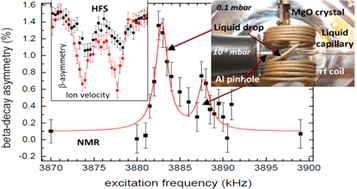
Before recording NMR resonances the ionic transition which gives the highest polarisation and resulting β asymmetry had to be found. This was achieved by changing the 31Mg+ beam energy and thus scanning through its hyperfine structure. In this way the behaviour of polarisation in different hosts and pressures could be checked. The fact that the β-asymmetry was lower with an empty liquid capillary than when a liquid drop was present at the end of it indicated that the beam indeed reached the liquid. This meant that the mean relaxation time increased when the drop was placed in the chamber and thus at least part of the recorded signals came from the drop (see figure 8).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Left: hyperfine structure of 31Mg+ observed in β-decay asymmetry with an empty liquid capillary (black) and with a drop of ionic liquid at the tip of the capillary (red). Centre: β-NMR resonances, see the text for details. Right: the interior of the NMR chamber [78]. The original figure was published in [78]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Download figure:
Standard image High-resolution image{kind=link}
A β-NMR spectrum was recorded within some minutes on only 107 ions of 31Mg in total, showing two resonances of about 2 kHz width and 1300 ppm chemical shift between them (see figure 8 centre). Due to a very limited access to the radioactive beam no signal could be recorded in the MgO crystal (reference for chemical shifts). The interpretation of the spectrum is that the most intense resonance corresponds to a signal from the liquid, while the weaker right resonance originates from the beam lost in the aluminum pipe forming the pinhole. The width of the resonances corresponds to ∼500 ppm, it is dominated by the RF power broadening and can be clearly reduced by using a lower RF (at the cost of lowering the resonance amplitude) [78].
4. Conclusions and outlook
In recent decades TDPAC spectroscopy has provided a number of interesting discoveries, exploiting the isotope production facilities, and benefitting from the highly qualified personnel and spectroscopic equipment at ISOLDE. As such, TDPAC spectroscopy is a very useful work horse, and looking ahead, we expect that this will also be the case in the future. Most of the published TDPAC work has focused on local molecular structure at the TDPAC probe site, while metal site dynamics has rarely been characterized. However, much interesting metallobiochemistry occurs on the nanosecond time scale, simply because many of the aqua ions of biologically relevant metal ions display ligand exchange processes on this time scale [95]. The TDPAC spectroscopic signal is affected by the dynamics on the nanosecond time scale (typically the time scale of the intermediate nuclear level half life in the decay), and thus, we expect that more work to explore the metal site dynamics in biomolecules over the next decade will be conducted.
β-NMR spectroscopy has been applied successfully for three decades in nuclear and solid state physics, but applications in chemistry are still scarce and none have been reported in biochemistry yet. In this context the ISOLDE proof-of-principle experiment and the intense developments which have followed it give exciting perspectives for the next decade, in particular, because several technological obstacles are close to being overcome.
Acknowledgments
We acknowledge the support provided by Federal Ministry of Education and Research (BMBF) through grants 05K13TSA and 05K16PGA and by the FCT project CERN-FIS-NUC-0004-2015 for the use of implantation equipment and infrastructure at ISOLDE/CERN, the EU for support via ENSAR, the Danish Council for Independent Research ∣ Natural Sciences, the Agency for Science, Technology and Innovation under the Ministry of Higher Education and Science, Denmark, for the NICE support, and the ISOLDE physics and technical teams. MS acknowledges CERN-KT Fund for funding. MK and SP acknowledge support by the European Research Council (ERC Starting Grant 640645).