

Abstract
Being the simplest molecule on the planet, H2, as well as its isotopes, have been the prototype systems for strong-field molecular physics for decades. Photoionization and dissociation of H2 have been extensively investigated. After single ionization, the electron left in the molecular ion and its microscopic localization around the two dissociating nuclei can be effectively manipulated using intense femtosecond laser fields with broken symmetry. In this paper, we review the recent progress made on the sub-cycle directional control of the dissociative ionization of hydrogen molecules by tailoring the waveform of femtosecond laser fields, including few-cycle pulses and 2-color fields polarized along the same direction (one-dimension) or different directions (two-dimension).
Export citation and abstract BibTeX RIS
1. Introduction
Electronic and nuclear motions taking place in the attosecond to femtosecond regimes play important roles in defining the fundamental environment of the Universe. With the advent of ultrafast laser technology, it is now possible to detect and manipulate electronic and nuclear dynamics in atomic and molecular systems. When interacting with femtosecond laser pulses with several optical cycles, the force exerted on an electron can be tailored on a sub-femtosecond timescale by manipulating the laser waveforms. Meanwhile, femtosecond laser pulses can provide enormously intense field strength after focusing which is comparable to the force that a bound electron experiences in the vicinity of the nucleus. The hydrogen molecule serves as a prototypical system for investigating intense field processes and has been widely studied [1–19]. Among these studies, the directional control of the electron or ion emission from hydrogen molecules has been realized in the early 1990s by using phase-controlled two-color laser pulses which exhibits asymmetric waveforms. Since then, enormous experimental and theoretical investigations have been carried out and there was a long debate about the 'preferred' emission direction in asymmetric laser fields. The 'non-intuitive' direction was found in early work, indicating that the ion is favored to be emitted into the direction opposite to the electric field. Theoretical calculations also predicted that the directionality of ion emission would be energy dependent [20–22]. With the development of laser technology and momentum-resolved detection techniques [23, 24], it is possible to measure the kinetic energy dependent electron/ion emission when the molecule interacts with intense laser fields. Directional ion emission from neutral hydrogen molecules [2, 5, 6, 10] and from molecular ions [4, 25] have been obtained in various laser fields with broken symmetry, such as phase-controlled two-color laser fields, few-cycle laser fields with carrier envelope phase (CEP)-stabilization or CEP-tagging [26, 27], and using attosecond XUV pump and few-cycle near infrared (NIR) probe techniques [7, 28]. Furthermore, it was recently demonstrated that directional bond breaking, governed by the laser phase at the instant of ionization, indeed exists in an optical symmetric multi-cycle laser pulse [8].
A general interpretation of the directional control on the electron localization in hydrogen molecule, revealed by Roudnev and Esry [29], involves interference of quantum pathways with opposite parities induced by intense laser fields. Both pathways result in the same dissociation energy. Most of the strong field directional control can be understood on the lowest two excited states of the hydrogen molecular ion, i.e. the 1sσg and 2pσu states [30, 31]. Another factor that influences the degree of directional control is the relative probability of the two coupling wave packets, which can be tuned by varying laser field parameters, such as intensity, pulse duration, wavelength and polarization. When interacting with an intense laser field, one electron can be ionized from the neutral molecule with a single electron left in the molecular ion. This electron, driven by the external field, oscillates back and forth between the two nuclei when they dissociate. This occurs until the inter-nuclear barrier rises sufficiently to block electron tunneling leading to localization of the electron at one of the atoms.
Theoretically, the hydrogen molecular ion with one electron and two nuclei is accessible with quantum calculations. For the neutral hydrogen molecule, approximations have to be included and discrepancies start to occur between experimental observations and numerical simulations [32]. Typically, the strongly-driven electron–nuclear dynamics in intense laser fields requires quantum dynamical models beyond the Born–Oppenheimer approximation, which is still challenging. Except for a few research groups using hydrogen molecular ion beams generated from accelerators, most experimental investigations start from neutral molecules. In intense femtosecond laser fields, the molecules can then be ionized producing an ensemble of molecular ions in intermediate states which will dissociate quickly in the field. Such a process usually involves complex coupling of ionization and dissociation steps increasing the difficulty for theoretical modeling. Recently pump–probe experiments using femtosecond laser pulses of various polarizations demonstrated that the complexity of the coupled ionization and dissociation processes can be disentangled by identifying the electrons and ions ejected from different pulses [33, 34]. On the other hand, to reach high intensity the laser beams are usually focused onto a beam of molecules and the volume averaging effect has to be taken into account which dramatically increase the capacity for calculations, especially for quantum simulations.
To directly observe the directional control of electron localization in the hydrogen molecule the inversion symmetry must be broken by using laser pulses with asymmetric waveforms. This can be satisfied by sufficiently short few-cycle laser pulses, or by coherently combining two-color femtosecond pulses. The waveforms of these laser fields can be dramatically changed by the phase. The first CEP-control of the directional dissociative ionization of D2 molecules was carried out by Kling et al using CEP-stabilized, NIR few-cycle pulses [2]. In that study, the CEP-dependent D+ ion emission was observed for kinetic energies (KE) of about 3–8 eV, originating from recollisional excitation. Since then, many experimental and theoretical investigations of the CEP-control of molecular dynamics have appeared [1, 4, 5, 10, 25, 35]. On the other hand, two-color femtosecond pulses synthesized in a phase-controlled way can also provide asymmetric laser fields. Compared to the few-cycle laser fields, a two-color field is easier to manipulate and more robust. Countless theoretical predictions and experiments have shown the potential of such fields in the directional control of molecular reactions [6, 9, 15, 20, 36–40]. Moreover, since the multi-color components can be monitored independently, versatile possibilities can be achieved in the manipulation of molecular dynamics. In this paper, we review on the recent progresses made on the sub-cycle directional control of the dissociative ionization of hydrogen molecules, in particular the one-dimension (1D) control by few-cycle pulses and two-dimension (2D) control by polarization shaped two-color multi-cycle laser pulses, to highlight the relative experimental and theoretical investigations and the underlying control mechanisms in the world's simplest molecule.
2. Experimental approach
2.1. Waveform controlled femtosecond laser fields
To achieve directional control on the dissociative ionization of hydrogen molecules, a waveform controlled laser field with broken symmetry is required. This can be achieved by using CEP-controlled few-cycle laser fields, and the coherent superposition of multi-color laser fields.
2.1.1. Few-cycle laser fields
Compression of high energy laser pulses below 5 fs in the NIR region has been realized at the end of the 20th century by spectral broadening in a hollow core fiber (HCF) and chirped mirror compression [41]. Techniques producing few-cycle pulses in the mid-infrared (MIR) spectral range have been developed based on, e.g. optical parametric chirped pulse amplification (OPCPA) [42–46]. A few-cycle femtosecond laser field can be expressed by E(t) = E0(t)cos(ωt + ϕ), where E0(t) is the envelope which is usually expressed by a Gaussian function, ω is the carrier frequency, and ϕ is the CEP which represents the phase offset between the envelope and the carrier field. With decreasing pulse durations, the CEP plays a more important role on the waveform. When we reach the few-cycle regime where only a couple of field oscillations exist within the pulse duration, as shown in figure 1, changing the CEP can dramatically alter the symmetry of the pulse waveform. For instance, a few-cycle pulse with CEP = 0 exhibits an asymmetric cosine waveform (with higher peak amplitudes in one direction versus the other), while the pulse with CEP = π/2 represents a symmetric sine waveform. Such sudden variation of the electric field shape can manipulate the instant force that exerts on a bound electron in the vicinity of a nucleus and therefore may affect electron motion on a sub-cycle timescale.

Figure 1. Waveforms of few-cycle laser pulses with different CEPs.
Download figure:
Standard image High-resolution image2.1.2. Two-color femtosecond laser fields
Another widely used laser field for the directional control of ultrafast dynamics arises from combining a couple of phase-controlled frequency components and generating waveform controlled multi-color femtosecond laser fields. The specific waveform is sensitive to the relative phase offset between the different frequencies. Here, we take the most commonly used two-color laser field as an example and introduce the characteristics of the superimposed waveforms and their potential applications in directional control of the dissociative ionization of hydrogen in both 1D and 2D. Usually, a beam of multi-cycle femtosecond laser pulses (at about 800 nm) is coherently combined with its second harmonic (SH) signal, which is generated by a nonlinear crystal (for instance, β-barium borate (BBO)). The SH signal after the BBO crystal polarizes perpendicular to the polarization direction of the fundamental wave (FW). A dual-wavelength waveplate can be used to rotate the polarization of the SH by 90° without changing the polarization of the FW. A calcite plate or α-BBO can be used to adjust the time delay (i.e. the phase offset) between the two colors. For a combination of the two components with collinear polarization, waveforms as shown in figures 2(a) and (b) can be obtained, of which by changing the relative phase offsets, the waveform can be dramatically modulated and resulting in either symmetric or asymmetric electric fields. Compared to the state-of-the-art few-cycle technique, the two-color femtosecond laser field is easier to generate and typically more stable. However, due to its multi-cycle characteristics, the ultrafast dynamics induced in different optical cycles may interfere, therefore the underlying physical processes are more complex. From this aspect, few-cycle pulses would be better suited for studies of single- to sub-cycle ultrafast dynamics. Both two-color fields and few-cycles pulses have shown their great potential in the field of ultrafast coherent control. Related works are shown in another recently published review and the references therein [47].

Figure 2. Multi-cycle two-color femtosecond laser fields consisting of the FW at 800 nm and its SH component at 400 nm. (a) Linearly polarized two-color femtosecond laser field with IFW:ISH = 2:1, ϕ = 0. (b) Similar to (a) but with ϕ = π/4. (c) Superposition of two beams of orthogonally polarized multi-cycle 2-color laser fields with IFW:ISH = 2:1, ϕ = 0. (b) Projection of the waveform in (c) onto the polarization plane (x–y plane).
Download figure:
Standard image High-resolution imageGenerally, a two-color femtosecond laser field can be expressed as
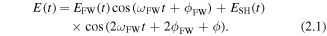
Here EFW/SH(t) represents the envelope of the FW (or SH) component which includes the information about pulse durations and field strengths; ωFW and ϕFW are the optical frequency and CEP for the FW; ϕ is the relative phase offset between the two color components. Such laser fields can be tuned in many degrees of freedom, such as the relative phase, ratio of the field strength, pulse durations etc. One more interesting parameter to be manipulated is the laser polarization. For instance, a multi-cycle two-color laser field by combining the 800 nm component with its orthogonally polarized SH signal is shown in figures 2(c) and (d). With such laser field electron localization in hydrogen can be steered in 2D [48], which also permits to distinguish sequential and non-sequential double ionization of molecules [49]. The electron excited by one color can be streaked by the other color in the perpendicular direction, therefore providing more degrees of freedom for the directional control.
2.2. Detection techniques
Momentum imaging is a favorable way for the exploration of ultrafast dynamics in strong field and matter interactions. Commonly used are velocity map imaging (VMI) [23, 50, 51] and reaction microscopy (REMI) or cold-target recoil-ion momentum spectroscopy (COLTRIMS) [24]. VMI and REMI/COLTRIMS have their advantages and disadvantages in momentum imaging. On one hand, REMI/COLTRIMS can image the three-dimensional (3D) momentum distributions of several fragments in coincidence, therefore can provide very rich information about the ultrafast dynamics under investigation. However, coincidence measurements are highly restrictive on the count rate with typically less than one event that should be detected for each laser shot. This factor dramatically elongates the data acquisition time for measurement and thus increases the requirement on the long-term stability of relative experimental conditions, such as the laser parameters. Where coincidence measurement of multiple fragments is not required, VMI shows enormous advantages. First, higher count rates can be accessed which are limited only by the avoidance of space charge effects and damage to the detector. The data acquisition time can be significantly shortened, by up to orders of magnitude compared to REMI/COLTRIMS. Nevertheless by using VMI it is difficult to clearly identify and resolve various dissociation channels which can be commonly distinguished in coincidence measurements, and in the most commonly used VMI experiments, the resulting momentum image is a projection of the 3D momentum distributions onto the detector plane, thus post inversion is required to restore the original 3D information [52]. Recently, tomographic reconstruction has been developed for VMI measurements such that by detecting the momentum distributions induced by laser fields in multiple polarization directions, 3D momentum distributions can be recovered [53]. All of these techniques can be combined with a stereo-ATI phase meter [54] for the investigation of CEP-dependent phenomena. In the CEP-tagging measurements, which can be performed with the phase meter, the laser is left in a free running mode such that the CEP of every consecutive laser shot is random. The phase information can be detected by the phase meter for each laser shot. Details about CEP-tagging can be found in [30, 31] and references therein.
3. Theoretical modeling
The influence of the CEP on the ultrafast dynamics has been widely studied for molecular hydrogen (H2, D2) and for the molecular ions [29, 35, 55–65]. After ionization, only one electron and two nuclei are left in the molecular ion and the system is accessible by ab initio calculations with high accuracy [55, 66–70]. As a typical example for such simulations on the hydrogen molecular ion, we review a quantum simulation on  conducted by solving the time-dependent Schrödinger equation (TDSE) [32]. In this simulation, the creation of
conducted by solving the time-dependent Schrödinger equation (TDSE) [32]. In this simulation, the creation of  ions in the intense laser field is treated in two steps. (i) Tunneling ionization occurs near the local maximum of the electric field with the tunneling ionization rate estimated according to the molecular Ammosov–Delone–Krainov theory [71]. The electron is released at time t0 and a nuclear wave packet (NWP) is created on the potential curve of
ions in the intense laser field is treated in two steps. (i) Tunneling ionization occurs near the local maximum of the electric field with the tunneling ionization rate estimated according to the molecular Ammosov–Delone–Krainov theory [71]. The electron is released at time t0 and a nuclear wave packet (NWP) is created on the potential curve of  (ii)
(ii)  dissociates in the remaining part of the laser field from either the ground state or the excited state which is populated by electron recollision at time tr. Here, the return time tr and the return energy Er can be obtained by a semi-classical simulation [72]. The time-dependent evolution of the NWP can be obtained by solving the coupled channel TDSE:
dissociates in the remaining part of the laser field from either the ground state or the excited state which is populated by electron recollision at time tr. Here, the return time tr and the return energy Er can be obtained by a semi-classical simulation [72]. The time-dependent evolution of the NWP can be obtained by solving the coupled channel TDSE:
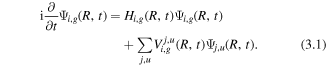
Here i, j stand for the index of the 1σ, 2σ, 3σ, and π states and g/u stands for the gerade/ungerade (even/odd) states.  is the interaction between the i, g and the j, u states which are coupled by the laser fields. When using few-cycle pulses with high peak intensity, highly excited states can be populated. The Hamiltonian can be written as
is the interaction between the i, g and the j, u states which are coupled by the laser fields. When using few-cycle pulses with high peak intensity, highly excited states can be populated. The Hamiltonian can be written as
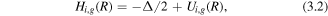
where Ui,g(R) is a given potential curve. For an initial state created at the time tr, the NWP is propagated in the NIR field by equation (3.1). The NWP at the end of the pulse can be expressed as
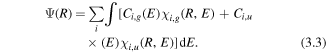
Here,  is the energy normalized continuum nuclear wave function for a given dissociation energy E and Ci,g is the amplitude. The final state of the NWP is determined by the initial condition, the laser parameters and the alignment angle between the molecular axis and the laser field polarization direction [55].
is the energy normalized continuum nuclear wave function for a given dissociation energy E and Ci,g is the amplitude. The final state of the NWP is determined by the initial condition, the laser parameters and the alignment angle between the molecular axis and the laser field polarization direction [55].
For the case that the laser pulse polarizes linearly along, i.e. up and down directions, the electron localization to the 'up' or 'down' direction around the two hydrogen nuclei can be expressed by
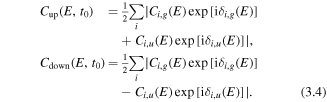
Here  is the phase offset for the gerade (g) and ungerade (u) states at a certain dissociation energy E. Coherent coupling of the state pairs with opposite parity will result in a preferential localization either in the 'up' or the 'down' direction. The
is the phase offset for the gerade (g) and ungerade (u) states at a certain dissociation energy E. Coherent coupling of the state pairs with opposite parity will result in a preferential localization either in the 'up' or the 'down' direction. The  ion will finally dissociate along the D(n = 1, 2) states and result in D+ + D, i.e. the D2(1, 0) channel. Here n is the principle quantum number for the hydrogen atom.
ion will finally dissociate along the D(n = 1, 2) states and result in D+ + D, i.e. the D2(1, 0) channel. Here n is the principle quantum number for the hydrogen atom.
The initial condition for equation (3.1) when the D2 molecule is ionized by the laser field at time t0, can be written as
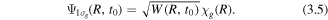
Here W(R, t0) is the tunneling ionization rate at a given nuclear separation R and at time t0 with the over-barrier correction [73],  is the vibrational ground state function of D2. The dissociation probability can be calculated from equation (3.1) and the dissociation of
is the vibrational ground state function of D2. The dissociation probability can be calculated from equation (3.1) and the dissociation of  to the up or down direction can be expressed as
to the up or down direction can be expressed as
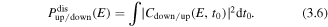
Note that the D+ ion will be emitted to the opposite direction of electron localization and equation (3.6) indicates the probability of the remaining electron localization direction in the system.
In the case of recollision-induced dissociation, the initial condition for equation (3.1) is
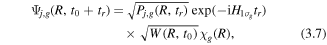
Here Pj,g(R, tr) is the electron impact excitation probability to the j, g states at the position R and  is the vibrational Hamiltonian associated with the
is the vibrational Hamiltonian associated with the  ground electronic state. Then the dissociation of D+ to the up or down direction can be expressed as
ground electronic state. Then the dissociation of D+ to the up or down direction can be expressed as
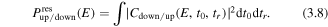
Therefore, for the recollisional excitation, the electron returning time tr will also affect the directional ion emission.
To illustrate the directionality of the emitted D+ ions as a function of CEP and kinetic energy E, we define an asymmetry parameter, A, which can be written as
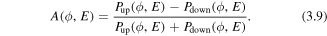
This parameter, depending on both CEP and kinetic energy, can be effectively manipulated by tuning the laser parameters of the few-cycle pulses. The CEP-dependence results in an asymmetry oscillation with a period of 2π. On the other hand, the phases of the NWP give rise to the kinetic energy dependence of the asymmetry, which is related to the phase shifts between the involved odd and even states.
4. Results and discussions
4.1. Directional control of the dissociative ionization in hydrogen with few-cycle laser pulses
In this section, the CEP-control of the dissociative ionization of hydrogen molecules in few-cycle laser fields is discussed. The sub-cycle control of electron localization in the dissociative ionization of molecular hydrogen was first shown with CEP-stabilized NIR few-cycle femtosecond laser fields in 2006 [2]. Controlling the CEP of the laser pulses can manipulate electron localization during dissociation, which is monitored experimentally by detecting the momentum distributions of the D+ ion emission that points to the opposite direction of the localized electron. Figure 3 shows the results obtained in the CEP-control of the dissociative ionization of D2. In figure 3(a), the 2D momentum distributions of D+ ion emission are averaged over all CEPs, where the laser is polarized in the y-direction. To illustrate the directional characteristics of the ion emission, the asymmetry parameter defined by equation (3.9) is analyzed which is obtained by integrating the ion yield within a certain angle range along the polarization direction. The corresponding asymmetry map as a function of CEP and ion kinetic energy is shown in figure 3(b). Clear oscillation of the asymmetry parameter with a period of 2π is observed for the energy region about 3–8 eV with an amplitude of about 20%. In this map, a positive asymmetry parameter stands for the cases where the D+ ion is preferentially emitted to the 'up' direction, corresponding to an electron localization in the 'down' direction. The high energy region is assigned to recollisional excitation due to the fact that the signal disappears when using circularly polarized few-cycle pulses, which omits rescattering processes. The oscillation amplitude of the asymmetry parameter, i.e. the degree of control over the directionality, can be adjusted by tuning the laser parameters such as intensity, pulse duration, etc. It is pointed out that the duration-dependence of the asymmetry amplitude more generally follows an exponential decay with increasing pulse duration [3]. By reducing the pulse duration close to the single-cycle regime [74], a higher degree of electron localization can be reached. As an example, reducing the pulse duration from 5 to 4 fs, the asymmetry oscillation amplitude can already be doubled [32].

Figure 3. (a) CEP-averaged momentum distributions for D+ from D2 in a 5 fs few-cycle laser field at an intensity of about 1 × 1014 W cm−2. (b) The experimental asymmetry parameter of the D+ ion emission as a function of kinetic energy and the CEP. Figure 3(a) adapted from [75]. CC BY 3.0. Figure 3(b) adapted from [2]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageWhen a hydrogen molecule interacts with intense few-cycle laser fields, typically an electron is liberated from the molecule, accompanied by launching of a NWP on the 1sσg state of the molecular ion, which can then dissociate via several processes indicated in figure 4. For example, the initially emitted electron can recollide with the molecular ion and excite the molecular ion from the 1sσg state to highly excited states (e.g. the 2pσu state). Laser-induced population transfer between the 1sσg and 2pσu states results in a superposition of these states. This allows control of the electron localization on one hydrogen nucleus or the other, which is determined by the laser field parameters. Quantum dynamical simulations can reveal the electron–nuclear coupled dynamics [76]. Figure 5(a) shows the electron expectation value along the molecular axis (z, where z = 0 represents the center of mass of the ions) in few-cycle NIR laser fields with CEP = 0. Here it is assumed that the molecule is ionized by the laser field's maximum at t = 0 fs, and the recollisional excitation occurs at about 2/3 optical cycles afterwards. The electron oscillates between the two hydrogen nuclei when the molecule dissociates, until a moment (approximately 12 fs) where the potential barrier between the two nuclei becomes too high for tunneling. Then the electron density remains localized on one hydrogen atom (in the +z direction) and a proton will be ejected to the opposite direction. Although ionization by the maximum of the electric field possesses high probability, ionization can also be induced by smaller peaks in the neighboring optical cycles, which leads to a different degree of control. As shown in figure 5(b), the calculated time-dependent expectation value of the electron along the z-coordinate is shown for three extrema (indicated by A, B, and C in different colors). During the first 2 fs, the electron dynamics initiated from A and C evolves in an opposite way compared to that of B. However, after about 12 fs all the three situations result in an electron localization in the +z direction, which indicates that the outcome cannot be simply predicted by the sign of the laser field at the moment of ionization. Nevertheless, the overall outcome (i.e. where the electron localizes) can be inversed by a CEP change of π. Recently, real-time electron localization has been observed for the first time in a few-cycle pump–probe scheme which revealed that electron localization in  is completed in about 15 fs at an inter-nuclear distance of about 8 au. [77].
is completed in about 15 fs at an inter-nuclear distance of about 8 au. [77].
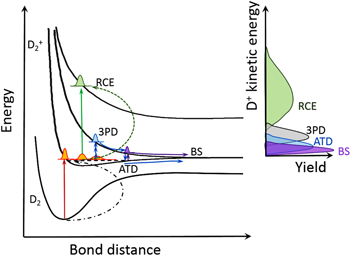
Figure 4. Schematic diagram showing different dissociative single ionization pathways of D2 (BS: bond softening, ATD: above threshold dissociation, 3PD: 3-photon dissociation and RCE: recollision excitation) yielding D+ + D fragments in NIR few-cycle laser fields. Double ionization pathways are not shown.
Download figure:
Standard image High-resolution image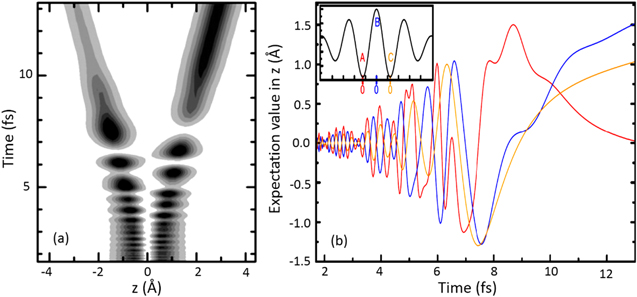
Figure 5. (a) Electron density as a function of time and the electron coordinate z (along the molecular axis) for 5 fs few-cycle pulses at 760 nm with CEP = 0. (b) Expectation values of electron in z as a function of time for different ionization moments (indicated by the red, blue, and yellow curves, respectively). Reproduced from [76]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageThe dissociative ionization of hydrogen can however occur via other pathways, complicating the whole picture. As shown in figure 4, these dissociation pathways can be illustrated on the potential curves including the ground state of the neutral molecule as well as the ground and excited states of the cation when interacting with NIR few-cycle pulses. Note, that we have omitted some pathways, including doubly excited neutral states and double ionization. Typically, after the ionization process, a NWP is started on the 1 sσg state of the hydrogen molecular ion. A dissociation pathway that is also not shown in figure 4 is the zero-photon dissociation channel, which produces slow protons with near-zero KE [78, 79]. Such process can occur due to the partial escape of a vibrational wave packet that is trapped in a laser-induced state. When using sufficiently broadband pulses, the NWP can be excited by one photon of the blue part of the spectrum and then down shift with one photon of the red part which results in very small kinetic energy release (KER) involving net-zero photons [39]. An important dissociation pathway shown in figure 4, involves the NWP propagating along the 1sσg state, where close to its outer turning point coupling to the 2pσu state occurs with absorption of one NIR photon resulting in dissociation. This channel is known as bond softening (BS) [31, 80]. In BS, photon-induced transitions between molecular states give rise to dissociation below the field-free threshold, resulting in fragment ions (here D+) with characteristic KE (see figure 4). For BS to occur requires that the cation stretches to a large bond separations, which takes about half of a vibrational period (about 12 fs for D2 [81]). This channel can therefore be strongly suppressed in few-cycle experiments if the pulse contrast is sufficiently high. The most commonly used few-cycle pulses are generated from compression of HCF-broadened multi-cycle pulses with chirped mirrors, typically resulting in only limited contrast and leaving a pedestal with up to about 10% in intensity lasting for tens of femtoseconds which can still induce dissociation via BS. Regarding the directional control, this channel will, in this case, not exhibit strong CEP-dependence. Kremer et al used slightly longer few-cycle pulses and investigated the CEP-dependent asymmetry parameter in the BS channel with CEP-controlled 6 fs NIR pulses. They showed an orientation dependence of the asymmetry oscillations [5], as depicted in figure 6. Asymmetry oscillations are observed for several alignment angle ranges which exhibits phase shifts between each other. The authors pointed out that switching on the control laser at the time when the wave packet approaches the coupling region can strongly enhance the population transfer and thus enhance the contrast of the asymmetry oscillation.
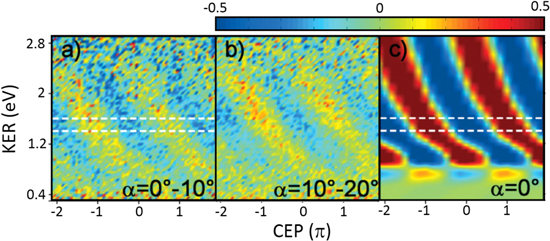
Figure 6. Asymmetry parameter as a function of CEP and KER for different emission angles with respect to the laser polarization axis for (a) 0°–10°, (b) 10°–20°. (c) The simulation results by solving the TDSE at 0°. Adapted figure with permission from [5], copyright 2009 by the American Physical Society.
Download figure:
Standard image High-resolution imageAccording to a series of studies, the CEP-control is expected to decrease exponentially with the number of optical cycles [3, 29] making it difficult with single few-cycle NIR pulses to enhance the directional control. To better match the dissociation time of molecules with few-cycle laser fields, fields with longer wavelengths can be used. Few-cycle pulses in the MIR region, centered at 2.1 μm, have been employed by Znakovskaya et al to investigate the directional control of the dissociative ionization of D2 [10]. At this wavelength, one optical cycle lasts for about 7 fs and the few-cycle pulse duration is comparable to the dissociation time in BS of the hydrogen molecule. Here, an initially prepared wave packet on the 1sσg state of the hydrogen molecular ion (populated through ionization of neutral hydrogen) moves towards the outer turning point of the potential curve, where efficient coupling to the 2pσu state occurs at the one-photon crossing. As a result, a higher degree of control can be achieved for the BS channel using MIR few-cycle pulses. As shown in figures 7(a) and (b), asymmetry oscillations in the BS channel with an amplitude of more than 20% is obtained in the measurement and can be reproduced convincingly with quantum calculations [10]. As shown in figure 7(c), the sign and amplitude of the asymmetry parameter depend on wavelength and the asymmetry amplitude is very small at 800 nm, while reaching large values at certain wavelengths (such as around 1.7 and 2 μm). With the blooming development of intense femtosecond MIR laser systems based on, e.g. optical parametric amplification (OPA) [82] and OPCPA [42–46] techniques, such control of ultrafast reactivity, including electron localization, deprotonation [83] and hydrogen migration [84–86], especially also in complex molecular systems [87, 88], can be explored in more detail.
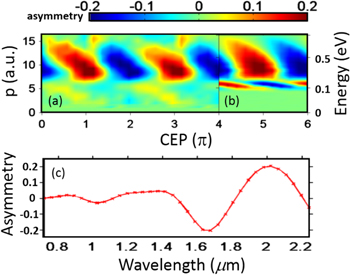
Figure 7. (a) Measured asymmetry parameter of the D+ ion emission from D2 in a 2 μm few-cycle laser field as a function of CEP and the fragment momentum. (b) Theoretical asymmetry parameter obtained for the BS channel, as a function of CEP and the fragment energy. (c) Calculated energy-integrated asymmetry amplitude in the BS channel for various wavelengths for 3.5-cycle pulses. Adapted figure with permission from [10], copyright 2012 by the American Physical Society.
Download figure:
Standard image High-resolution imageAnother dissociation pathway which generates hydrogen fragment ions with slightly higher kinetic energy is the above threshold dissociation (ATD) channel (two-photon pathway) [89] and the three photon dissociation (3PD) channel (also known as the three-photon ATD channel [67, 90]). As shown in figure 4, the ATD channel involves coupling between the 1sσg and 2pσu states by absorption of three photons and emitting one photon after a certain time interval. The 3PD channel involves coupling to the 2pσu state by absorption of three photons. These two dissociation pathways, differing by one photon, interfere and can cause electron localization. In an experimental study by Xu et al [1], a strong asymmetry oscillation of about 40% for the directional emission of H+ ions from H2 following this mechanism was observed for 6 fs few-cycle pulses, see figure 8. In general, as also pointed out in that study, the two most sensitive parameters for the control of CEP effects are the peak intensity and pulse duration. To achieve the highest directional control, these two parameters need to be optimized.
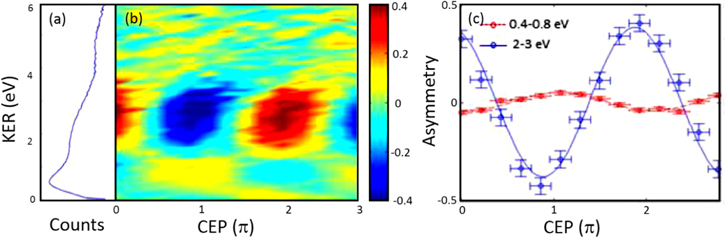
Figure 8. (a) CEP-averaged KER spectrum for protons ejected from H2 in 6 fs NIR few-cycle laser fields. (b) Asymmetry parameter as a function of CEP and KER. (c) Energy-integrated asymmetry oscillations as a function of CEP for the indicated regions. Adapted from [1]. © IOP Publishing Ltd. CC BY 3.0.
Download figure:
Standard image High-resolution imageIn a linear polarized NIR few-cycle laser field, higher energy (above 3 eV) H+/D+ fragments from H2/D2 with broad angular distribution were observed. This channel, which disappears in circularly polarized fields, arises from recollision excitation [2], and is noted as the RCE channel. The first experimental evidence of electron localization induced by few-cycle pulses was found in this region with an amplitude of about 20% (see figure 3). Systematic experimental investigations as functions of pulse durations [3, 29, 76] and peak intensities [32] showed that the amplitude of the asymmetry parameter can be optimized by using shorter pulses with proper peak intensity. In a more recent experiment [32], using shorter pulses of around 4 fs and CEP-tagging allowing for improved signal-to-noise ratio, asymmetric emission at higher KE at and above 10 eV was observed, which originates from dissociation from highly excited states of 
The observation of CEP-control for various dissociation channels usually exhibits characteristic phase offsets and differences in the amplitudes of the asymmetry oscillation with CEP. By careful analysis of the CEP-dependence of the momentum distributions of D+ ions from D2, and comparison to quantum simulations, it is possible to resolve multiple dissociation pathways and reveal the underlying population mechanisms. As an example, we show results from an intensity-dependent study [32] on the directional D+ emission from D2 in 4 fs NIR laser fields (see figure 9). In this near single-cycle regime, the dissociative ionization of D2 is explored for a series of intensities in the range of 1.0–2.8 × 1014 W cm−2. By using near single-cycle laser pulses, it is possible to study the electron localization in hydrogen molecules in a high intensity regime before the onset of double ionization, which through Coulomb explosion will cause symmetric ion emission decreasing the degree of CEP-control. The study revealed D+ ions from the dissociative ionization of D2 with KE up to 12 eV. These high KE signals are from dissociation via the highly excited 3σ state. Ion signals at such high KE have been observed in MIR few-cycle laser fields [10] but the directional control of the ion emission for such high energy region was only realized in the study with near single-cycle NIR pulses at high intensities (likely because double ionization becomes a more likely channel for MIR laser fields). Two distinct channels can be distinguished for the RCE region, as shown in figure 9. One channel centered around 7 eV (marked as RCE-I channel) and the other higher KE channel centered around 10 eV (marked as RCE-II channel) near the cutoff region. The energy integrated asymmetry oscillation (shown in figures 9(g)–(i)) show clear differences on both amplitude and phase offsets for these two channels. Quantum dynamical simulations can reproduce the results quantitatively. Further analysis shows that two mechanisms are responsible for the generation of ions in the RCE-I and II regions. First, for relatively low intensities (<1.8 × 1014 W cm−2) the NWP ignited after ionization can be excited to lower excited states (such as 1σ, 1π and 2σ) through electron recollision, followed by further excitation to the 3σ state induced by the laser field. This will result in ions observed within the RCE-I channel. Second, the NWP can be directly excited to the 3σ state by electron recollision, which dominates at higher intensities above 1.8 × 1014 W cm−2 (RCE-II).

Figure 9. (a)–(c) Measured asymmetry as a function of CEP and kinetic energy of D+ ion from D2 at intensities of 1.0, 1.8 and 2.8 × 1014 W cm−2 for 4 fs few-cycle NIR fields. (d)–(f) Simulated asymmetry maps for the corresponding experimental conditions. (g)–(i) Kinetic energy integrated asymmetry oscillations over the regions of RCE-I (black triangle for experiments and lines for simulations) and RCE-II (red bullets for experiments and lines for simulations). Adapted from [32]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageThe above mentioned mechanisms can be further illustrated by careful analysis of the momentum distributions of the CEP-dependent ion signal and comparison to theoretical calculations. Figure 10 presents angular distributions of the asymmetry amplitude and phase obtained by the following fitting process. For each position in momentum space, the CEP-dependent D+ yield is fitted with a cosine function expressed below:
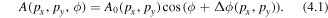
Here px and py are the coordinates in momentum space, A0 and Δϕ are the amplitude and the phase offset of the yield for the CEP-dependent ion emission, ϕ is the measured CEP (with a phase meter, which exhibits a constant offset with respect to the absolute CEP). The resulting momentum maps for the amplitudes (A0) and the phase shifts (Δϕ) can usually help to distinguish different channels. In these maps, the contributions around 12 a.u. in radial momenta (defined as  ) are from the BS channel with a small amplitude and narrow angular distribution. As the intensity increases, the ATD channel shows up in the low momentum region above the BS channel. The RCE channel occupies the region with pr > 25 a.u. with very wide angular distributions. We can see that the asymmetry amplitude is almost zero in the orthogonal direction to the polarization (y-direction), which indicates no asymmetry can be induced in such direction since an external field is required for the coupling of gerade and ungerade states [29] which is absent in this direction. A depletion along the polarization axis is noticeable at higher radial momenta with narrow angular distribution along the polarization axis in figures 10(c) and (d) at the laser intensity of 2.5 × 1014 W cm−2. This contribution is assigned to a sequential process involving recollisional excitation with subsequent laser-induced transition to the 3σ states. This assumption is supported by the calculated angular distribution of the dissociation yield of D+, which is shown in figure 10(e). The yield of 3σ state maximizes at about 15°, while the yield of 1π state changes almost oppositely. When the NWP is excited from the 1π state to the 3σ state, the yield of D+ ions from the 1π start is reduced while the one for dissociation via 3σ state is increased. Also, as evident from this study an asymmetry with an amplitude of up to about 43% can be obtained for the RCE-I channel at an intensity of 1.3 × 1014 W cm−2 in 4 fs NIR fields (see figure 9).
) are from the BS channel with a small amplitude and narrow angular distribution. As the intensity increases, the ATD channel shows up in the low momentum region above the BS channel. The RCE channel occupies the region with pr > 25 a.u. with very wide angular distributions. We can see that the asymmetry amplitude is almost zero in the orthogonal direction to the polarization (y-direction), which indicates no asymmetry can be induced in such direction since an external field is required for the coupling of gerade and ungerade states [29] which is absent in this direction. A depletion along the polarization axis is noticeable at higher radial momenta with narrow angular distribution along the polarization axis in figures 10(c) and (d) at the laser intensity of 2.5 × 1014 W cm−2. This contribution is assigned to a sequential process involving recollisional excitation with subsequent laser-induced transition to the 3σ states. This assumption is supported by the calculated angular distribution of the dissociation yield of D+, which is shown in figure 10(e). The yield of 3σ state maximizes at about 15°, while the yield of 1π state changes almost oppositely. When the NWP is excited from the 1π state to the 3σ state, the yield of D+ ions from the 1π start is reduced while the one for dissociation via 3σ state is increased. Also, as evident from this study an asymmetry with an amplitude of up to about 43% can be obtained for the RCE-I channel at an intensity of 1.3 × 1014 W cm−2 in 4 fs NIR fields (see figure 9).
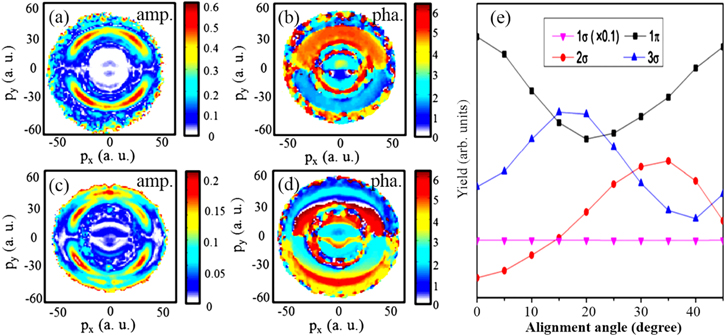
Figure 10. Momentum maps of the (a), (c) asymmetry amplitude and (b), (d) phase offset of the CEP-dependent part of the D+ ion emission at a laser intensity of (a), (b) 1 × 1014 W cm−2, and (c), (d) at 2.5 × 1014 W cm−2. (e) Simulated dissociation yields for different  states as a function of alignment angle with respect to the laser polarization axis at 1.8 × 1014 W cm−2. Adapted from [32]. © IOP Publishing Ltd. All rights reserved.
states as a function of alignment angle with respect to the laser polarization axis at 1.8 × 1014 W cm−2. Adapted from [32]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution image4.2. Directional control of the dissociative ionization of hydrogen with two-color femtosecond laser fields
In addition to using sufficiently short few-cycle pulses, which possess an asymmetric waveform for the directional control of molecular dissociative ionization, another experimental technique can be used, where femtosecond pulses of multiple frequencies are coherently combined. The most commonly used synthesized field is the 2-color field, which is generated from a superposition of the fundamental frequency component with its SH. Two-color fields can induce rich processes in molecular dynamics due to its multi-color perspective, especially with a flexible field configuration of the two frequency components. In general, the underlying mechanisms induced in two-color fields are similar to those in few-cycle fields. However, photons of more color components are involved and the ultrafast dynamics may occur in consecutive optical cycles superimposed coherently, therefore resulting in more complex, and entangled processes (which is why the two control scenarios bear similarities and can both be regarded as Brumer–Shapiro type phase control [91] but are not identical). Typically, few-cycle fields generate results, which result from the interaction with the field within about one optical cycle making their theoretical modeling simpler. Nevertheless, two-color laser fields have shown great potential in the control of molecular dissociative ionizations. A most recent, remarkable result is the possibility to control the asymmetry of proton emission from H2 in 2D [48, 92], a highlight we will summarize further below.
The control mechanism for the asymmetric ion emission, i.e. electron localization, in hydrogen molecules in two-color femtosecond laser fields is shown in figure 11. In the two-color fields the directional dissociation of hydrogen molecules is usually induced by the coupling between the 1sσg and 2pσu states. The NWP on the 1sσg state can be excited to the 2pσu state in multiple ways and the coupling between them induced by the asymmetric laser field causes the directional proton emission. These pathways include the BS channel, the ATD channel, and the RCE channel caused by recollisional excitation. Furthermore, due to the fact that most 2-color fields consist of multiple optical cycles, molecular bonds can be stretched during the initial part of the laser pulse until a critical nuclear separation is reached, where charge resonance enhanced ionization (CREI) [16, 93] occurs, which excites the wave packet to the dicationic state. This can be attributed to field-induced transitions between charge-resonant state pairs (existing at large bond length). However, this channel will generate two hydrogen ions in a Coulomb explosion and they will be emitted with equal probability in the two directions thus exhibiting no asymmetry.
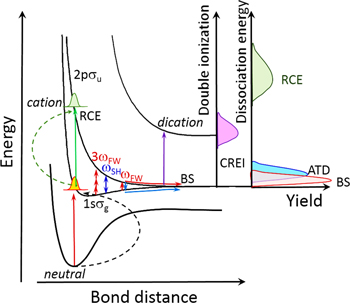
Figure 11. Schematic diagram of the ionization and dissociation processes for hydrogen molecules in a 2-color laser field. Reprinted figure with permission from [6], Copyright 2009 by the American Physical Society.
Download figure:
Standard image High-resolution image4.2.1. 1D directional control of the H+ emission in two-color laser fields
Observations of asymmetric ion yield from the dissociative ionization of molecular hydrogen in two-color laser fields have been reported decades ago [37]. In collinearly polarized two-color laser fields, energy dependent directional emission of D+ ions from D2 was observed when manipulating the relative two-color phase [6]. The reaction pathways were assigned on the basis of the analysis of asymmetry parameters and comparison to quantum simulations. The observations together with major reaction channels are shown in figure 12.
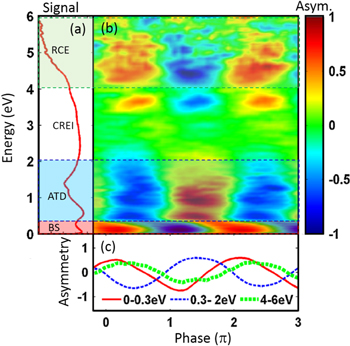
Figure 12. (a) Log plot of the phase-averaged D+ yield as a function of energy. (b) Asymmetry parameter of D+ ion emission from D2 in a two-color field as a function of the relative phase and ion kinetic energy. (c) Energy-integrated asymmetry oscillations for three regions. Adapted figure with permission from [6], copyright 2009 by the American Physical Society.
Download figure:
Standard image High-resolution imageClear asymmetry oscillations with two-color phase are observed in figure 12 and different channels can be identified. These are the one-photon BS channel, the ATD channel, and the RCE channel, as indicated in figure 12. The directionality for the BS channel arises from pathway interference involving (i) absorbing one FW photon and dissociating via the u-state, or (ii) absorbing one SH photon and then emit one FW photon and dissociate via the g-state. Similarly, the ATD channel asymmetry is formed by interference between two dissociation pathways, either by absorbing three FW photons and emitting one later with dissociation along the u-state, or by absorbing one SH photon and dissociation along the g-state. The maximum asymmetries for all these channels occur at different phases, which becomes clearer when inspecting the energy-integrated asymmetry in figure 12(c). The strong ion yield centered around 3 eV arises from CREI with formation of the dication and reduces the asymmetry parameter for the neighboring dissociation channels, which could contribute in this region.
It is also possible to realize probability (instead of directional) control by manipulating the interference between two pathways differing by an even number of photons [29]. In a recent two-color experiment utilizing the combination of a FW at 1.8 μm and its third harmonic frequency (TH) at 600 nm, the probability of the dissociative ionization of H2 was modulated by varying the relative phase between the two-color (ω + 3ω) field [40]. Large yield oscillations with phase (with a period of 2π) and an amplitude around 50% were achieved, as shown in figure 13. The H+ signal at low KER in the region of 0–2 eV is from dissociation along the H2+ states. In the higher KER region, i.e. 3–7 eV, the signal is dominated by CREI, where H2+ is ionized near the critical inter-nuclear distance which results in a pair of H+ ions. Strong modulation in the normalized ion yield was observed for both channels (see figure 13). For the CREI channel, the modulation arises from the combined field strength, which reaches a maximum for every 2π phase shifts between the two frequency components. The yield modulation for the low KER region exhibits a KER-dependence with different phases and amplitudes. This indicates the dissociation probability control to originate from quantum interference between multiple dissociation pathways rather than a simple field strength effect (as for the CREI channel). Such KER-dependent yield modulation can also be achieved with CEP-control [1, 4, 25] and with 2-color fields using 800 nm for the FW [1].
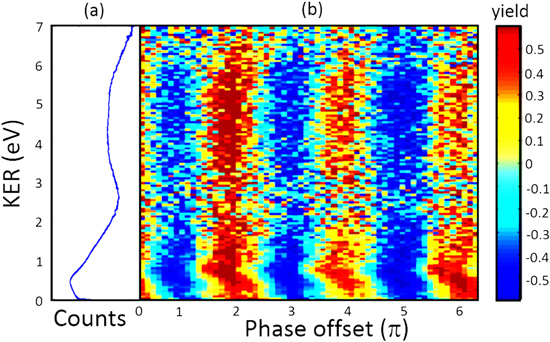
Figure 13. (a) Measured KER spectrum for the H+ ion in ω + 3ω laser fields with the FW at 1.8 μm. (b) Normalized ion yield of H+ as a function of relative phase between the two color component and KER. Reprinted figure with permission from [40], copyright 2016 by the American Physical Society.
Download figure:
Standard image High-resolution imageFor most of the 2-color experiments, the observations can be explained with a simple two-level model. Theoretically, quantum calculations can take into account most features of the physical processes but still have to rely on certain approximation methods, such as the Born–Oppenheimer approximation, which ignores the coupling between electronic and nuclear degrees of freedom. However, coupling between electron and nuclear dynamics can also affect the final results. In laser fields with pulse durations of tens of femtoseconds, complex photoionization and dissociation processes can be induced, and could exhibit dependencies on the molecular orbital shapes for various molecules [94, 95]. This requires development of theoretical models, which can benefit from comparison to experimental data. On the other hand, experimental techniques resolving complex processes can help to simplify the modeling. Recently, an experimental scheme utilizing femtosecond pump and probe pulses with various combinations of polarizations and frequencies was introduced, which can distinguish the ionization and dissociation processes in the strong-field dissociative ionization of multi-electron systems [33, 34]. This is an example for a powerful tool in the investigation of complex molecular reactions in strong laser fields.
4.2.2. 2D directional control of the H+ emission in two-color laser fields
When combining laser pulses of two frequency components, it is possible to dress the directional control with more degrees of freedom since the laser parameters can be tuned independently for both colors. Recently, 2D directional proton emission was realized in the dissociative ionization of H2 in orthogonally polarized and circularly polarized two-color femtosecond laser fields [48, 92], which opened a new avenue for the control of molecular reactions in multiple dimensions.
In an orthogonal two-color (OTC) femtosecond field, significant directional emission is obtained in the emission angle between the polarization axes of the two colors for multiple energy ranges [48]. Dissociation pathways in the OTC field, similar to the cases shown in figure 11, take place with one more degree of freedom, i.e. the emission angle. The momentum distribution for H+ emission from H2 in the polarization plane of the OTC field with zero phase delay between the two colors is shown in figure 14(a) [48]. Rich structure is seen for the angular distributions (as shown in figure 14(b)). A butterfly structure is obtained, as indicated by the white dashed curve in figure 14(a), which only exists when the two colors are overlapped in time. Electrons being driven by one color can be streaked by the other frequency component in the perpendicular direction and result in complex electron trajectories. Two distinct energy regions are recognized, i.e. the low energy region (0–0.4 eV) and the high energy region (0.5–1.2 eV). These two region are assigned to one-photon and two-photon processes, respectively.

Figure 14. (a) A schematic drawing of the OTC field and the resulting momentum distribution of H+ from H2. FW stands for fundamental wave, and SH stands for second harmonics. (b) The H+ ion yield as a function of the emission angle  and ion kinetic energy. The right panel shows the kinetic energy spectrum of H+ on log scale. Adapted figure with permission from [48], copyright 2014 by the American Physical Society.
and ion kinetic energy. The right panel shows the kinetic energy spectrum of H+ on log scale. Adapted figure with permission from [48], copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution imageMore interestingly, when the asymmetry parameters are analyzed for all emission angles in the polarization plane, oscillations are found for both low and high energy channels which maximize at different angles. As shown in figure 15, both experiment and simulation show that the asymmetry for the low energy channel favors an angle close to the polarization of the FW (i.e. 0° and 180°) while that for the high energy channel maximizes at an angle closer to the polarization of the SH wave (i.e. ±90°) [48]. This is related to the 2D steering of the NWP in the OTC fields. This experimental strategy introduced a new degree of freedom for directional control, such that the asymmetric proton emission can be steered in directions between the two polarization axes. This goes beyond the classical 1D-control scheme and opens new possibilities for the steering of molecular reactions.
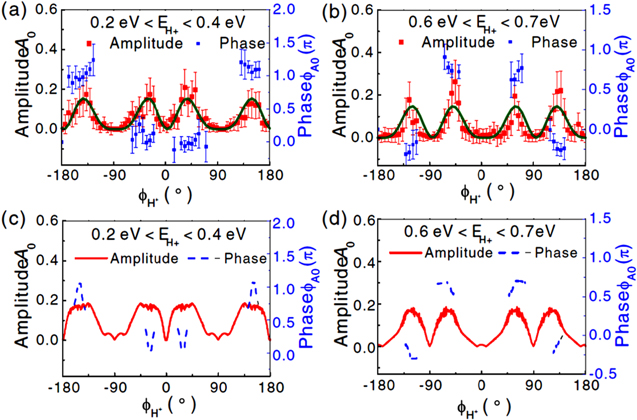
Figure 15. (a), (b) Measured and (c), (d) calculated asymmetry amplitude A0 and phase  as a function of the emission angle of H+ ions for the low energy (0.2–0.4 eV, (a) and (c)) and high energy (0.6–0.7 eV, (b) and (d)) regions, respectively. Adapted figure with permission from [48], copyright 2014 by the American Physical Society.
as a function of the emission angle of H+ ions for the low energy (0.2–0.4 eV, (a) and (c)) and high energy (0.6–0.7 eV, (b) and (d)) regions, respectively. Adapted figure with permission from [48], copyright 2014 by the American Physical Society.
Download figure:
Standard image High-resolution imageActually, the way of tuning waveforms of two-color fields is not limited to the presented methods. Recently, circularly polarized two-color femtosecond pulses with the same or opposite helicities have been applied to the steering of 2D directional bond breaking of H2 [92]. Such ultrashort laser fields exhibit trefoil or semilunar waveforms, which can be tuned effectively by the relative intensity ratio between the two frequencies. As shown in figure 16, unique asymmetry patterns can be extracted in the polarization plane. These asymmetry patterns will rotate with the relative phase between the two color components. The unique trefoil waveform has also been used for the manipulation of electron rescattering since electrons can recollide with the parental core multiple times within one optical cycle from different impact directions and related physical processes such as non-sequential double ionization can be enhanced [96–98].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 16. (a), (b) The electric field of counter- and co-rotating two color laser fields with zero two-color phase. (c), (d) Measured 2D asymmetry patterns of the directional emission of H+ in the polarization plane of the field of (a) and (b), respectively. Reproduced from [92] © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution image{kind=link}
5. Summary and outlook
The previous experimental and theoretical explorations on electron localization in hydrogen molecules addressed the control mechanisms, the reaction pathways and the optical parameter dependence of the control. Until now, major parts of the ultrafast dissociative ionization processes have been revealed for hydrogen, which serves as a prototype model system for the investigation of molecular reactions in intense laser fields. However, new processes and mechanisms could still be explored by utilizing broader experimental conditions. Obviously, intensity serves as a control parameter, which can be easily adjusted. However, the combined effects of the exponentially increasing ionization rate and saturation significantly limit applicable intensities in NIR fields. Using few-cycle pulses, already a broader intensity region could be explored, where more highly excited states were populated, which is obscured with multi-cycle pulses due to the onset of double ionization processes. On the other hand, moving to the MIR spectral region with longer wavelengths the control becomes more efficient with lower limitation of saturation and better matching between the optical cycle and molecular dissociation time. Under the same intensity compared to NIR pulses, the deep tunneling regime can be reached for MIR pulses. The wavelength-dependent investigations can benefit from the development of intense, ultrashort laser sources in the MIR region and beyond.
From a theoretical perspective, the strong-field interaction with the hydrogen molecular ion can be treated exactly with quantum computations. However, starting from neutral hydrogen molecules, the exact modeling becomes difficult due to the complex entanglement between the ionization and dissociation processes induced by the strong laser fields, which are hard to separate in experiments. Experimental schemes were developed to disentangle the role of laser coupling in the ionization and dissociation steps [33, 34, 49]. This exemplifies that there is still large room for the improvement of experimental techniques. For instance, using pre-aligned/oriented molecular ensembles the degree of control can be increased [57]. The experimental schemes for the directional control of dissociative ionization of hydrogen molecule and its isotopes in few-cycle and two-color laser fields presented in this review are a basis for further studies and are already also heavily applied to much more complex systems [83, 85, 99, 100].
Importantly, in the presented overview the control dealt with dissociation reactions, while the control in tailored laser fields (few-cycle or multi-cycle multi-color) can also affect other types of reactions. Recent studies have shown that the directionality of isomerization processes in hydrocarbons can be manipulated by tuning the CEP of few-cycle pulses [85, 99]. The underlying mechanism, revealed by quantum dynamical simulations, is the manipulation of the phases of a superposition of vibrational degrees of freedom (rather than electronic states). The directional control with asymmetric laser fields can go beyond the type of control for hydrogen shown in this review. In the future, it may be feasible to control both electronic and nuclear degrees of freedom such that restructuring even complex molecules at will becomes feasible and new compounds can be light-synthesized.
Acknowledgments
We acknowledge our coworkers and collaborators that have contributed to the research results described in this review. This work is supported by the National Natural Science Fund of China (Grants No. 11425416, No. 11374103, No. 61690224, and No. 11621404), and the 111 project of China (Grant No. B12024). HL acknowledge support by Shanghai Sailing Program (Grants No. 17YF1404000). XMT was supported by the Grand-in-Aid for Scientific Research (16K05495) from the Japan Society for the Promotion of Science. Numerical calculations were partly performed using COMA at the Center for Computational Sciences, University of Tsukuba. MFK acknowledges support by the Max Planck Society, the EU through the ERC grant ATTOCO (No. 307203), and the DFG via LMUexcellent. MFK and RdV acknowledge support by the DFG via the center of excellence: Munich Centre for Advanced Photonics.