

Abstract
The selective modification of individual components in a biosensor array is challenging. To address this challenge, we present a generalizable approach to selectively modify and characterize individual gold surfaces in an array, in an in situ manner. This is achieved by taking advantage of the potential dependent adsorption/desorption of surface-modified organic molecules. Control of the applied potential of the individual sensors in an array where each acts as a working electrode provides differential derivatization of the sensor surfaces. To demonstrate this concept, two different self-assembled monolayer (SAM)-forming electrochemically addressable ω-ferrocenyl alkanethiols (C11) are chemisorbed onto independent but spatially adjacent gold electrodes. The ferrocene alkanethiol does not chemisorb onto the surface when the applied potential is cathodic relative to the adsorption potential and the electrode remains underivatized. However, applying potentials that are modestly positive relative to the adsorption potential leads to extensive coverage within 10 min. The resulting SAM remains in a stable state while held at potentials <200 mV above the adsorption potential. In this state, the chemisorbed SAM does not significantly desorb nor do new ferrocenylalkythiols adsorb. Using three set applied potentials provides for controlled submonolayer alkylthiol marker coverage of each independent gold electrode. These three applied potentials are dependent upon the specifics of the respective adsorbate. Characterization of the ferrocene-modified electrodes via cyclic voltammetry demonstrates that each specific ferrocene marker is exclusively adsorbed to the desired target electrode.
Export citation and abstract BibTeX RIS
1. Introduction
Multiplexed biosensing arrays are of great interest as they enable the rapid and selective detection of two or more analytes in a complex mixture. Controlling the functionalization of the individual sensor surfaces of the array is however complex, and when achieved is generally performed in a serial fashion using additional apparatus [1]. Techniques used to modify one sensor in the presence of many others include microcontact printing [2, 3], dip-pen nanolithography [4] and modified ink-jet printing [5, 6]. Here we present a technique to functionalize the individual components in an array of gold electrodes with different functionalities by using selective potential-assisted electrochemical deposition and inhibition of deposition. Two different electroactive ferrocene markers (Fc–C11–SH and Fc–CO–C11–SH) attached at the ω-position to a C11-alkyl thiol are used to demonstrate this concept.
The adsorption of alkanethiols on single crystal and polycrystalline gold have been shown to be potential-dependent [7–11]. A modest positive potential (e.g. >200 mV versus Ag/AgCl) can increase the rate of adsorption of alkanethiols on polycrystalline gold so that excellent coverage can be achieved within minutes [7, 12, 13]. Applying a potential during self-assembled monolayer (SAM) formation results in a well-defined SAM in terms of coverage. On the other hand cathodic potentials (<−200 mV versus Ag/AgCl) slows the rate of chemisorption of alkythiols on polycrystalline gold and indeed at potentials <−600 to <−1100 mV (depending on chain length [14] and crystallographic orientation [15]), chemisorbed alkythiols are reductively desorbed [11, 16–18]. The reductive desorption reaction is described in equation (1)
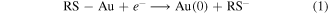
and the oxidative adsorption reaction in equation (2)
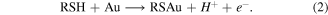
The electrochemical removal of a functional monolayer followed by full coverage selective adsorption of a second molecule has been demonstrated [19, 20]. Collman et al [20] functionalized two adjacent gold electrodes with the same adsorbate. Subsequent reductive desorption at one electrode removed the chemisorbed thiol. Exposure to a second adsorbate resulted in derivatizion of only the electrochemically cleaned electrode, as the other electrode remained unchanged under the experimental conditions. This approach uses the selective reductive desorption of an alkythiol SAM to allow for introduction of a second adsorbate. This process requires complete monolayer functionalization at each step, as otherwise cross-functionalization and binary SAMs will result.
In some applications, a partially coated sensor surface is desired [21–23]. Peterson et al [24] have shown that surface-tethered single stranded DNA, when in a low surface density regime, is desired as probe hybridization proceeds with relatively fast kinetics. In comparison, high probe surface leads to a decrease in both hybridization efficiency and relatively slower kinetics. Many other studies have shown that sub-monolayer coverage of tethered DNA probes leads to optimal hybridization efficiencies for DNA binding on gold [25, 26], on gold nanowires [27], or for protein binding [28]. Nagai et al [23], using microcantilever sensors, demonstrated that maximal surface stress changes between single stranded oligonucleotides and hybridized probes are achieved at a probe density of ca.  (i.e. 30% surface coverage) in the absence of other adsorbates. There is however, to our knowledge, no potential-assisted method described in the literature which results in the selective in situ modification of multiple electrodes each with submonolayer coverage of the probe molecules.
(i.e. 30% surface coverage) in the absence of other adsorbates. There is however, to our knowledge, no potential-assisted method described in the literature which results in the selective in situ modification of multiple electrodes each with submonolayer coverage of the probe molecules.
A method is presented here which produces submonolayer adsorbate coverage on two spatially proximal gold electrodes in situ each with a different electroactive ferrocene alkanethiol. This selectivity is achieved by controlling the applied potential of the gold electrodes in the presence of the two different adsorbates. Three distinct potentials are required: Eads (adsorption), Edes (desorption) and Ehold (holding). The potential Eads promotes the chemisorption of an alkanethiol on the gold surface. The potential Edes maintains the electrode in the reductive desorption state and inhibits further alkanethiol adsorption from occurring. Lastly, Ehold is an intermediate potential that is neither reductive enough to desorb an already formed monolayer nor sufficiently anodic to promote the adsorption of new alkanethiols via the electrochemically promoted exchange reaction.
It is important to note that Ma et al [7] and Paik et al [12] have demonstrated that reproducible SAM formation can occur over time scales of a few minutes by using potential-assisted deposition. This technique enables the multiple electrode experiments described here. In the absence of potential-assisted adsorption of the electroactive ferrocenylalkylthiols, the rates of both adsorption and desorption are highly dependent upon the value of the open circuit potential. The rest is invariably poor reproducibility of the extent of coverage. The long incubation times necessary for SAM formation without potential assistance will also lead to considerable exchange of one alkylthiol derivatize for another.
The functionalization technique described here yields submonolayer coverage on a gold surface, in situ modification, and is scalable in that any number of electrode sensor surfaces can be functionalized with different molecules with the use of a multichannel potentiostat.
2. Experimental section
2.1. Materials
11-(ferrocenyl)undecanethiol (Sigma Aldrich, USA) was dissolved in absolute ethanol to a concentration of 1 mM. 11-(ferrocenyl)-carbonyl undecanethiol was synthesized as per literature methods (see Supporting Information). Alkanethiol SAM formation involves the use of a 1:1 solution of 11-(ferrocenyl)undecanethiol (Fc–C11–SH) and 11-(ferrocenyl)-carbonyl undecanethiol (Fc–CO–C11–SH) in 100 mM LiClO4 (absolute ethanol). Electrochemical cleaning was performed in a 50 mM KClO4 solution. All other cyclic voltammograms were recorded in 100 mM NaClO4. The following reagents were purchased and used without further purification: potassium perchlorate (>99%, Sigma Aldrich, USA), lithium perchlorate (>95%, Sigma Aldrich, USA), sodium perchlorate (>98%, Sigma Aldrich, USA) and absolute ethanol (>99.8%, Sigma Aldrich, USA).
2.2. Gold surface preparation
Silicon wafers were diced into small (0.5 × 1 cm) pieces and solvent-cleaned with acetone, isopropanol and methanol before sequential thermal evaporation of Ti and Au was performed. A 2 nm Ti adhesion layer was evaporated at a constant rate of 0.9 Å s–1 followed by a 100 nm thick Au layer at a constant rate of 1 Å s–1 (pressure <1 × 10−6 mBar, room temperature). Samples were stored under ambient conditions until needed. To define the electrochemical active area of the exposed gold in solution, a thin layer of Eccobond 286 (Emerson & Cuming, USA) is applied to the base of the gold surface leaving an exposed macroscopic area of ∼1.0 mm2.
2.3. Electrochemical cleaning
All samples were electrochemically cleaned prior to each experiment. Samples were cycled between −0.8 and 1.4 V (versus Ag/AgCl (sat. KCl)) in 50 mM KClO4 at a scan rate of 20 mV s–1 until a stable voltammogram was obtained. The gold electrode served as the working electrode, a platinum wire (1 mm diameter, Alfa Aesar, USA) as the counter electrode, a standard Ag/AgCl (sat. KCl) reference electrode (BASi, USA) as the reference. Electrochemical measurements were performed using a CHI 1030A (CH Instruments, USA) potentiostat. For all experiments two gold electrodes are recorded simultaneously by using each electrode as a separate working electrode sharing one counter and one reference electrode (Bipotentiostat).
2.4. Measurement of ferrocene coverage
The area of the gold electrode was determined from the magnitude of the gold oxide reductive stripping peak in the cyclic voltamogram performed from −0.8 to 1.4 V. The quantity of surface oxide formed during the anodic excursion is determined by integrating the gold oxide reduction peak in the cathodic scan, Qred. Assuming a standard charge value of 400 μC cm–2 for polycrystalline gold [29], the microscopic surface area can be calculated by:
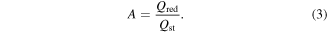
The geometric surface area is determined for each gold electrode via optical methods. The ratio of the electrochemical surface area to the geometric area yields roughness factor values of 1.1–1.6 for the electrodes used in this study.
The electrochemical area determined using equation (3) is then used to determine the ferrocenylalkythiol coverage by integrating the ferrocene-associated oxidation peak in the CV obtained in 100 mM NaClO4. The surface coverage of the electroactive ferrocene group is given by:
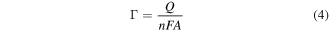
with  charge obtained by integrating the current peak, n is the number of electrons transferred (n = 1 for the ferrocene couple), F the Faraday constant, and A is the electrochemical surface area of the gold electrode [30]. To determine the fractional surface coverage of the ferrocene-modified alkanethiol on the gold electrode, the theoretical maximal coverage of 4.5 × 10−10 mol cm–2 (equivalent to 2.7 × 1014 molecules cm–2) is divided by the calculated coverage [31]. The theoretical maximal coverage is calculated by assuming a spherical ferrrocene head group adopting a hexagonally close-packed geometry with a diameter of 6.6 Å [32].
charge obtained by integrating the current peak, n is the number of electrons transferred (n = 1 for the ferrocene couple), F the Faraday constant, and A is the electrochemical surface area of the gold electrode [30]. To determine the fractional surface coverage of the ferrocene-modified alkanethiol on the gold electrode, the theoretical maximal coverage of 4.5 × 10−10 mol cm–2 (equivalent to 2.7 × 1014 molecules cm–2) is divided by the calculated coverage [31]. The theoretical maximal coverage is calculated by assuming a spherical ferrrocene head group adopting a hexagonally close-packed geometry with a diameter of 6.6 Å [32].
2.5. SAM formation
The gold surfaces were selectively modified with the two different ferrocene derivatives by varying the applied potential as described in detailed below. Modification was performed in either 1 mM Fc–CO–C11–SH or Fc–C11–SH (1:1) with 100 mM NaClO4 solutions for 10 min at a constant applied potential. After electrodeposition, the surfaces were rinsed with ethanol and Milli-Q water before a CV was recorded in 100 mM NaClO4 from 0.2 to 0.8 V (versus Ag/AgCl (sat. KCl)).
3. Results and discussion
3.1. Determination of applied potentials required for electrode derivitization
The first step toward a successful potential-assisted electrode modification is to determine the ferrocenylalkythiol coverage as a function of the applied potential. Prior to each experiment, the evaporated gold electrodes are electrochemically cleaned before exposing them to the ferrocene derivatize [22]. Measurement of the extent of potential-dependent adsorption involved exposure of the gold electrode to a 1 mM Fc C11–SH/100 mM LiClO4 solution while applying a series of cathodic potentials (versus Ag/AgCl (sat. KCl)) ranging from −1.4 to −0.9 V, for 10 min at each potential. After each incubation step, the sample was rinsed with ethanol and Milli-Q water before a CV was recorded in 100 mM NaClO4 at 20 mV s–1. The resulting ferrocene oxidation peak was integrated to calculate the ferrocene surface coverage (equation (4)). Coverage as a function of applied potential (figure 1(A)) reveals that no apparent ferrocene derivative adsorption occurs at potentials <−0.9 V. Potentials of <−0.9 V (red area) thus serve to maintain a gold electrode free of chemisorbed ferrocenylalkanethiol.
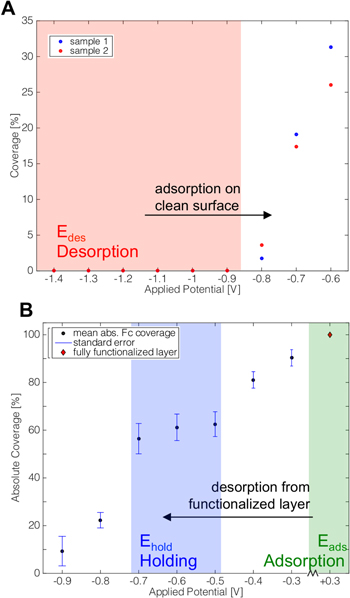
Figure 1. Coverage dependent on the applied potential to the gold electrode. The electrode was exposed to 1 mM Fc–C11–SH, 100 mM NaClO4 for 10 min at designated potentials. After each incubation at a specific potential, the electrode was rinsed and a CV was recorded in 100 mM NaClO4 at 20 mV s–1 (versus Ag/AgCl (sat. KCl)). The area of the ferricinium peak is integrated to calculate the ferrocene surface coverage, using equation (4). (A) shows the adsorption of Fc–C11–SH onto a clean electrode, as a function of increasing applied potentials. In (B), a fully functionalized electrode is systematically desorb by applying a step-wise decreasing potential.
Download figure:
Standard image High-resolution imageOn the other hand, application of a potential of 0.3 V (versus Ag/AgCl (sat. KCl)) to a clean gold electrode results in Fc–C11–SH coverage of >50% after 10 min incubation.
Ehold, the potential range over which the ferrocenylalkylthiol SAMs are stable in regard to both desorption and thiol-for-thiol exchange is important. To measure the desorption potential of the ferrocene alkanethiol, the modified surface was exposed to the ferrocenyl solution (1 mM) while changing the potential from −0.3 to −0.8 V. The corresponding ferrocene coverage is shown in figure 1(B). To compare the coverage achieved under different experimental regimes, the coverage is expressed relative to the full coverage case (100%). Starting at −0.3 V and proceeding cathodically, the ferrocene surface coverage decreases indicating that goldthiol bond is destabilized and ferrocenylalkylhiol desorbs from the surface to increasing extent. The holding potential, described as a potential where an existing SAM is stable towards additional alkythiol adsorption or desorption for the time necessary to modify another electrode (5 min) is determined to be >−0.6 V.
This potential-dependent ferrocene coverage establishes the three operational potentials of interest to this study: Eads, Edes, and Ehold. Eads (>0.3 V) is the potential at which adsorption of the two ferrocenyl C11-alkanethiols occurs. Edes is the potential at which alkanethiol adsorption onto a clean gold electrode is not measurable (<−0.9 V). Ehold (−0.6 V) is at the potential at which a pre-formed ferrocenyl C11 alkylthiol SAM is held to prevent both further thiol desorption, adsorption and thiol-for-thiol exchange. These potential values provide operational potentials at which one can selectively functionalize two different alkylthiol ferrocene markers onto different but spatially proximal gold electrodes.
3.2. Potential-assisted modification protocol
These three potential regimes allow one to control the functionalization of two proximal electrodes with two different species. Two different ferrocene derivatives, Fc–CO–C11–SH and Fc–C11–SH demonstrate the effectiveness of the selective functionalization protocol based on the desorption and adsorption. The two probes have the same alkyl chain length (C11) and the same chain terminus (thiol), yet one has an added carbonyl group to the ferrocene moiety. This carbonyl shifts the corresponding redox potential anodically by 250 mV with respect to the Fc–C11–SH [33–36]. The Fc–C11–SH exhibits an oxidation peak at Ep = 0.34 V (versus Ag/AgCl), whereas the Fc–CO–C11–SH exhibits the redox peak at Ep = 0.59 V (versus Ag/AgCl). If a mixed SAM is formed on one electrode, two distinct peaks are observed.
These two different ferrocenes are used to experimentally demonstrate the two-step protocol of selective functionalization of the electrodes shown in figure 2. In the first step, both gold electrodes are simultaneously exposed to the same probe by exposure to 1 mM Fc–CO–C11–SH (100 mM LiClO4) for 10 min. During the functionalization process, electrode 1 is held at 0.3 V (Eads), whereas electrode 2 is held at −1.4 V (Edes). After incubation, the electrodes are rinsed with ethanol and Milli-Q water and placed in aqueous 100 mM NaClO4. A cyclic voltammogram verifies the extent of derivatization of each electrode. Each electrode is rinsed with ethanol at the completion of the functionalization process. In a second step, each electrode is placed in the same solution containing the second adsorbate (2 ml, 1 mM Fc–C11–SH, 100 mM LiClO4) and held for 10 min at specific potentials. During the functionalization process, electrode 1 is held at −0.6 V (Ehold) while electrode 2 is held at +0.3 V (Eads) to modify it with the second ferrocene probe.
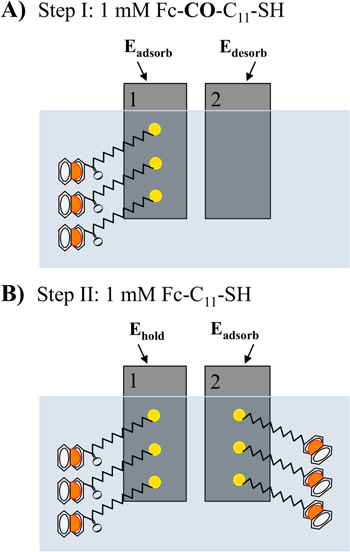
Figure 2. Scheme showing the two-step selective in situ surface modification on two gold electrodes. (A) Both electrodes are immersed in 1 mM Fc–CO–C11–SH, 100 mM LiClO4 for 10 min. Different potentials are applied to the two electrodes, so that the alkanethiol is only deposited onto electrode 1. (B) The solution is changed to the second adsorbate, 1 mM Fc–C11–SH, 100 mM LiClO4 and the two electrodes are immersed for 10 min while the applied potentials targets the Fc–C11–SH to the second electrode exclusively.
Download figure:
Standard image High-resolution imageThe resulting cyclic voltammogram recorded simultaneously on each gold electrode after the second modification step is shown in figure 3. Electrode 1 (blue) is modified with Fc–CO–C11–SH and Electrode 2 (red) is modified with Fc–C11–SH. Oxidation peaks at Ep = 0.59 V (Fc–CO–C11–SH) and Ep = 0.34 V (Fc–C11–SH) provide for the determination of the respective ferrocene coverage. For the Fc–CO–C11–SH derivitazed electrode 1, the overall surface coverage is 43.4 ± 0.4%, corresponding to an alkanethiol density of 1.951 × 10−10 ± 0.016 × 10−10 mol cm–2. On the other hand, electrode 2 derivitazed with Fc–C11–SH has a coverage of 32.7 ± 0.3% (density: 1.474 × 10−10 ± 0.015 × 10−10 mol cm–2).

Figure 3. Cyclic voltammograms recorded simultaneously for each electrode in 100 mM NaClO4 (versus Ag/AgCl (sat. KCl) at 20 mV s–1. The peak at Ep = 0.34 V corresponds to the Fc–C11 peak and the peak at Ep = 0.59 V corresponds to the Fc–CO–C11.
Download figure:
Standard image High-resolution image3.3. Cross-coverage
Measurable quantities of Fc–C11 are observed on the Fc–CO–C11-derivatized electrode (figure 4). The latter peak results from the competitive deposition of the undesired alkylferrocene in the presence of desired adsorbate. Quantification of the cross-coverage establishes that electrode 1 (modified with Fc–CO–C11–SH) has a coverage of Fc–C11–SH of 1.1 ± 0.2% (surface density: 4.727 × 10−12 ± 0.748 × 10−12 mol cm–2) and electrode 2 (modified with Fc–C11–SH) shows a cross-coverage of 2.4 ± 0.2% (surface density: 1.071 × 10−11 ± 0.066 × 10−11 mol cm–2) of Fc–CO–C11–SH. The coverage and cross-coverage values associated with three independent experiments are summarized in figure 4. The previously analyzed experiment is shown in C. A and B are carried out using the same protocol. The variance in the coverage likely arises from the polycrystalline nature of the gold electrodes and the relatively short incubation times (10 min) of each step. The effect of roughness has not been correlated with the coverage values. The histogram shows that electrode 1 undergoes high Fc–CO–C11–SH coverage whereas electrode 2 shows high Fc–C11–SH modification. The cross-coverage at each electrode is less than 4% in all experiments.

{kind=link}
{kind=link}
{kind=link}
Figure 4. Cross-coverage for three different modification regimes (A–C). Electrode 1 is modified with Fc–CO–C11–SH (+0.3 V) while a negative potential is applied to electrode 2. In the second step, electrode 2 is modified with Fc–C11–SH (+0.3 V) while electrode 1 is held in the protective state (−0.6 V). The final surface coverage is plotted. A cross-coverage value of less than 4% is observed in all experiments.
Download figure:
Standard image High-resolution image{kind=link}
Differentiation has to be made between the two cross-coverage values, as they have different origins. In case C, the cross-coverage value of 2.4% of Fc–CO–C11–SH on electrode 2 results from deposition on a clean electrode held at −1.4 V during the first step. The second cross-coverage value is lesser (with a value of 1.1% of Fc–C11–SH on electrode 1) and results from the second modification step while the partially modified surface is being held at  . Adsorption on a clean non-functionalized electrode can cause complications if additional surfaces are being modified, as each modification step will increase the degree of undesired adsorption. Due to the in situ electrochemical system, electrochemical cleaning can be performed on a set of non-functionalized arrays after every nth modification. This step will ensure that minimal contamination on new blocked surfaces occurs. On the other hand, undesired adsorption that occurs at Ehold is not likely to increase significantly during further modification steps, as the adsorption kinetics are slower at higher coverage.
. Adsorption on a clean non-functionalized electrode can cause complications if additional surfaces are being modified, as each modification step will increase the degree of undesired adsorption. Due to the in situ electrochemical system, electrochemical cleaning can be performed on a set of non-functionalized arrays after every nth modification. This step will ensure that minimal contamination on new blocked surfaces occurs. On the other hand, undesired adsorption that occurs at Ehold is not likely to increase significantly during further modification steps, as the adsorption kinetics are slower at higher coverage.
4. Conclusions
The work presented here describes an in situ method to selectively functionalize gold electrodes with different probe adsorbates at submonolayer coverages. Application of specific potentials to the gold electrodes during SAM formation demonstrates that the extent of deposition of electrochemically active molecules can be controlled. All modification processes are under potential control and exhibit reproducible SAM formation within the 10 min process. In this study, two spatially proximate gold electrodes were functionalized, each with an electrochemically distinct alkylferrocene adsorbate: Fc–CO–C11–SH and Fc–C11–SH. Potential-assisted SAM formation is achieved by using three different operating potentials: Eads (+0.3 V) held at slightly cathodic potentials enhances the alkanethiol adsorption rate and excellent coverage is achieved within 10 min [7, 8]; Edes (−1.4 V) inhibits adsorption by maintaining the electrode in a reductive (alkylthiol) desorption state; Ehold (−0.6 V) maintains a modified ferrocene alkanethiol electrode in a stable state by hindering the adsorption of new alkanethiols vie electrochemically promoted exchange reactions. Ehold is critical to this work as it is also the potential at which the functionalization of SAMs with submonolayer coverage can be effected.
Cyclic voltammogram measurements in 100 mM NaClO4 show two distinct electrochemical peaks indicating the successful selective modification on the two spatially proximal gold electrodes. From these measurements, cross-coverage values of 4% of the full coverage are determined. The presented potential values are optimized for probes that feature a thiol group for binding to surfaces. Any thiolated probe can be used in combination with this technique. A re-evaluation of the potential values for the three states might be necessary if extending the modification principle to other linekers.
Previous reports have only shown potential-assisted functionalization relying on a full SAM coverage. For sensors modifying oligonucleotides however, full coverage is not desired but rather submonolayer coverage [21–28]. The method presented here can selectively modify electrodes at submonolayer coverage by implementing a hold potential (Ehold) that maintains a functionalized electrode in its current state and prevents further adsorption or desorption within the time frame of the experiments. The technique can be applied in situ and is scalable so that large arrays of gold electrode sensors can be modified and can thus be used for a wide variety of metal-based sensors.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Regroupement quebecois sur les materiaux de pointe (RQMP), the Centre for Self-Assembled Chemical Structures (CSACS). Ann-Lauriene Haag thanks the Swiss National Science Foundation (SNF) for a DOC.Mobility Fellowship.