
Abstract
We present detailed comparisons between the results of embedded atom model (EAM) and density functional theory (DFT) calculations on defected Ni alloy systems. We find that the EAM interatomic potentials reproduce low-temperature structural properties in both the γ and  phases, and yield accurate atomic forces in bulk-like configurations even at temperatures as high as ∼1200 K. However, they fail to describe more complex chemical bonding, in configurations including defects such as vacancies or dislocations, for which we observe significant deviations between the EAM and DFT forces, suggesting that derived properties such as (free) energy barriers to vacancy migration and dislocation glide may also be inaccurate. Testing against full DFT calculations further reveals that these deviations have a local character, and are typically severe only up to the first or second neighbours of the defect. This suggests that a QM/MM approach can be used to accurately reproduce QM observables, fully exploiting the EAM potential efficiency in the MM zone. This approach could be easily extended to ternary systems for which developing a reliable and fully transferable EAM parameterisation would be extremely challenging e.g. Ni alloy model systems with a W or Re-containing QM zone.
phases, and yield accurate atomic forces in bulk-like configurations even at temperatures as high as ∼1200 K. However, they fail to describe more complex chemical bonding, in configurations including defects such as vacancies or dislocations, for which we observe significant deviations between the EAM and DFT forces, suggesting that derived properties such as (free) energy barriers to vacancy migration and dislocation glide may also be inaccurate. Testing against full DFT calculations further reveals that these deviations have a local character, and are typically severe only up to the first or second neighbours of the defect. This suggests that a QM/MM approach can be used to accurately reproduce QM observables, fully exploiting the EAM potential efficiency in the MM zone. This approach could be easily extended to ternary systems for which developing a reliable and fully transferable EAM parameterisation would be extremely challenging e.g. Ni alloy model systems with a W or Re-containing QM zone.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Ni-based superalloys are high performance structural materials that exhibit excellent strength and creep resistance at high temperatures, even in chemically aggressive environments [1, 2]. They are manufactured in polycrystalline form for turbine disks and in single crystal form for turbine airfoils. In both cases, the high strength of the alloy is due to the precipitation of a  -ordered phase phase, known as
-ordered phase phase, known as  , within the Nickel fcc matrix [3]. Defects such as vacancies and impurities are also thought to affect dislocation motion, and the inclusion of chemical impurities such as Re and/or W in the γ matrix is a standard commercial practice known to be extremely important to improve the plasticity response. However, there is currently no detailed understanding of the atomic-scale mechanisms underlying these processes. The hypothesis of Rhenium clustering, originally proposed in pioneering atom probe experiments [4, 5], has been dismissed after more refined experiments (three-dimensional atom probe technique [6, 7] and extended x-ray absorption fine spectroscopy [8, 9]) and theoretical atomistic investigation [10]. Slow-diffusing atoms in the matrix were thought to hinder dislocation climb processes at the interface [11]. However, the reductions of the vacancy diffusion coefficient predicted from first-principle calculations is not large enough to provide the creep strengthening effect experimentally observed [12]. Such understanding could provide fundamental insight and allow the design of materials with improved properties, such as longer lifetimes or higher energy efficiency enabled by higher operational temperatures.
, within the Nickel fcc matrix [3]. Defects such as vacancies and impurities are also thought to affect dislocation motion, and the inclusion of chemical impurities such as Re and/or W in the γ matrix is a standard commercial practice known to be extremely important to improve the plasticity response. However, there is currently no detailed understanding of the atomic-scale mechanisms underlying these processes. The hypothesis of Rhenium clustering, originally proposed in pioneering atom probe experiments [4, 5], has been dismissed after more refined experiments (three-dimensional atom probe technique [6, 7] and extended x-ray absorption fine spectroscopy [8, 9]) and theoretical atomistic investigation [10]. Slow-diffusing atoms in the matrix were thought to hinder dislocation climb processes at the interface [11]. However, the reductions of the vacancy diffusion coefficient predicted from first-principle calculations is not large enough to provide the creep strengthening effect experimentally observed [12]. Such understanding could provide fundamental insight and allow the design of materials with improved properties, such as longer lifetimes or higher energy efficiency enabled by higher operational temperatures.
The problem is formidable if approached at the atomistic level: the typical accuracy level of first principles methods such as density functional theory (DFT) [13] is needed in order to accurately describe atoms close to dislocation cores or chemically complex interfaces, and at the same time model systems must be large enough to accommodate the strain gradients typical of long-range elastic interactions. Embedded atom models allow sufficiently large system sizes, and have been tremendously successful at modelling many properties of metallic materials [14, 15]. However, they are usually restricted to at most two different atomic species and reliably transferable to a very limited number of structural phases. Recently, ternary potentials have been developed to address the problem of chemical complexity, but they seem to be even less transferable to structures they were not fitted to, and no general consensus has been reached in the community upon their reliability. Non-uniform precision techniques such as the 'learn on the fly' (LOTF) multiscale quantum mechanical/molecular mechanical (QM/MM) scheme [16, 17] appear promising, although they have not yet been extensively applied to metallic systems [18–20]. Augmenting this approach by learning algorithms that allow performing QM calculations only where and when they are necessary could further reduce the overall computational cost [21]. Before such a methodology can be applied to the present class of problems, however, it is of crucial importance to validate the existing EAM potentials with respect to DFT, to confirm whether these potentials might suffice, or determine where and when QM-based augmentation is necessary (see [22] for oxides). The present work addresses this issues for systems of increasing chemical complexity, starting from simple bulk models for the matrix phase and the precipitate in section 2 and moving to systems including point defects such as vacancies and chemical impurities and, finally, to extended dislocation cores in section 3.
2. Validation of the interatomic potential
2.1. Zero temperature properties
We initially test the EAM potential against DFT data, to verify its reliability in describing the properties of Nickel alloys. We use an EAM model developed by Mishin in 2004 [23], generally considered to be amongst the most reliable interatomic potentials for modelling  interfaces and plasticity-related phenomena in Ni-based alloys. This is due to its accuracy in reproducing the correct lattice parameters for both phases as well as quantities directly related to dislocations such as the intrinsic stacking fault energy of Nickel. A new EAM potential for simulating Ni–Al alloys was made available in 2009 [24]. It is however less accurate for describing
interfaces and plasticity-related phenomena in Ni-based alloys. This is due to its accuracy in reproducing the correct lattice parameters for both phases as well as quantities directly related to dislocations such as the intrinsic stacking fault energy of Nickel. A new EAM potential for simulating Ni–Al alloys was made available in 2009 [24]. It is however less accurate for describing  interface geometries due to its underestimation of the lattice parameter for
interface geometries due to its underestimation of the lattice parameter for  . The potential is fitted to the properties of the B2 NiAl phase, and the only quantity related to
. The potential is fitted to the properties of the B2 NiAl phase, and the only quantity related to  included in the potential is its formation energy.
included in the potential is its formation energy.
To gain more insight in the issue of transferability of EAM potentials in Ni systems, we preliminarily compare DFT and EAM results for a selection of structural properties. For a more complete study and a comparison with experimental data, refer to [23]. Our electronic structure calculations have been performed using the VASP package [25], adopting the PBE/GGA approximation for the exchange and correlation energy [26]. Brillouin zone (BZ) sampling was carried out using Monkhorst–Pack (MP) grids [27] with different meshes for different simulation cells, as specified for each case below. We used a 300 eV plane-wave energy cutoff, the projector augmented wave (PAW) method [28], and Fermi energy smearing with a Gaussian (Methfessel–Paxton) broadening of 0.1 eV [29].
As a first step, we evaluated the equilibrium lattice parameter for both the γ and  phases using four-atom cubic elementary cells for different volumes (11 configurations, ±5% from experimental value). A
phases using four-atom cubic elementary cells for different volumes (11 configurations, ±5% from experimental value). A  MP grid is used for BZ sampling. The resulting energies were fitted to the Murnaghan equation of state. Both DFT and EAM predict equilibrium lattice parameters of 3.52 Å and 3.57 Å for γ and
MP grid is used for BZ sampling. The resulting energies were fitted to the Murnaghan equation of state. Both DFT and EAM predict equilibrium lattice parameters of 3.52 Å and 3.57 Å for γ and  respectively. Bulk moduli obtained from fitting are reported in table 1.
respectively. Bulk moduli obtained from fitting are reported in table 1.
Table 1. Elastic properties (in GPa) for bulk models of Ni and  systems. Comparison between DFT and EAM data.
systems. Comparison between DFT and EAM data.
| DFT (γ) | EAM (γ) | DFT 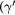 ) ) |
EAM (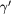 ) ) |
|
|---|---|---|---|---|
| B | 192 | 181 | 177 | 176 |
| C11 | 268 | 237 | 236 | 236 |
| C12 | 161 | 151 | 147 | 154 |
| C44 | 127 | 125 | 126 | 127 |
Elastic moduli are important quantities for a QM/MM simulation, as any mismatch between the two descriptors would result in a discontinuity at the interface between the classical and the quantum regions [17]. The ability to reproduce the correct elastic constants (once the lattice constants are aligned by parameter tuning) is thus an important feature for an interatomic potential. We evaluated the elastic constants with a linear fit to the stress/strain curve, using stress tensors calculated using the virial (derivative of potential energy with respect to cell vectors). For these calculations, we use a 400 eV plane-wave cutoff energy, necessary for obtaining accurate diagonal elements of the stress tensor for the  phase. The resulting data are presented in table 1, and show good agreement between the DFT and EAM approaches.
phase. The resulting data are presented in table 1, and show good agreement between the DFT and EAM approaches.
Vacancies are crucial defects to enable plasticity behaviour through dislocation climb. The EAM potential used here was explicitly fitted to reproduce the experimental values for vacancy formation and diffusion energy [23]. It is therefore important to check the consistency of these values with the DFT predictions. We used a  supercell of the conventional 128 atom cubic system for the γ phase, with one atom removed to model an isolated vacancy. The system was relaxed to its structural minimum within a 1 meV energy tolerance using a conjugate gradient algorithm and
supercell of the conventional 128 atom cubic system for the γ phase, with one atom removed to model an isolated vacancy. The system was relaxed to its structural minimum within a 1 meV energy tolerance using a conjugate gradient algorithm and  MP
MP  -point grid. We then calculated the vacancy formation energy and migration barrier, the latter using the NEB method, which involved the optimisation of 3 intermediate images by a RMM-DIIS quasi-Newton, variable metric algorithm for the QM calculations and the FIRE algorithm [30] for the MM calculations. Our calculated results for the γ phase are reported in table 2. The agreement between EAM and DFT is overall good, with the interatomic potential overestimating the reference DFT values properties by only around 10%.
-point grid. We then calculated the vacancy formation energy and migration barrier, the latter using the NEB method, which involved the optimisation of 3 intermediate images by a RMM-DIIS quasi-Newton, variable metric algorithm for the QM calculations and the FIRE algorithm [30] for the MM calculations. Our calculated results for the γ phase are reported in table 2. The agreement between EAM and DFT is overall good, with the interatomic potential overestimating the reference DFT values properties by only around 10%.
Table 2. Formation and migration energies for a vacancy and intrinsic stacking fault energy per unit surface in the γ phase of Ni.
| DFT | EAM | |
|---|---|---|
 |
1.45 | 1.57 |
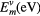 |
1.08 | 1.19 |
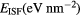 |
0.85 | 0.84 |
Close agreement between the calculated values of intrinsic stacking fault energy (ISF) using DFT and EAM is also important, as perfect dislocations in fcc metals are always dissociated into Shockley Partials (SP) and the distance between them is inversely proportional to the ISF energy. To investigate this issue, we constructed a model system by orienting the crystal with the z direction perpendicular to the normal to the  close packed family of planes. The x and y directions were chosen perpendicular to the
close packed family of planes. The x and y directions were chosen perpendicular to the ![$\left[\bar{1}\,1\,0\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn022.gif) and the
and the ![$\left[1\,1\,\bar{2}\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn023.gif) crystal directions, respectively. A twelve
crystal directions, respectively. A twelve  layer supercell was initially created as a bulk reference. We then removed one layer, and rescaled the simulation cell in order to produce a periodic model system containing a single stacking fault defect, where the distance between planes with hcp stacking and their nearest periodically repeated images is ∼20 Å. For the structural optimisation we used a conjugate gradient algorithm. A MP grid of
layer supercell was initially created as a bulk reference. We then removed one layer, and rescaled the simulation cell in order to produce a periodic model system containing a single stacking fault defect, where the distance between planes with hcp stacking and their nearest periodically repeated images is ∼20 Å. For the structural optimisation we used a conjugate gradient algorithm. A MP grid of 
 -points was used to sample the BZ for QM calculations. The ISF energy value was obtained as a direct subtraction of the bulk reference energy (renormalised for the correct number of atoms) from the energy of the defected system fully relaxed up to 1 meV tolerance, after dividing by the area of a single
-points was used to sample the BZ for QM calculations. The ISF energy value was obtained as a direct subtraction of the bulk reference energy (renormalised for the correct number of atoms) from the energy of the defected system fully relaxed up to 1 meV tolerance, after dividing by the area of a single  layer. Our results are reported in table 2, and once more indicate close match between EAM and DFT results.
layer. Our results are reported in table 2, and once more indicate close match between EAM and DFT results.
The generally good agreement between EAM and DFT results for systems in their structural minima provides no indication of the performance of the EAM potential when the system is displaced from equilibrium. Perhaps the simplest test configuration for non-zero forces geometries is obtained by displacing a single atom from its equilibrium structure, and checking the consistency of the forces predicted by the two models. We therefore performed a set of calculations using a  supercell with one of the atoms displaced along the
supercell with one of the atoms displaced along the ![$\left[1\,1\,0\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn029.gif) direction i.e. towards one of its nearest neighbours. The forces acting on both atoms are presented in figure 1(a) as a function of the applied displacement, revealing a significant discrepancy between the two approaches when bond lengths are modified by 10% or more. A similar behaviour has been reported in [31] where the EAM ansatz of an exponentially decreasing charge density was studied for different deformations of a fcc crystal by measuring the QM charge density in a (vacant) octahedral site. This work found that when a second nearest neighbour of the vacant site moves towards it, the site loses charge in favour of the region comprised between the approaching atom and its nearest neighbours, which violates the EAM approach ansatz.
direction i.e. towards one of its nearest neighbours. The forces acting on both atoms are presented in figure 1(a) as a function of the applied displacement, revealing a significant discrepancy between the two approaches when bond lengths are modified by 10% or more. A similar behaviour has been reported in [31] where the EAM ansatz of an exponentially decreasing charge density was studied for different deformations of a fcc crystal by measuring the QM charge density in a (vacant) octahedral site. This work found that when a second nearest neighbour of the vacant site moves towards it, the site loses charge in favour of the region comprised between the approaching atom and its nearest neighbours, which violates the EAM approach ansatz.
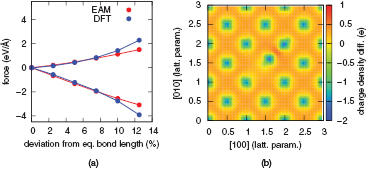
Figure 1. (a) EAM and DFT forces calculated for a Ni atom displaced from its equilibrium position along the [1 1 0] direction. (b) Charge difference with respect to isolated Ni atoms in a 1 0 0 plane for a bulk system with a single atom displaced towards one of its neighbours along the [1 1 0] direction by 12% of the equilibrium bond length.
Download figure:
Standard image High-resolution imageThe plot of the charge density distribution for a 12% bond deformation is displayed in figure 1(b). The density associated with isolated atoms was subtracted in this  plane plot, so that the charge depletion from atomic sites and its redistribution into the interatomic space, typical of the metallic bonding, is immediately apparent. A charge transfer pattern similar to the one described in [31] is observed, indicating that the failure of the EAM to describe the charge rearrangement can lead to the system visiting unphysical configurations during a constant temperature MD simulations, or at the very least that the distribution of visited configurations may not correctly sample the canonical ensemble consistent with the reference QM energetics. While these discrepancies still occur as relatively rare events for bulk systems simulated at moderate temperatures, we find that large force errors are incurred when using the EAM model in the defected systems containing vacancies or dislocations, as described in section 3.
plane plot, so that the charge depletion from atomic sites and its redistribution into the interatomic space, typical of the metallic bonding, is immediately apparent. A charge transfer pattern similar to the one described in [31] is observed, indicating that the failure of the EAM to describe the charge rearrangement can lead to the system visiting unphysical configurations during a constant temperature MD simulations, or at the very least that the distribution of visited configurations may not correctly sample the canonical ensemble consistent with the reference QM energetics. While these discrepancies still occur as relatively rare events for bulk systems simulated at moderate temperatures, we find that large force errors are incurred when using the EAM model in the defected systems containing vacancies or dislocations, as described in section 3.
2.2. Finite temperatures
We next test the ability of the EAM potential to reproduce the DFT forces in configurations produced by finite temperature MD simulations, as would be necessary to correctly predict thermodynamic observables [32]. We start from simple bulk structures, and investigate the EAM force error temperature dependence. A  , 108 atom supercell was used for modelling both the γ and
, 108 atom supercell was used for modelling both the γ and  phases. Temperature effects were introduced in the system by means of classical MD simulations carried out at fixed volumes in each case consistent with thermal expansion. The system was initially equilibrated for 5 ps, using a 2 fs time step. This was followed by a further 10 ps period during which configuration snapshots were collected at 1 ps intervals. The DFT forces were then calculated on the collected structures (using calculation parameters as in the previous sections) and compared with the classical values.
phases. Temperature effects were introduced in the system by means of classical MD simulations carried out at fixed volumes in each case consistent with thermal expansion. The system was initially equilibrated for 5 ps, using a 2 fs time step. This was followed by a further 10 ps period during which configuration snapshots were collected at 1 ps intervals. The DFT forces were then calculated on the collected structures (using calculation parameters as in the previous sections) and compared with the classical values.
The error distribution plots produced by these calculations are presented in figure 2, and the associated average force errors reported in table 3. These results suggest that for perfect bulk systems the EAM interatomic potential is remarkably accurate, with more than 70% of the absolute errors contained below the 0.1 eV  threshold for room temperature γ phase simulations. However, the average error increases for higher temperatures as presented in figures 2(a) and (b) and approximately doubles for T = 1200 K (table 3). The error distribution of the relative errors is shown in figures 2(c) and (d), showing no appreciable difference between the plotted curves for different temperature values. This indicates that the errors incurred by the EAM predicted forces are approximately proportional to the forces, which implies an acceptable performance in simulating bulk systems even at relatively high (while still below melting) temperatures.
threshold for room temperature γ phase simulations. However, the average error increases for higher temperatures as presented in figures 2(a) and (b) and approximately doubles for T = 1200 K (table 3). The error distribution of the relative errors is shown in figures 2(c) and (d), showing no appreciable difference between the plotted curves for different temperature values. This indicates that the errors incurred by the EAM predicted forces are approximately proportional to the forces, which implies an acceptable performance in simulating bulk systems even at relatively high (while still below melting) temperatures.
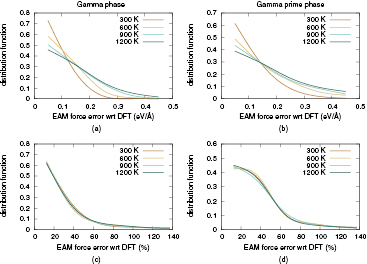
Figure 2. Error distribution, absolute ((a), (b)) and relative ((c), (d)) of EAM forces with respect to DFT reference calculations at different temperatures for γ Ni ((a), (c)) and 
 ((b), (d)).
((b), (d)).
Download figure:
Standard image High-resolution imageTable 3. Average force component error of the EAM interatomic potential computed as deviation from the reference DFT calculations.
| T (K) | 300 | 600 | 900 | 1200 |
|---|---|---|---|---|
Force error γ (eV 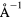 ) ) |
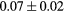 |
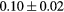 |
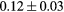 |
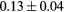 |
Force error 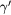 (eV (eV 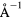 ) ) |
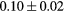 |
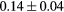 |
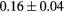 |
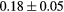 |
Note: Averages over ten independent γ (Ni) and  (
( ) 108 atom model system configurations, at different temperatures.
) 108 atom model system configurations, at different temperatures.
Our computed results for the  phase are similar, but reveal that here EAM is overall less accurate: only about 40% of the forces are accurate within 25% of the reference DFT values. This is probably due to a greater difficulty in describing the interaction between different chemical species, consistent with the fact that fitting accurate classical potential forms so that accuracy is preserved while the number of chemical species increases is notoriously challenging [33].
phase are similar, but reveal that here EAM is overall less accurate: only about 40% of the forces are accurate within 25% of the reference DFT values. This is probably due to a greater difficulty in describing the interaction between different chemical species, consistent with the fact that fitting accurate classical potential forms so that accuracy is preserved while the number of chemical species increases is notoriously challenging [33].
3. Defects in the γ-matrix
3.1. Vacancies
The main long-term objective of this work is to identify the working simulation parameters needed to run multiscale QM/MM simulations of plasticity-related processes in Ni-based superalloys. In these simulations there is no requirement for an EAM potential to correctly reproduce forces on atoms in all conditions. Providing that local configurations for which these potentials are accurate can be identified a priori, all that is needed is to make sure that the MM zone of the simulation remains within this 'comfort zone'. A QM description can then be used whenever more chemically complex configurations (e.g. involving bond breaking and reforming) are encountered, where the potential cannot be assumed to be accurate enough.
Accordingly, we next set out to explore the QM region requirement associated with our EAM potential in the proximity of a vacant lattice site of the γ matrix. We note that vacancies are not just the most common intrinsic point defects found in metal lattices: in the present context, at phase boundaries, they are also a necessary ingredient for the dislocation climb-assisted glide mechanisms generally thought to enable creep deformation in Ni-based superalloy blades, particularly in the low-stress, high-temperature conditions where plasticity remains mostly confined in the γ channels of the alloy microstructure. Our vacancy system was obtained from a  γ phase bulk cell (108 atoms), upon removal of one atom. This system is small enough that fully converged DFT reference results are readily obtainable, and large enough to make the interaction between the vacancy and its repeated images negligible for the present purpose. As in section 2.2, temperature effects were introduced by carrying out constant temperature EAM MD simulations, this time setting the temperature to 1200 K, in line with with typical operating temperatures of superalloy turbines.
γ phase bulk cell (108 atoms), upon removal of one atom. This system is small enough that fully converged DFT reference results are readily obtainable, and large enough to make the interaction between the vacancy and its repeated images negligible for the present purpose. As in section 2.2, temperature effects were introduced by carrying out constant temperature EAM MD simulations, this time setting the temperature to 1200 K, in line with with typical operating temperatures of superalloy turbines.
Our goal is to investigate the accuracy of the EAM potential by comparing the atomic forces it produces with those obtained from DFT calculations. If discrepancies indicate that using a QM region is required, we wish to determine both the size of this QM region and the width of the associated 'buffer' region necessary for decoupling core quantum atoms from the surface states to ensure accurate QM forces are obtained [17].
Plots of the EAM error, both absolute and relative, with respect to DFT references are reported in figures 3(a) and (c). The different line colours correspond to different distances from the vacancy: 2.5  on average for nearest neighbour (n.n.) atoms and 3.5 Å for next-nearest neighbours (n.n.n). Results for atoms from the same frames located more than 4.3 Å from the vacancy, and thus expected to be 'bulk-like', are also included for reference, and for these atoms about 50% of the data points have an absolute force error below 0.1 eV
on average for nearest neighbour (n.n.) atoms and 3.5 Å for next-nearest neighbours (n.n.n). Results for atoms from the same frames located more than 4.3 Å from the vacancy, and thus expected to be 'bulk-like', are also included for reference, and for these atoms about 50% of the data points have an absolute force error below 0.1 eV  and only 20% exceed 0.2 eV
and only 20% exceed 0.2 eV  . Here, the error is rarely larger than 0.4 eV
. Here, the error is rarely larger than 0.4 eV  , in line with the results of section 2.2. For the atoms neighbouring the vacancy the force error distribution is broader. While in about 40% of the data points the error is still below 0.1 eV
, in line with the results of section 2.2. For the atoms neighbouring the vacancy the force error distribution is broader. While in about 40% of the data points the error is still below 0.1 eV  a much larger population of forces deviating by 0.2 eV
a much larger population of forces deviating by 0.2 eV  appears (about 35%), with the maximum errors being as large as 0.8 eV
appears (about 35%), with the maximum errors being as large as 0.8 eV  . Inspecting the relative error distribution function also makes clear that the relative error for these atoms is likely to be much larger than for bulk-like atoms. The error plot measured for next-nearest neighbours show an intermediate behaviour.
. Inspecting the relative error distribution function also makes clear that the relative error for these atoms is likely to be much larger than for bulk-like atoms. The error plot measured for next-nearest neighbours show an intermediate behaviour.

Figure 3. Error distribution, absolute (a) and percentage (c), of the EAM potential with respect to DFT reference calculations for Ni atoms neighbouring a vacancy and comparison with bulk. Convergence of the quantum forces with respect to the size of the cluster (b), (d). The shaded areas in the left panels correspond to the range of errors observed in DFT cluster calculations, and the horizontal grey lines in right panels show the average EAM force error.
Download figure:
Standard image High-resolution imageWe note that the EAM potential examined here is in fact by design accurate enough for modelling the dynamics of vacancies in the γ phase of Nickel alloys, as both the formation and the migration energies are included in the fitting database used to develop the potential [23]. However, although the EAM forces integrate the correct migration barrier, we find here that the local description of the potential energy surface (of which the calculated forces are simply tangent vectors) is not as accurate for the n.n. of vacancies as it is for the bulk-like atoms. As a result, the potential could easily fail to describe the interaction between a vacancy and other defects such as dislocations, for which the potential has not been fitted, so that a more accurate treatment is required. We therefore proceed to define a quantum region surrounding vacancies for accurate QM/MM simulations.
Implementing an accurate QM/MM treatment of this system requires a separate analysis of the convergence of the quantum forces with respect to the size of the QM cluster. This is presented in figures 3(b) and (d) including results for the isolated vacancy and the bulk system for comparison. For the bulk, we started by selecting our target QM atoms to be those with forces exceeding 0.5 eV  and performed DFT calculations for Ni clusters of increasing size (denoted in the figure by buffer width radius) centred on each of these atoms. The cluster radii were chosen to lie between neighbour shells to reduce number fluctuations associated with thermal disorder. The amount of vacuum surrounding clusters was set to 10 Å in all calculations, after verifying that this is sufficiently large for the interactions between clusters and their periodic images to be negligible. Force errors were averaged over ten configurations.
and performed DFT calculations for Ni clusters of increasing size (denoted in the figure by buffer width radius) centred on each of these atoms. The cluster radii were chosen to lie between neighbour shells to reduce number fluctuations associated with thermal disorder. The amount of vacuum surrounding clusters was set to 10 Å in all calculations, after verifying that this is sufficiently large for the interactions between clusters and their periodic images to be negligible. Force errors were averaged over ten configurations.
For systems including a vacancy calculations were performed on larger clusters centred on the defect itself. In this case, we defined our QM regions as the first shell of neighbours of the vacant sites. For each of the ten system snapshots considered, we examined QM atoms with forces exceeding 0.5 eV  , and averaged our convergence results to produce the plots reported in figure 3. The QM convergence behaviour is similar for the the vacancy and bulk cases. Good convergence is already obtained when the width of the buffer zone surrounding the QM atoms in the clusters is about 4.2 Å or bigger. These values correspond to including the first three shells of neighbours of a fcc crystal in the quantum total calculation. The radius of the corresponding cluster is about 7 Å for the system centred on the vacancy. While the error is flat when further increasing the cluster radius in the bulk-like system, the last point for the vacancy system still deviates slightly from the converged value (while being below our accuracy threshold), possibly due to residual image effects in the reference periodic calculation.
, and averaged our convergence results to produce the plots reported in figure 3. The QM convergence behaviour is similar for the the vacancy and bulk cases. Good convergence is already obtained when the width of the buffer zone surrounding the QM atoms in the clusters is about 4.2 Å or bigger. These values correspond to including the first three shells of neighbours of a fcc crystal in the quantum total calculation. The radius of the corresponding cluster is about 7 Å for the system centred on the vacancy. While the error is flat when further increasing the cluster radius in the bulk-like system, the last point for the vacancy system still deviates slightly from the converged value (while being below our accuracy threshold), possibly due to residual image effects in the reference periodic calculation.
3.2. Screw dislocation
As a next step, we considered a screw dislocation in the γ phase. The crystal was oriented with orthorhombic cell axes ![$x=\left[1\,1\,\bar{2}\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn060.gif) ,
, ![$y=\left[1\,1\,1\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn061.gif) ,
, ![$z=\left[\bar{1}\,1\,0\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn062.gif) , leading to a six-atom unit cell. A
, leading to a six-atom unit cell. A  supercell was then formed, and an
supercell was then formed, and an  screw dislocation was introduced by imposing the correct elastic displacement [34]. The resulting system is periodic along the dislocation line direction (z, also parallel to the Burgers vector). The dislocation core was tracked using the Nye tensor analysis [35]. Replication of the system along the line direction z allows carrying out averages of Nye tensor components during a molecular dynamics simulation at high temperature. This is useful to average out the local thermal fluctuations, making it easier to locate the partial dislocation cores and define the (approximately) cylindrical regions where a QM treatment is necessary. The z-averaged surface integral of the tensor can also be more easily checked to add up to the relevant Burgers vector.
screw dislocation was introduced by imposing the correct elastic displacement [34]. The resulting system is periodic along the dislocation line direction (z, also parallel to the Burgers vector). The dislocation core was tracked using the Nye tensor analysis [35]. Replication of the system along the line direction z allows carrying out averages of Nye tensor components during a molecular dynamics simulation at high temperature. This is useful to average out the local thermal fluctuations, making it easier to locate the partial dislocation cores and define the (approximately) cylindrical regions where a QM treatment is necessary. The z-averaged surface integral of the tensor can also be more easily checked to add up to the relevant Burgers vector.
A vacuum region was introduced along the x and y directions to prevent dislocation emission at the stacking fault, due to imposing periodic boundary conditions Atoms within 80 Å of initial dislocation core positions were free to move, while those outside this region were kept fixed during both the relaxation and the dynamics, in order to prevent the dislocation reaching the surface. The system was relaxed using a conjugate gradient algorithm using a 0.03 eV  force convergence threshold. The atomic positions were then rescaled to account for thermal expansion, and the dynamics of the system simulated in the NVT ensemble. The system was initially equilibrated for 5 ps, after which five different system snapshots were considered, taken at intervals of 5 ps to minimise correlation. Atoms were classified into three categories according to the value of the zz component of the Nye tensor:
force convergence threshold. The atomic positions were then rescaled to account for thermal expansion, and the dynamics of the system simulated in the NVT ensemble. The system was initially equilibrated for 5 ps, after which five different system snapshots were considered, taken at intervals of 5 ps to minimise correlation. Atoms were classified into three categories according to the value of the zz component of the Nye tensor:
- (i)n.n. of the dislocation core: defined as the atoms for which the screw component of the Nye tensor is above 0.04
 . Few of these atoms are present in each layer, but they contribute about 70% of the integrated Burgers vector. These atoms are usually grouped together around each partial core.
. Few of these atoms are present in each layer, but they contribute about 70% of the integrated Burgers vector. These atoms are usually grouped together around each partial core. - (ii)n.n.n. of the dislocation core: atoms with a smaller, but non-zero (0.02) screw component of the Nye tensor, typically neighbouring the atoms of the previously defined region.
- (iii)Bulk atoms: all other atoms located within 40 Å from the dislocation core(s).
Force accuracy was investigated in convergence tests only for the first two categories, as the faraway atoms' main role is to provide the elastic embedding for the region surrounding the dislocation, which was assumed in all cases achievable by classical potential (EAM) modelling. Full DFT calculations cannot be performed on the whole system for the system sizes required here, to provide a reference target values. However, the convergence of QM forces near the dislocation cores can still be monitored as before, by performing calculations on clusters of increasing size. As above, the forces acting on the central atom of each cluster are the quantity of interest in each calculation, while the remaining atoms in the QM cluster just act as a buffer [17].
Our force convergence results with respect to the cluster radius are displayed in figures 4(b) and (d). The reference forces are taken from a larger cluster, not shown in the plot, including one more atomic shell than those shown (i.e. nine neighbour shells, or 177 atoms). As for the vacancy systems, we performed ten sets of calculations for atoms which are n.n. to the core and averaged over these results. The resulting average error is monotonically decreasing for increasing sizes of the system, and falls below out accuracy threshold of 0.1 eV  with a cluster radius of 5.5 Å. We note here that an additional neighbour shell is required for QM/MM modelling of dislocations, compared with the bulk and vacancy cases. However, further testing shows that the force convergence can be further improved by increasing the Fermi energy smearing width from 0.1 to 0.5 eV in the DFT calculations. This reduces the force errors for smaller clusters, so that accurate QM forces can be obtained with a three shell cluster for the as for the bulk and vacancy cases.
with a cluster radius of 5.5 Å. We note here that an additional neighbour shell is required for QM/MM modelling of dislocations, compared with the bulk and vacancy cases. However, further testing shows that the force convergence can be further improved by increasing the Fermi energy smearing width from 0.1 to 0.5 eV in the DFT calculations. This reduces the force errors for smaller clusters, so that accurate QM forces can be obtained with a three shell cluster for the as for the bulk and vacancy cases.
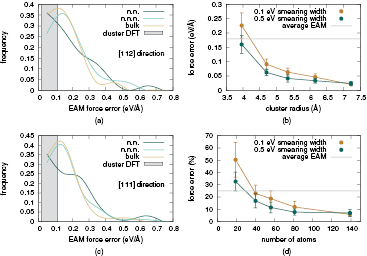
Figure 4. ((a), (c)) Force error distributions for Ni atoms surrounding a dislocation core displayed as plots of the average EAM force deviation from the reference DFT values for two different crystal direction. Convergence of the quantum forces, absolute (b) and percentage (d), with respect to cluster radius for different values of smearing width along glide direction.
Download figure:
Standard image High-resolution imageThe results described allow to determine which atoms require QM corrections, once more by direct comparison with the EAM predictions. We picked a set of 30 atoms from each system snapshot obtained above, and performed a DFT calculation for each of them using a well converged 5.5 Å cluster radius. The deviation of the EAM forces from the DFT forces evaluated this way were analysed separately for two crystallographic directions, one corresponding to the ![$\left[1\,1\,\bar{2}\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn070.gif) dislocation glide direction and the other to the
dislocation glide direction and the other to the ![$\left[1\,1\,1\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn071.gif) climb direction. We find that the EAM potential performs reasonably well in both cases, with 40% and 35% of the forces deviating by more than 0.2 eV
climb direction. We find that the EAM potential performs reasonably well in both cases, with 40% and 35% of the forces deviating by more than 0.2 eV  for the glide and climb directions, respectively (the corresponding value for the bulk is 25%). Errors are, however, higher along the direction of motion for a gliding dislocation, and the presence of vacancies possibly yielding the formation of jogs during the diffusion will very likely increase the force error along the
for the glide and climb directions, respectively (the corresponding value for the bulk is 25%). Errors are, however, higher along the direction of motion for a gliding dislocation, and the presence of vacancies possibly yielding the formation of jogs during the diffusion will very likely increase the force error along the ![$\left[1\,1\,1\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn073.gif) climb direction. Moreover, in both directions, there is a significant probability of incurring force deviations as large as 0.6 eV
climb direction. Moreover, in both directions, there is a significant probability of incurring force deviations as large as 0.6 eV  for both the nearest neighbours and the next nearest neighbours of the dislocation core. The next nearest neighbours are associated with larger EAM force errors in the
for both the nearest neighbours and the next nearest neighbours of the dislocation core. The next nearest neighbours are associated with larger EAM force errors in the ![$\left[1\,1\,\bar{2}\right]$](https://content.cld.iop.org/journals/0965-0393/24/4/045012/revision1/msmsaa1d22ieqn075.gif) direction, i.e. the one linking the two partial dislocations across the stacking fault region where the atomic linkage less closely reproduces the perfect crystal bonding geometry.
direction, i.e. the one linking the two partial dislocations across the stacking fault region where the atomic linkage less closely reproduces the perfect crystal bonding geometry.
3.3. Impurities in the γ matrix
Modern Ni-based superalloys include different atomic species known to improve the plasticity properties [2]. In particular, the so-called 'rhenium effect' has not been fully explored and understood at the atomic scale, mainly due to the lack of accurate tools for describing both the chemical bonding and the elastic properties for systems larger than a few hundred atoms. One of the biggest limitations of EAM (and generally) potentials is their lack of straightforward transferability to chemical environments not fully represented in the fitting database, for instance when describing a different phase or when introducing a new atomic species in the simulated system is required.
Since re-fitting a potential in such a way that its domain of application is generalised while the previous performance is in no way affected can require generating sizeable new fitting databases and is generally a non-trivial process, there is scope to inspect the convergence of QM/MM methods which can, in general, be immediately applied to new problems, due to the intrinsic generality/transferability of first-principles methods.
We thus devote this section to quantifying the size of a quantum region surrounding an impurity atom (specifically, rhenium or tungsten) in the γ phase, to determine the QM cluster radii required to obtain converged DFT forces. As for the vacancy analysis, we model the γ matrix with a  supercell, this time substituting one atom with an impurity atom. Atomic positions are in all cases rescaled to match the lattice parameter predicted for pure Nickel by thermal expansion, assuming a target temperature of 1200 K. In contrast with the system preparation procedures described above where classical MD was used, here the relevant systems configurations were generated by DFT dynamical simulations, since no suitable EAM parameters are available for impurities.
supercell, this time substituting one atom with an impurity atom. Atomic positions are in all cases rescaled to match the lattice parameter predicted for pure Nickel by thermal expansion, assuming a target temperature of 1200 K. In contrast with the system preparation procedures described above where classical MD was used, here the relevant systems configurations were generated by DFT dynamical simulations, since no suitable EAM parameters are available for impurities.
The DFT calculation parameters are the same as described previously for cells of the same shape and size. The MD simulations were carried out in the NVT ensemble using a 2 fs time step and a Nosé Hoover thermostat with mass corresponding to an 80 fs oscillation period. The system was first equilibrated at 1200 K for 2 ps, and a snapshot configuration was afterwards sampled every 200 fs during the next 2 ps. In order to monitor the chemical effects affecting the QM region size region necessary in the presence of impurity atoms, we created an auxiliary system for each frame, in which the central atom was substituted back with a Nickel atom. This allows a comparison between the total charge density and magnetisation fields between the two systems.
We used Bader analysis [36, 37] to determine the atom-resolved charge and magnetisation. Our results are presented in figures 5(a) and (b). The behaviour is similar for the Re and W impurity species: the first shell of neighbours gains a small amount of charge (the net charge loss of the Rhenium atom is about 0.05 e for rhenium and 0.1 e for tungsten, shared between 12 neighbouring atoms). The second nearest neighbours lose charge to compensate for this effect, and atoms further away are unchanged (i.e. any local charge fluctuations are compatible with the temperature-induced symmetry breaking). The magnetisation of the impurity atom is zero in both cases, and the magnetic moment for the neighbouring Nickel atoms is also considerably reduced. The local magnetisation is practically converged to the bulk value (0.63  ) by the second and third shells of neighbours for rhenium and tungsten impurities, respectively. This is consistent with the larger charge transfer effects observed in the tungsten system.
) by the second and third shells of neighbours for rhenium and tungsten impurities, respectively. This is consistent with the larger charge transfer effects observed in the tungsten system.

Figure 5. Locality of the effect of an impurity on surrounding Ni atoms. Charge (a) and magnetisation (b) difference with respect to an auxiliary pure Nickel system of same atomic positions, plotted as a function of the distance from the Re or W impurity. Force difference, absolute (c) and percentage (d), between the same systems as a function of the distance from the impurity atom.
Download figure:
Standard image High-resolution imageTo further probe the local effects of the impurity on the surrounding metal matrix atoms, we measure the average force difference between the defected structures and the corresponding auxiliary ones. When this difference vanishes, Nickel atoms are no longer affected by the presence of the close-by defect. Our results are presented in figures 5(c) and (d). The force differences are above the 0.1 eV  threshold only for the first neighbouring shell, and vanish at 6 Å from the defect. These first principles results indicate that the QM zone must contain at least the first shell of neighbours for Rhenium, and possibly also the second one in model systems containing Tungsten atoms.
threshold only for the first neighbouring shell, and vanish at 6 Å from the defect. These first principles results indicate that the QM zone must contain at least the first shell of neighbours for Rhenium, and possibly also the second one in model systems containing Tungsten atoms.
The standard test for determining the minimal size of the QM region involves a direct comparison between the DFT forces and those predicted by the interatomic potential, allowing to terminate the QM region as soon as the MM description is sufficiently accurate. Unfortunately, as mentioned above, no parametrisation has been made available extending this EAM potential to atomic species other than Nickel and Aluminium. An attempt to produce a ternary (Ni–Al–Re) interatomic potential based on the MEAM method for modelling processes at the  interface has been reported in [38]. However, this ternary potential is not yet fully reliable as it has been reported to overestimate the lattice parameters and to predict spurious Rhenium clustering in the γ phase not observed by extended x-ray absorption fine structure and atom probe tomography [7, 8] and later ruled out using DFT calculations [10]. Another ternary potential was constructed by Du et al [39] based on a previous model for generic fcc structures [40]. In the present work we have tested this potential against DFT forces and found large deviations when modelling substitutional rhenium impurities in γ. This is probably due to the non-transferability of the potential (otherwise accurate in describing properties of non-defected ordered phases) to the isolated impurity system in high temperature conditions of interest here.
interface has been reported in [38]. However, this ternary potential is not yet fully reliable as it has been reported to overestimate the lattice parameters and to predict spurious Rhenium clustering in the γ phase not observed by extended x-ray absorption fine structure and atom probe tomography [7, 8] and later ruled out using DFT calculations [10]. Another ternary potential was constructed by Du et al [39] based on a previous model for generic fcc structures [40]. In the present work we have tested this potential against DFT forces and found large deviations when modelling substitutional rhenium impurities in γ. This is probably due to the non-transferability of the potential (otherwise accurate in describing properties of non-defected ordered phases) to the isolated impurity system in high temperature conditions of interest here.
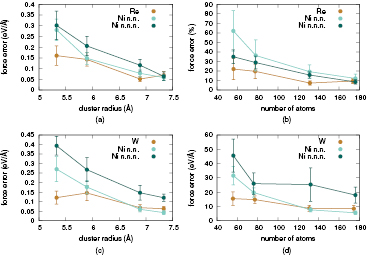
Once the size of the QM region containing the atomic forces that will be used in a QM/MM dynamical simulation is determined, the next step is to determine the thickness of the surrounding buffer region required to effectively decouple it from spurious surface effects. The two combined requirements will give the size (radius) of the overall cluster used in QM computations. We carried out convergence tests using clusters of different radii centred on the impurity, and the results are presented in figure 6. These reveal that forces on impurity atoms accurately converge to their reference periodic DFT values for a radius values larger than ∼6.5 Å. Converging the QM forces on all QM region atoms requires a ∼7.5 Å radius (∼180 atoms) cluster, while a 6.8 Å (∼130 atoms) cluster calculations already yields forces accurate enough to match the overall very good accuracy achieved by the interatomic potential in the surrounding (MM) region.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Convergence with respect to the cluster radius of the quantum forces on an impurity atom for Re ((a), (b)), W ((c), (d)), and the first two shells of neighbouring Ni atoms.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
We have presented detailed comparisons between the result of embedded atom model and density functional theory-based approaches to modelling defect systems in Ni and  (
( ) systems. While the agreement between the classical and the QM description is generally good for zero temperature properties, and the potential can be fitted to exactly reproduce the reference QM intrinsic stacking fault energy, we find that the EAM remains unable to correctly reproduce the interatomic forces of the QM model when the crystal is perturbed beyond the quasi-harmonic regime or in the presence of defects, e.g. in configurations containing vacancies and/or dislocations at the high temperatures relevant for Ni superalloy applications. The discrepancies between atomic forces produced by the two models are, however, localised close to the examined defects, and vanish a few Angstroms away where the non-defected bulk-like behaviour is recovered in the physical systems. This makes the defect systems considered here ideal candidates for QM/MM multiscale simulation approaches. Finally, We find that this 'fast defect-screening character' of the γ matrix extends to Re and W impurities for which a suitably accurate EAM potential is currently not available.
) systems. While the agreement between the classical and the QM description is generally good for zero temperature properties, and the potential can be fitted to exactly reproduce the reference QM intrinsic stacking fault energy, we find that the EAM remains unable to correctly reproduce the interatomic forces of the QM model when the crystal is perturbed beyond the quasi-harmonic regime or in the presence of defects, e.g. in configurations containing vacancies and/or dislocations at the high temperatures relevant for Ni superalloy applications. The discrepancies between atomic forces produced by the two models are, however, localised close to the examined defects, and vanish a few Angstroms away where the non-defected bulk-like behaviour is recovered in the physical systems. This makes the defect systems considered here ideal candidates for QM/MM multiscale simulation approaches. Finally, We find that this 'fast defect-screening character' of the γ matrix extends to Re and W impurities for which a suitably accurate EAM potential is currently not available.
Acknowledgments
We acknowledge funding from the Thomas Young Centre-US Air Force Research Laboratory Collaboration funded by the European Office of Aerospace Research and Development, the Engineering and Physical Sciences Research Council (EPSRC, under grants EP/L014742/1 and EP/L027682/1) and the US Office of Naval Research Global. We acknowledge PRACE for awarding us access to resource Fermi based in Italy at Cineca and Juqueen based in Germany at Jülich Supercomputing Centre. An award of computer time was provided by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357. Additional computing facilities were provided by the Centre for Scientific Computing of the University of Warwick with support from the Science Research Investment Fund. The raw DFT data used to generate figures 1–6 has been deposited at http://wrap.warwick.ac.uk/id/eprint/78256. We would like to thank A Mottura and E Bitzek for useful discussions.